

Supplemental Digital Content is available in the text
Abstract
Temozolomide (TMZ) is an oral alkylating agent with established effects on the central nervous system of glioblastoma (GBM) patients. Clinical trials have demonstrated a significant impact on overall survival (OS) with TMZ. Ever since, several TMZ regimens have been designed to improve treatment efficacy by increasing the cumulative dose per cycle. We report a meta-analysis to systematically evaluate different treatment schedules of TMZ in GBM patients.
All searches that were conducted in the Cochrane library, Science Direct, and PubMed Databases, and 3 randomized controlled trials (1141 patients) were included. OS and progression-free survival (PFS) were the primary outcomes to be pooled.
Unexpectedly, this analysis did not reveal any OS or PFS advantage for the high cumulative dose (HCD) regimen compared with the normal cumulative dose regimen (1141 total patients; hazard ratio [HR] 1.07, 95% CI 0.94–1.22, P = 0.31). Then after analyzing the characteristics of the results from each trial, we found that the regimen with a higher peak concentration during a short-term period (daily doses ≥150 mg/m2/d within ≤7 days/cycle) always had a more superior clinical benefit. So we generated a new pooled HR of 1.10 with a 95% CI of 0.96–1.25 (P = 0.17), which prefers the high peak concentration schedule even without a significant difference. The adverse outcome also indicates a significant increased risk of leukopenia (risk ratio 1.59, 95% CI 1.03–2.46, P = 0.04) among the HCD group.
Our study suggests that increasing the cumulative dose per cycle is not an ideal way to improve the efficacy of TMZ, and it will lead to increased risk for leukopenia. Future trials should be designed to examine schedules of higher peak concentration rather than the cumulative dose per cycle.
INTRODUCTION
As the most common primary malignant brain tumor, even in modern times of high-precision brain surgery and irradiation, glioblastoma (GBM) is always associated with a poor prognosis.
TMZ is an orally administered, DNA alkylating agent with excellent effects on the central nervous system. Clinical trials have demonstrated a significant impact on overall survival (OS) with TMZ.1
The cytotoxic effect of temozolomide (TMZ) is mediated primarily via methylation at the O6-position of guanine, and the overexpression of O6-methylguanine-DNA methyltransferase (MGMT) is the predominant mechanism of tumor resistance to TMZ. A few publications have proven that MGMT status can predict the efficacy of chemotherapy.2
Although MGMT can repair TMZ-mediated DNA damage, MGMT is consumed during this process.3 With the idea that MGMT is potentially depleted from tumor cells as a result of the overwhelmed cells’ inability to synthesize MGMT, several TMZ treatment schedules have been designed to verify whether they can overcome the resistance to TMZ. The concept of MGMT depletion was validated in peripheral blood by Tolcher et al4 during treatment with TMZ for 7 consecutive days every 14 days (2100 mg/m2/circle) or for 21 consecutive days every 28 days (2100 mg/m2/circle). The cumulative dose was nearly twice as much as the standard dosing of 200 mg/m2 1–5/28 (1000 mg/m2/circle) and the metronomic schedule with 50 mg/m2/d (1400 mg/m2/circle).4
However, it is still unclear whether a regimen with a higher cumulative TMZ dose per cycle can deplete MGMT in tumors and overcome MGMT-mediated resistance. Additionally, the efficacy and safety of such a TMZ treatment plan is the subject of much controversy.
This study is a meta-analysis to systematically evaluate the 7 to 14 and 21 to 28 day schedule (2100 mg/m2/circle) versus the standard or metronomic schedule (1000 mg/m2/circle) in GBM patients. The objective of this study was to verify whether a double cumulative dose per cycle can achieve a better survival benefit without an increasing incidence and severity of toxicity profile.
METHODS
Included
Type of Studies
All studies included are randomized controlled trial (RCTs).
Type of Participants
All patients had histologically confirmed GBM.
Type of Interventions
Schedules with different cumulative doses (Figure 1) of TMZ per cycle after conventional radiation planning are compared.
FIGURE 1.
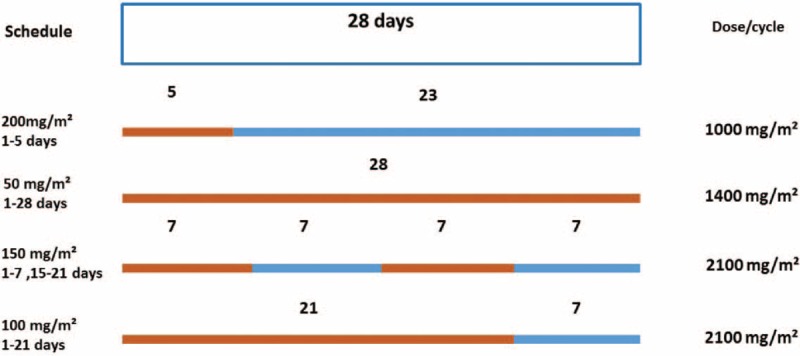
Schematic outline of various TMZ dosing regimens. Orange, TMZ; blue, no drug application. TMZ = temozolomide.
Type of Outcomes
Primary Outcome
OS was defined as the time from diagnosis or randomization to the date of death or last follow up.
Secondary Outcomes
Progression-free survival (PFS): the time from diagnosis or randomization to the date of progression defined by the Macdonald criteria,5 death, or the last follow-up without progression.
Toxicity was defined according to the National Cancer Institute Common Terminology Criteria.
Literature Search
All searchings were conducted carefully in Cochrane library, Science Direct, and PubMed databases were searched by 2 independent investigators with the following search strings: “glioma,” “glioblastoma,” “malignant glioma,” and “high grade glioma”; “dose-dense,” “treatment schedule,” and “regimen”; “temozolomide,” and “Temodar.” The flow diagram of study selection is presented in Figure 2.
FIGURE 2.
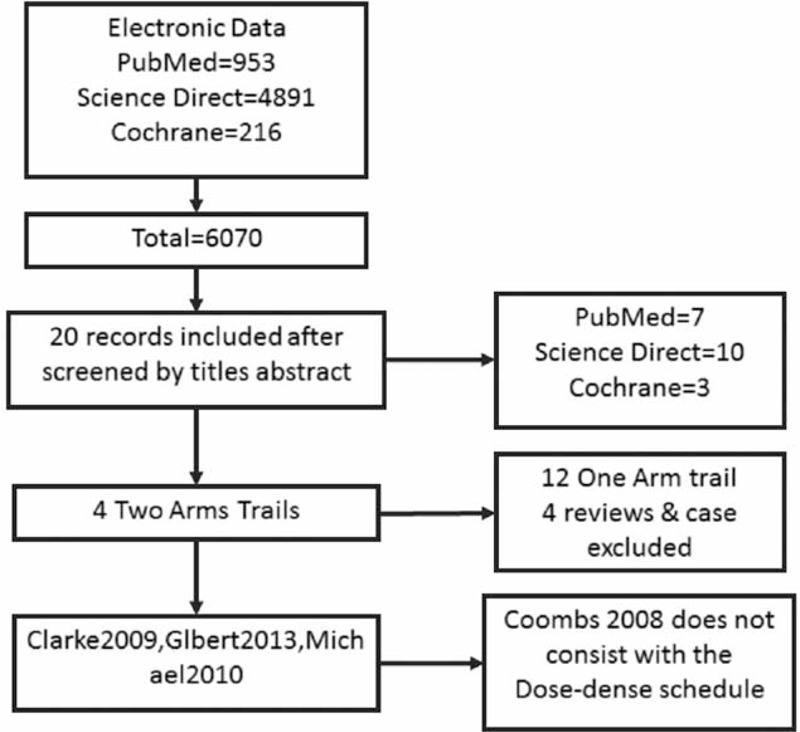
Flow diagram of study selection.
Study Selection and Data Extraction
All studies included met the following criteria:
Must be random, prospective clinical RCTs.
No language restriction was applied, and the nationality and race of research subjects were not restricted.
Must compare 2 different TMZ schedules. They must include the TMZ regimen with a lower cumulative dose per cycle for the control group and the dose of the comparison group(s) per cycle must be higher than the control group.
The primary endpoint was OS, and secondary endpoints were time to tumor progression and overall response rate (ORR).
Studies containing the following criteria have been excluded:
The study did not meet the inclusion criteria.
The research failed to provide the required information, such as the total number of patients, the median OS time, ORR, and 1-year survival rate.
The study had poor quality, serious bias, and too little literature information.
Assessing the Risk of Bias in the Eligible Studies
The risk of bias was assessed at time-to-event outcome level by 2 independent investigators (H.S. and G.L.) using the domain-based Cochrane Collaboration's tool.6 Disagreements were resolved through consensus.
Statistical Analysis
The hazard ratios (HRs) for OS and PFS were statistically pooled to evaluate the efficacy, with a value <1 favoring the regimen with a higher cumulative dose (HCD) per cycle. If the HR was not given directly, then the value was calculated based on the Kaplan–Meier survival curves.
For the safety analysis, the risk ratio (RR) was utilized to compare normal cumulative dose (NCD) and HCD, with a value>1 indicating an increased risk of adverse events in the HCD group.
The inverse-variance (HR pooling) and the Mantel–Haenszel (RR pooling) methods were utilized to combine the data from each study, and either the fixed-effect model or the random-effect model was applied based on the heterogeneity of the included studies.
Sensitivity analyses were conducted by considering the risk of bias of the studies and variables that were not closely related to our topic.
Publication bias was described by the funnel plots and by analytic methods (eg, Egger test).7
All of the analyses were performed by Review Manager V5.3 (the Cochrane Collaboration, Software Update, Oxford, UK).
RESULTS
Characteristics of the Studies
According to the inclusion criteria, we identified 3 RCTs that compared the NCD and the HCD groups with GBM (2 phase II RCTs: Clarke et al8 and Brada et al9; 1 phase III RCT: Gilbert et al)10 and comprised 1141 patients. The characteristics of these RCTs are summarized in Table 1.
TABLE 1.
Characteristics of All Identified Studies
Among the 3 RCTs, the schedule of the NCD group is 50 mg/m2 1 to 28 days (1400 mg/m2) compared with the HCD with 150 mg/m2 1 to 7 days and 15 to 21 days (2100 mg/m2) in the study by Clarke et al,8 and TMZ 200 mg/m2 1 to 5 days (1000 mg/m2) compared with TMZ 100 mg/m2 1 to 21 days (2100 mg/m2) in the Brada et al9 and Gilbert et al10 trials.
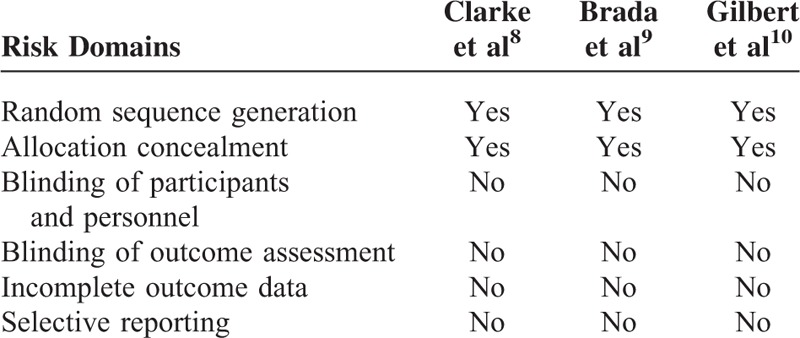
The risk of bias in the included studies was assessed using the standard Cochrane Collaboration's tool (Table 2). All 3 RCTs with straight random principle were suggested to have low risk despite no performance of double blinding, which is considered to not bias the assessment of OS and PFS. The individual prognostic factors (eg, age, sex, performance status, extent of surgery) were all well balanced within these studies.
TABLE 2.
Risk of Bias in the Included Studies Was Assessed Using the Standard Cochrane Collaboration's Tool
Overall Survival
There was no heterogeneity (χ2 = 3.14, P = 0.21, I2 = 36 %) among the 3 RCTs (Figure 3A); therefore, the HR and 95% CI were calculated by the fixed effects model (1141 total patients, HR 1.07, 95% CI 0.94–1.22, P = 0.31). This meta-analysis does not suggest a significant reduction in the risk of death in the HCD regimen. On the contrary, the standard regimen with higher peak TMZ concentration during short-term treatment (daily doses ≥150 mg/m2/d within ≤7 days/cycle) seemed to be more effective according to the statistic. To verify whether the peak drug concentration is the main reason for improvement in treatment effect, we swapped the control and the experimental group of the study by Clarke et al8 to keep consistency with the HR (low peak drug concentration vs high peak drug concentration) of the studies by Gilbert et al10 and Brada et al9 before we calculated a new pooled HR (supplementary Table 1, http://links.lww.com/MD/A271). The new analysis yielded a new pooled HR of 1.10 with a 95% CI of 0.96–1.25 (P = 0.17) (HR with value >1 prefer the regimen with high peak drug concentration) (Figure 3B). Unfortunately, there is no conclusive evidence for the superiority of the peak TMZ concentration regimens.
FIGURE 3.
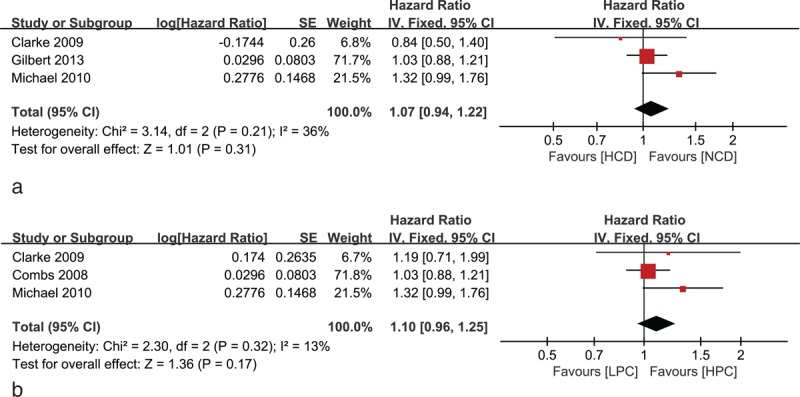
Forest plot of comparison of (A) HCD regimen versus NCD regimen: the primary analysis for OS; (B) HPC regimen, LPC regimen: the analysis for OS. HCD = high cumulative dose, HPC = higher peak concentration, LPC = lower peak concentration, NCD = normal cumulative dose, OS = overall survival.
Progression-Free Survival
Two studies9,10 were included for the PFS analyses due to the lack of statistics in the study by Clarke et al8 (Figure 4). The HR was calculated using the random-effect model (1056 total patients, HR 1.08, 95% CI 0.69–1.69, P = 0.31). No therapeutic benefits on PFS were detected for the HCD regimen.
FIGURE 4.

Forest plot of comparison of HCD versus NCD: the primary analysis for PFS. HCD = high cumulative dose, NCD = normal cumulative dose, PFS = progression-free survival.
Adverse Outcomes
For the safety analysis, we directly extract the data from trails included in our study, and the RR was utilized to compare NCD and HCD, with a value>1 indicating an increased risk of adverse events in the HCD group. Safety data were collected among all these 3 studies and are demonstrated in Table 3.
TABLE 3.
Toxicity Comparison Between the HCD and NCD Groups
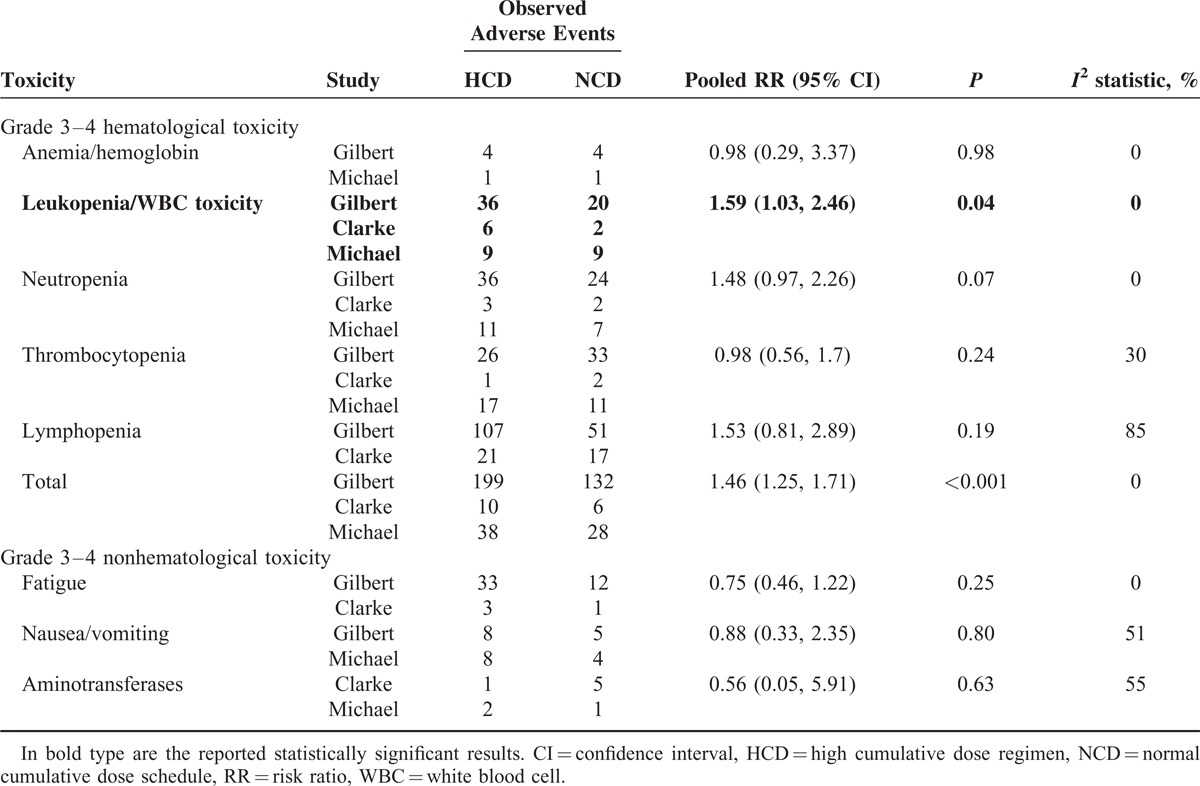
Chemotherapy-related hematological adverse events (HAEs) such as anemia/hemoglobin, leukopenia/white blood cell toxicity, neutropenia, and thrombocytopenia were the major safety concerns. The results did not indicate a significant increased risk of grade 3–4 (G3–4) HAEs among the patients with the HCD regimen except for leukopenia (RR 1.59, 95% CI 1.03–2.46, P = 0.04). Other nonhematological toxicity like fatigue, nausea/vomiting, and aminotransferases were similar between the groups (supplementary Figures 1 and 2, http://links.lww.com/MD/A271).
Therefore, our study demonstrates that the HCD regimen would lead to increased risk for leukopenia during the treatment period. No significant difference in nonhematological toxicities, such as fatigue/asthenia, nausea/vomiting, and aminotransferases, were observed.
DISCUSSION
The studies included in this meta-analysis are limited to RCTs for good homogeneity. Table 3 indicates that all of the studies had a low risk of bias. The primary outcome of these 3 RCTs indicated no therapeutic benefits for patients under the HCD regimen.
The primary analysis of all 3 RCTs has not indicated a significant difference between the 2 regimens. The studies by Gilbert et al10 and Michael et al9 demonstrated an unexpected result that the HCD group seemed to be inferior to the normal 5-day TMZ schedule. Only the study by Clarke et al8 indicated an improvement in OS with the HCD regimen. The discrepancy might be explained by the bias of the peak TMZ concentration among the regimens in each trial. After analyzing the characteristics of the treatment schedule among all 3 trials, there is a common tendency that higher peak drug concentration was always associated with a better clinical benefit.
We conducted another pooled HR analysis (Figure 3B), which demonstrates that improving the peak TMZ concentration rather than increasing cumulative dose per cycle would enhance the treatment effectiveness even without a statistically significant different. To date, only a few studies had been conducted to compare the efficacy between different TMZ regimens. More studies should be conducted to verify this hypothesis.
The adverse outcomes also demonstrate an increased risk of leukopenia among the HCD regimen group. As the leukopenia was always associated with a high risk of infection, our study indicated that the HCD regimen is not superior to the standard 5-day regimen at this point.
TMZ is among the most promising substances to achieve tumor control; few other chemotherapeutic remedies are able to cross the blood–brain barrier. Above all, as the one most promising treatment for GBM patients, TMZ is irreplaceable. Adjusting the treatment schedule is an ideal way to improve the therapeutic effect, although we do not conclude a robust significant difference between the 2 groups. More large randomized controlled clinical trials to evaluate different regimens are warranted.
In pharmacokinetic terms, it may be the real drug exposure achieved with a higher daily dose rather than higher cumulative dose that is the main determinant of cytotoxicity. The data of this meta-analysis has supported this theory, and future TMZ trials should test schedules using higher daily doses ≥150 mg/m2/d with intensification to achieve higher peak concentrations.
This study has a few limitations. First, the cumulative dose per cycle of each of the 2 compared groups in the Clarke et al8 study is slightly different than the other studies. Second, only the Gilbert et al10 study has demonstrated the treatment effectiveness with the MGMT promoter status. None of the clinical trials included in our study, however, provides data on MGMT level of patients treated by different TMZ regimens. So we cannot conduct a further analysis to verify whether the benefit is associated with MGMT depletion. More trials, which tested the MGMT status in peripheral blood, are needed to provide clinical evidence for this hypothesis.
CONCLUSION
This meta-analysis has demonstrated the inferiority of the HCD regimen to the standard 1000 mg/m2/cycle to some extent. Furthermore, the HCD regimen may be associated with increased risk for leukopenia for patients with Glioma. The results of this meta-analysis suggest that regimens aiming to prolong exposure to TMZ or to increase cumulative dose cannot replace the standard 5-day regimen in clinical practice. Future trials should be designed to examine schedules of higher peak concentration rather than the cumulative dose per cycle.
Footnotes
HS and SD contributed equally to this work.
This study was supported by the National Natural Science Foundation of China (81302385).
The authors have no conflicts of interest to disclose.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Website (www.md-journal.com).
REFERENCES
- 1.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352:987–996. [DOI] [PubMed] [Google Scholar]
- 2.Reifenberger G, Hentschel B, Felsberg J, et al. Predictive impact of MGMT promoter methylation in glioblastoma of the elderly. Int J Cancer 2012; 131:1342–1350. [DOI] [PubMed] [Google Scholar]
- 3.Christmann M, Nagel G, Horn S, et al. MGMT activity, promoter methylation and immunohistochemistry of pretreatment and recurrent malignant gliomas: a comparative study on astrocytoma and glioblastoma. Int J Cancer 2010; 127:2106–2118. [DOI] [PubMed] [Google Scholar]
- 4.Tolcher AW, Gerson SL, Denis L, et al. Marked inactivation of O6-alkylguanine-DNA alkyltransferase activity with protracted temozolomide schedules. Br J Cancer 2003; 88:1004–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Macdonald DR, Cascino TL, Schold SJ, et al. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol 1990; 8:1277–1280. [DOI] [PubMed] [Google Scholar]
- 6.Anonymous. Research and Markets: Cochrane Handbook for Systematic Reviews of Interventions, 2008 [Google Scholar]
- 7.Egger M, Davey SG, Schneider M, et al. Bias in meta-analysis detected by a simple, graphical test. BMJ 1997; 315:629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clarke JL, Iwamoto FM, Sul J, et al. Randomized phase II trial of chemoradiotherapy followed by either dose-dense or metronomic temozolomide for newly diagnosed glioblastoma. Journal of Clinical Oncology 2009; 27:3861–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brada M, Stenning S, Gabe R, et al. Temozolomide versus procarbazine, lomustine, and vincristine in recurrent high-grade glioma. J Clin Oncol 2010; 28:4601–4608. [DOI] [PubMed] [Google Scholar]
- 10.Gilbert MR, Wang M, Aldape KD, et al. Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. Journal of Clinical Oncology 2013; 31:4085–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]