

Abstract
The acyl-CoA:diacylglycerol acyltransferase (DGAT) enzymes DGAT1 and DGAT2 catalyze the final step in triglycerides biosynthesis. This study examined the relationships of baseline phenotypes and the common polymorphisms in DGAT1 and DGAT2 with the lipid responses to niacin.
Lipid responses in Chinese patients with dyslipidemia treated with the extended release (ER) niacin/laropiprant combination 1000/20 mg for 4 weeks and then 2000/40 mg for 8 weeks (n = 121, the primary study) or with ER niacin 1500 mg for at least 4 weeks (n = 68, the replication study) were analyzed according to genotypes of DGAT1 rs7003945 T>C and DGAT2 rs3060 T>C polymorphisms.
Treatment with ER niacin improved all lipid parameters in both studies. Absolute and percentage changes in lipids were related to their baseline levels, particularly for low-density lipoprotein cholesterol (LDL-C). The DGAT2 rs3060 T>C polymorphism was associated with lower baseline LDL-C, apoB, high-density lipoprotein cholesterol (HDL-C), and apoAI in patients on statin therapy in the primary study. Subjects with the DGAT2 rs3060 T>C variant had less reduction in LDL-C in the primary study and smaller changes in triglyceride and HDL-C in the replication study but these associations became non-significant after adjusting for baseline lipid values. The DGAT1 rs7003945 T>C polymorphism was not related to lipid baseline values or changes in either study. Concomitant statin therapy and lower body weight were also associated with greater reduction in LDL-C.
Baseline lipid levels were the main determinants of lipid responses especially for LDL-C. The DGAT2 rs3060 polymorphism might influence the lipid responses depending on baseline phenotype, but this association did not persist after adjustment for the baseline lipid levels.
INTRODUCTION
Nicotinic acid, or niacin, is one of the naturally occurring B vitamins (vitamin B3), and dietary deficiency results in pellagra. Pharmacological doses of niacin have favorable effects on all traditionally measured lipid parameters, including increasing high-density lipoprotein cholesterol (HDL-C) and decreasing low-density lipoprotein cholesterol (LDL-C), triglycerides and lipoprotein (a).1,2 Niacin treatment was associated with decreased total mortality in the 15-year follow-up of patients in the Coronary Drug Project originally performed at a time when statins were not available.3,4 However, 2 recent large outcome studies found that the addition of extended release (ER) niacin or the combination of ER niacin and laropiprant (a prostaglandin D2 receptor antagonist developed to reduce niacin-induced flushing) to intensive statin therapy had no significant benefit in further reduction of the cardiovascular event endpoints.5,6
In addition to lipid-regulating actions, niacin has a broad range of additional effects and some of these may offset the potentially beneficial effects on the lipid profile; it increases serum concentrations of glucose, insulin, and uric acid and long-term treatment with niacin is associated with increased free fatty acid levels, although these are reduced in the short-term.7 The cutaneous flushing side effect induced by niacin also occurs in most patients. Although the exact mechanisms for the wanted and unwanted effects of niacin are still not fully elucidated, it appears that some of them may be mediated directly via the niacin receptor, hydroxycarboxylic acid receptor 2, previously known as G protein-coupled receptor 109A. However, a recent animal study found that the lipid-lowering effects of niacin were independent of the niacin receptor.8
The acyl-CoA:diacylglycerol acyltransferase (DGAT) enzymes DGAT1 and DGAT2 catalyze the final and the only committed step in triglyceride synthesis.9,10 Recent in vitro and animal studies have shown that niacin has a direct and noncompetitive inhibitory effect on hepatic DGAT2, and it has been suggested that this may be involved in the lipid-lowering effects of niacin.11,12DGAT1 and DGAT2 are both expressed in many of the same tissues among mammals, especially those that produce large amounts of triglycerides, eg, small intestine, adipose tissues, liver and mammary gland, and so on.9,10 A functional single nucleotide polymorphism (SNP) in DGAT1, 79T>C (rs7003945) was reported to affect promoter activity in cultured cells, and the C allele was associated with 25% to 50% increased DGAT1 expression compared with the T allele in adipocytes, intestinal cells, and hepatocytes.13 This polymorphism was associated with higher body mass index, lower HDL-C levels, and lower blood pressure in Turkish women,13 but it did not affect the obesity-related phenotypes examined in obese subjects in France.14 Polymorphisms in DGAT2 were associated with hepatic triglyceride changes, but no changes in body weight or fat or insulin resistance during lifestyle intervention in patients with fatty liver.15 We recently demonstrated that niacin significantly reduced hepatic triglyceride content in a DGAT2 genotype-dependent manner in a small group of Chinese patients with dyslipidemia.16 This pilot study also showed that the DGAT2 rs3060 or the linked rs101988116 polymorphism tended to be associated with less reduction of plasma triglycerides in response to niacin.
Pharmacogenetic studies may help to identify subgroups that might benefit from niacin based on their genetic makeup. However, so far there is no study reporting the pharmacogenetics of niacin, which may be partly due to the fact that the mechanisms of the lipid-modifying actions of niacin are not well understood and niacin itself is not usually used as first-line therapy but may be used in combination with statins in some patients with high triglycerides or uncontrolled LDL-C levels. This study provides the first report of the effect of phenotypic factors and the common polymorphisms in DGAT1 and DGAT2 on the lipid responses to niacin in clinical studies with the ER niacin/laropiprant combination and with ER niacin alone. The hypothesis of the study is that certain phenotypic factors and polymorphisms in DGAT1 and DGAT2 may influence the lipid response to niacin.
METHODS
Study Design
The Primary Study
The primary study involved Chinese patients with dyslipidemia, who were attending Out-patient Clinics in the Prince of Wales Hospital in Hong Kong, and they were treated with ER niacin/laropiprant (Merck & Co Inc, Whitehouse Station, NJ) 1000/20 mg for 4 weeks and then ER niacin/laropiprant 2000/40 mg for an additional 8 weeks in a pharmacogenetic study of the ER niacin/laropiprant combination therapy from December 2010 to December 2012. This was an investigator-initiated, uncontrolled, single center study designed to examine genetic determinants of the lipid responses and flushing symptoms induced by ER niacin/laropiprant.17 In brief, these patients were Hong Kong Chinese adults aged 18 to 85 years with primary hypercholesterolemia or mixed dyslipidemia with or without lipid-lowering therapy other than niacin. Patients had to be naïve to all lipid-lowering therapy or taking a stable dose of lipid-modifying therapy for at least 4 weeks for statins and ezetimibe or 6 weeks for fibrates.17,18 Lipid profiles and laboratory safety tests were measured before starting the treatment (week 0), after 4 weeks treatment with ER niacin/laropiprant 1000/20 mg (week 4), and after 4 and 8 weeks treatment with ER niacin/laropiprant 2000/40 mg (week 8 and week 12). Anthropometric measurements, including body weight, body height, waist circumference, hip circumference, and percentage of total body fat using an impedance device (TANITA Body Composition Analyzer BF-350, TANITA Corporation, Tokyo, Japan) were obtained at each study visit. Among 166 of the patients who participated in the 12-week study, 130 patients completed the study with good drug compliance (>80%), and genotyping was performed in 121 patients who were included in the genetic analysis of lipid response to ER niacin in the present study. The study protocol was approved by the local clinical research ethics committee. All patients provided written informed consent. The study was performed in accordance with the Declaration of Helsinki and Good Clinical Practice standards (Clinical Trial Registration No. on WHO-ICTRP: ChiCTR-ONC-10001038).
The Replication Study
The replication study included data from Chinese patients with dyslipidemia who participated in either of 2 clinical studies, which examined the effect of ER niacin on liver fat or erectile dysfunction, respectively, in Chinese patients with dyslipidemia who were attending Out-patient Clinics in Prince of Wales Hospital from August 2008 to March 2011.16,19 Both studies were approved by the local clinical research ethics committee. In the study that examined the effect of ER niacin on liver fat, 39 patients completed the treatment with ER niacin (Niaspan, Abbott Laboratories, Hong Kong) once daily at bedtime in increasing doses of 375, 500, and 750 mg (each for 1 week); 1000 and 1500 mg (each for 4 weeks); and 2000 mg (for 12 weeks). Patients had to be naïve to all lipid-lowering therapy or taking a stable dose of lipid-modifying therapy other than niacin and fibrates (eg, statins and ezetimibe) for at least 6 weeks before enrollment and throughout the entire duration of the study.16 Lipid profiles and laboratory safety tests were performed at baseline, after 4-weeks treatment with 1000, 1500, and 2000 mg and at the end of the study. All patients gave written informed consent for genetic analysis of the niacin effect. In the other study in 160 Chinese men with erectile dysfunction and dyslipidemia who were randomized in a one-to-one ratio to receive up to 1500 mg oral niacin daily (500 mg for 2 weeks, 1000 mg for 4 weeks, and 1500 mg for 6 weeks) and or placebo for 12 weeks, 61 patients in the niacin group completed the study. Lipid profile was assessed at baseline and after treatment. Written informed consent for genetic analysis of lipid response to niacin was obtained from 29 patients including 9 patients receiving long-term stable doses of statins or fibrates.19 The data on the lipid response to ER niacin 1500 mg in these 2 studies were merged for genetic analysis.
Measurement of Lipids and Lipoproteins
The lipid profile before and after treatment in the 2 studies was assessed using routine methods in the Department of Chemical Pathology Laboratory at the Prince of Wales Hospital, which has international laboratory accreditation. Total cholesterol level was measured by the enzymatic method (Centrichem Chemistry System, Baker Instruments Co. Allentown). HDL-C level was determined by using fractional precipitation with dextran sulfate and manganous ion. Triglycerides levels were measured by the glyceryl dehydrogenase reaction following the hydrolysis of the triglyceride (Centrichem Chemistry System, Baker Instruments Co.). LDL-C level was estimated using Friedewald formula20 or directly measured if triglycerides levels were greater than 4.5 mmol/L. Apolipoprotein (apo) AI and apoB levels before and after treatment in the primary study were measured using immunoturbidimetric assays (Sekisui Medical, Co. Ltd. Tsukuba, Japan) and were assessed by BioMajesty Series JCA-BM6050 (JEOL Ltd, Tokyo, Japan).21
Genotyping
A 10 mL EDTA blood sample was collected from subjects for DNA extraction and genotyping for the DGAT1 rs7003945 T>C and DGAT2 rs3060 T>C polymorphisms, which are common and may be functional based on the previous findings.13,16 The genotyping for the DGAT2 rs3060 T>C polymorphism was performed using the Taqman SNP genotyping assays (assay ID: C_8750930_10; Applied Biosystems, Foster City, CA), and the DGAT1 rs7003945 T>C polymorphism was assayed with custom Taqman SNP genotyping assay which was designed based on the sequence and the target site (rs7003945 T>C “TTCAACCCGCCCGCGTC [T/C]GCCCGTCGGCCTCAAGGA” provided (Applied Biosystems). Genotypes were detected using the ABI Prism 7700 sequence detection system (Applied Biosystems).
Statistical Analysis
Changes in lipid parameters from pretreatment level at the end of the studies among the DGAT1 rs7003945 T>C or DGAT2 rs3060 T>C genotype groups were assessed by ANOVA for normally distributed variables or Kruskal–Wallis test for skewed variables. χ2 tests were used to test Hardy–Weinberg equilibrium and to compare the genotype distribution in the primary and replication studies. Multivariate linear regression analysis was performed to determine the genotypic and phenotypic factors that may influence the lipid response to niacin. P < 0.05 was considered to be statistically significant. All statistical analyses were performed using SPSS Version 17.0 (SPSS Inc, Chicago, IL).
The sample size of this study was estimated based on our previous small study examining the effect of the DGAT2 rs3060 T>C polymorphism on the liver fat and plasma lipids in 39 Chinese patients with dyslipidemia (TT:TC:CC = −42.1% ± 36.1%: −31.9% ± 40.8%:−22.7% ± 34.5%, P < 0.05 for trend).16 The post-hoc power analysis showed that this study had over 80% power to detect a 12% difference in plasma triglycerides in response to ER niacin between patients with the DGAT2 homozygous wild-type allele versus those with the variant allele with a type I error probability of 5%.
RESULTS
The baseline characteristics of the 121 patients in the primary study and the 68 patients in the replication study are shown in Table 1. At the end of the primary study, ER niacin/laropiprant 2000/40 mg daily significantly (P < 0.001 for all) increased HDL-C and apoAI by 23.3% ± 22.7% and 4.4% ± 9.6%, respectively, and reduced triglycerides, LDL-C, and apoB by −31.8% ± 22.7%, −19.8% ± 26.2%, and 22.4% ± 15.2%, respectively. At the end of the replication study, ER niacin 1500 mg increased HDL-C by 20.4% ± 24.1% and reduced triglycerides by −24.7% ± 32.7% (P < 0.001 for both), but only had a marginal effect on plasma LDL-C (−2.7% ± 29.5%, P < 0.05), which may be related to the lower pretreatment LDL-C level in the replication study patients (Table 1).
TABLE 1.
Baseline Characteristics of the Study Subjects in the Primary and the Replication Studies
The absolute and percentage reductions in LDL-C were greater in patients with higher pretreatment LDL-C levels in both studies (Figure 1). The cubic regression equation generated from the primary study revealed that when pretreatment LDL-C was about 1.8 mmol/L or below, the LDL-C was unchanged or increased with niacin treatment. Likewise, the percentage changes in triglycerides were greater in patients with higher pretreatment triglycerides (r = −0.270, P < 0.001) and the increases in HDL-C were greater in patients with lower pretreatment HDL-C (r = −0.179, P < 0.05). Similarly, pretreatment apoB and apoAI levels also tended to be associated with percentage changes in apoB and apoAI in response to ER niacin/laropiprant (r = −0.224, P < 0.05 and r = −0.175, P = 0.055, respectively). The percentage change in triglycerides was significantly negatively associated with the percentage change in HDL-C, but was not related to changes in LDL-C, in both primary and replication studies (r = −0.472 and −0.554, respectively, P < 0.001 for both).
FIGURE 1.
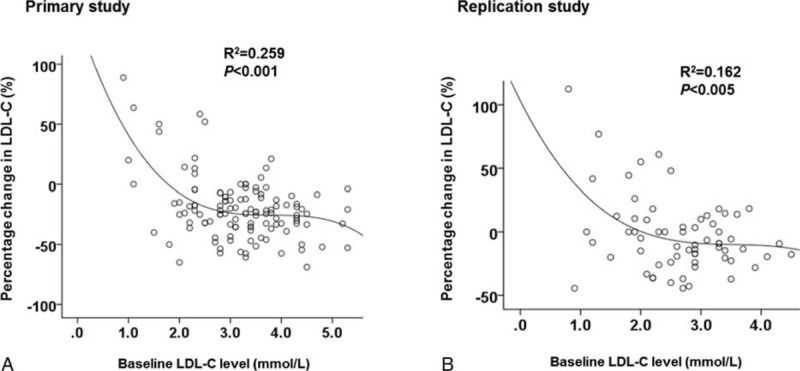
Association between the baseline plasma LDL-C level and the percentage change in LDL-C with ER niacin/laropiprant in the primary study (modified and reprinted with permission18) (A) and ER niacin in the replication study (B) in univariate analysis.
At the end of the study, there was a small but significant reduction in body weight in the primary (73.1 ± 15.5 kg to 72.5 ± 15.2 kg, P < 0.001) and replication studies (80.9 ± 16.8 kg to 79.7 ± 16.7 kg, P < 0.001). There was no significant association between body weight changes and lipid response to niacin in both studies.
In the primary study, there were 75 patients receiving statins or other lipid-lowering therapy (eg, ezetimibe, fibrates, or bile acid sequestrants). Among the 55 patients receiving statin therapy, 48 patients were receiving simvastatin and these patients had greater reductions in triglycerides (−38.8% ± 24.9% vs −27.2% ± 30.9%, P < 0.05) and tended to have greater reductions in LDL-C (−22.9% ± 31.7% vs −17.8% ± 21.9%, P > 0.05) with niacin than the other patients including those receiving other lipid-lowering drugs. The association between statin usage and LDL-C response to niacin became significant (P < 0.05) after adjustment for the pretreatment levels of LDL-C.
The DGAT1 rs7003945 T>C and DGAT2 rs3060 T>C polymorphisms were successfully genotyped in all patients (Table 1). The genotype distributions of these 2 polymorphisms were in Hardy–Weinberg equilibrium (P > 0.05), and there were no significant differences in the genotype distribution in the 2 studies (P > 0.05).
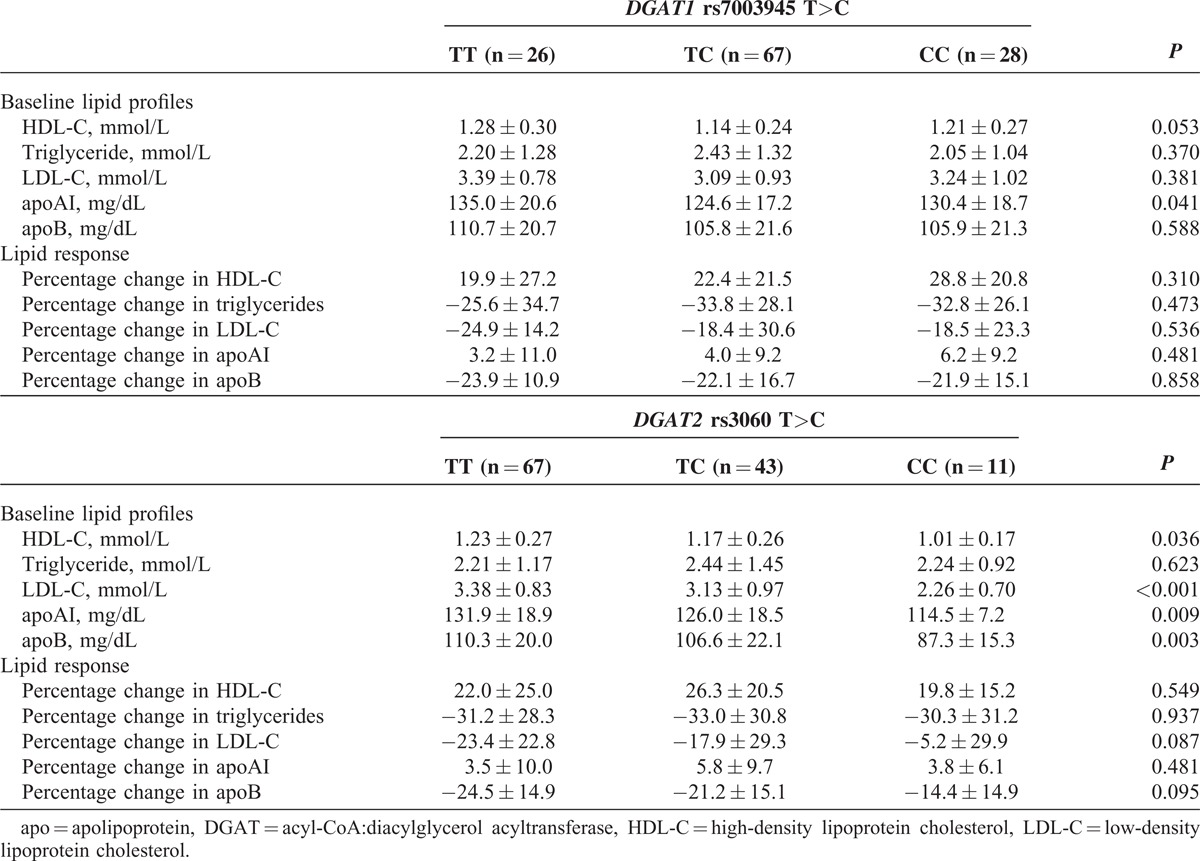
In the primary study, having the DGAT2 rs3060 T>C variant was associated with lower baseline LDL-C, apoB, HDL-C, and apoAI levels in a gene-dose dependent manner (Table 2). Further analysis showed that the associations between the DGAT2 rs3060 T>C polymorphism and baseline LDL-C, apoB, HDL-C, and apoAI levels were only observed in patients receiving lipid-lowering drugs, which were mainly statins, (n = 75), and not in those lipid treatment-naïve patients (n = 46). The DGAT2 rs3060 T>C polymorphism had no significant effect on the triglyceride or HDL-C responses to ER niacin/laropiprant (Table 2), but was associated with the LDL-C response in a gene-dose dependent manner (TT:TC:CC = −23.3% ± 22.6%:−17.9% ± 29.3%:−5.2% ± 29.9%, P < 0.05 for trend). However, this association became nonsignificant after adjustment for the pretreatment LDL-C levels.
TABLE 2.
Associations Between the Polymorphisms in DGAT1 and DGAT2 and the Lipid Response to ER Niacin/Laropiprantat the End of the Study in the Primary Study
In the replication study, the DGAT2 rs3060 T>C polymorphism had no significant association with baseline lipids or the LDL-C response (Table 3), but was associated with the HDL-C and triglyceride responses to ER niacin only in a recessive model, in which the TT and TC carriers were combined for comparison with those with the CC genotype (n = 11) (P < 0.05 for both, Figure 2). However, this association was only significant for the HDL-C response but not for the triglyceride response after adjusting for their respective pretreatment levels.
TABLE 3.
Associations Between the Polymorphisms in DGAT1 and DGAT2 and the Lipid Response to ER Niacin at the End of the Study in the Replication Study
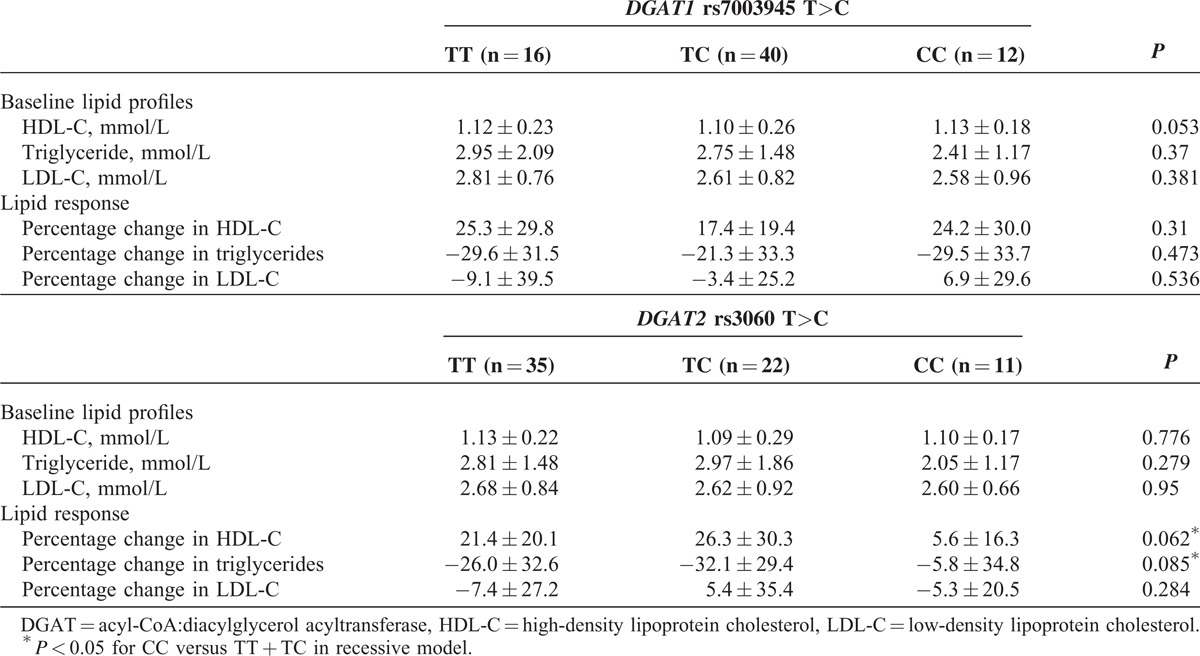
FIGURE 2.
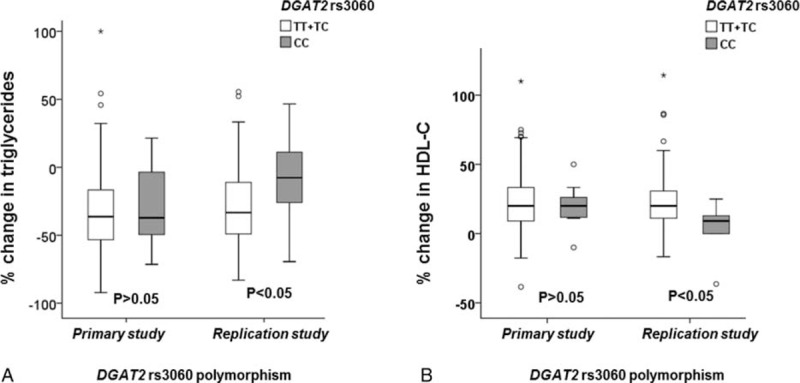
Effect of DGAT2 rs3060 polymorphism on the triglyceride (A) and HDL-C (B) response to niacin in the primary and the replication study.
The DGAT1 rs7003945 T>C polymorphism showed no association with pretreatment lipid levels or lipid responses in both the primary and replication studies (Tables 2 and 3).
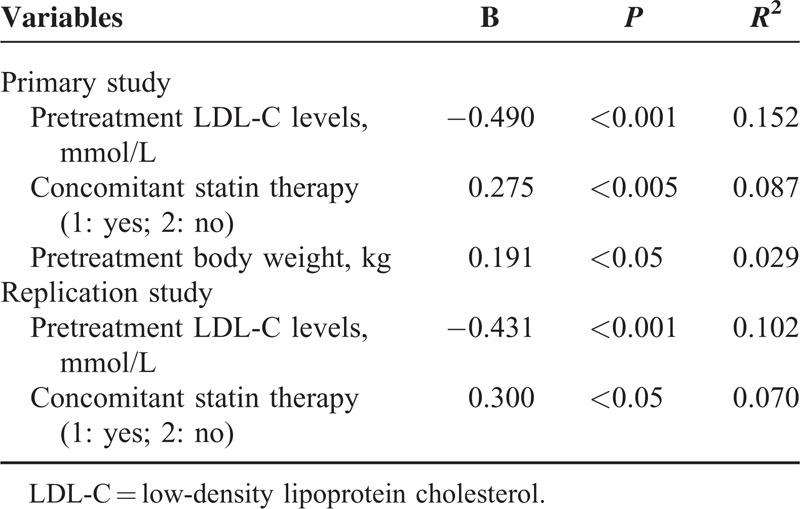
Multivariate stepwise linear regression analysis showed that higher pretreatment LDL-C level, concomitant statin therapy, and lower baseline body weight, but not the genetic polymorphisms, were significantly associated with greater percentage reductions in LDL-C, and these factors explained 26.8% of the variance in the LDL-C response to ER niacin/laropiprant in the primary study (Table 4). The associations between the LDL-C response to ER niacin and the pretreatment LDL-C level (r = −0.431, P < 0.001) and concomitant statin therapy (r = −0.300, P < 0.05) were also observed in the replication group (Table 4). The baseline triglyceride level was the only significant factor associated with the triglyceride response to ER niacin/laropiprant and ER niacin in the study subjects and contributed to approximately 11% of the variance in the triglyceride response in both studies.
TABLE 4.
Multivariate Linear Regression Analysis of Predictors of LDL-C Response to Niacin in the Primary and the Replication Studies
Neither the DGAT1 rs7003945 nor the DGAT2 rs3060 T>C polymorphism showed significant associations with body mass index, body fat, blood pressure, baseline levels of free fatty acid and fasting glucose, or the changes in these parameters during the study (data not shown).
DISCUSSION
The DGAT enzymes DGAT1 and DGAT2 play an important role in triglyceride biosynthesis. Niacin inhibits hepatic DGAT2 in cell lines and in animal studies11,12 and this may contribute to its lipid-modifying effects so the DGAT2 rs3060 polymorphism was considered an appropriate primary candidate genotype to examine the pharmacogenetics of the lipid responses to niacin. DGAT1 is also important for triglyceride synthesis although there are conflicting data from studies in mice over-expressing liver DGAT1 on whether this influences hepatic very low-density lipoprotein production22,23 and an in vitro study showed that niacin did not appear to affect hepatic DGAT1 activity.11 We found that the functional DGAT1 79T>C polymorphism had no effect on the lipid responses to niacin or baseline lipid values in the 2 separate studies, and there was still no effect when males and females were analyzed separately (data not shown).
The DGAT2 rs3060 T>C polymorphism tended to be associated with a reduced LDL-C response in the primary study but this trend no longer existed after adjustment for the baseline LDL-C levels before niacin therapy. The association of the DGAT2 rs3060 T>C polymorphism and the lower LDL-C, apoB, HDL-C, and apoAI levels before niacin therapy may be just a chance finding as the associations were only observed in 75 patients receiving lipid-lowering drugs (mainly statins) in the primary study, and their true baseline LDL-C levels without any lipid-lowering therapy were not available. The DGAT2 rs3060 T>C polymorphism was not found to be associated with plasma lipid traits in previous genome-wide association studies.24–26 On the contrary, this finding could indicate that the DGAT2 rs3060 T>C polymorphism might influence the lipid response to statins. However, polymorphisms in DGAT2 were not found to affect statin response in the previous genome-wide analyses of lipid responses to statins or in Chinese patients with hypercholesterolemia.27–31
DGAT2 catalyzes the final step of triglyceride synthesis in hepatocytes. Animal studies showed that mice over-expressing DGAT2 were associated with severe hepatic steatosis and increased hepatic triglyceride content,22,32 and antisense oligonucleotide reduction of DGAT2 expression improved hepatic steatosis and reduced plasma triglycerides in obese mice.33 We previously demonstrated that ER niacin significantly reduced the liver fat content in Chinese patients with dyslipidemia, and this effect was influenced by the DGAT2 rs3060 polymorphism. It has been proposed that inhibition of DGAT2 may be involved in the lipid-lowering effect of niacin. Le Bloc’h et al12 reported that niacin decreases apoB100-containing lipoprotein levels by reducing hepatic very-low-density lipoprotein production in obese dogs. They showed niacin treatment reduced DGAT2 expression by 60% ± 8% (P < 0.05), but had no effect on the expression of the other key genes involved in the metabolism of apoB100-containing lipoproteins examined in the study, suggesting inhibition of DGAT2 by reducing enzyme expression may be the mechanism for niacin-induced plasma triglyceride reduction.
In our previous study, the DGAT2 rs3060 polymorphism tended to be associated with reduced plasma triglyceride response to ER niacin (TT:TC:CC = −41.9% vs −33.1% vs 20.0%, P < 0.05 for trend) in the 39 patients with dyslipidemia.16 However, when we increased the sample size by combining the 39 patients with the 29 patients with dyslipidemia and erectile dysfunction in the present study, there was no significant trend but patients with the CC genotype did have a smaller triglyceride and HDL-C response to ER niacin when compared with those with the TT and TC genotypes in a recessive model (P < 0.05 for both). This association was not observed in the primary study. The discrepancy between the primary and the replication study may be related to the different study design and baseline phenotypes of the patients included. For example, the primary and the replication study patients have different baseline characteristics, particularly the baseline lipid profiles that appear to determine the lipid response to niacin. The primary and replication studies also used different doses of niacin, but we would not expect that to influence the effect of genetic polymorphisms on the lipid responses to niacin within each study. However, multivariate regression analysis suggested that the baseline triglyceride level but not the rs3060 polymorphism was the only determinant for the triglyceride response to ER niacin. Further large studies are needed to verify whether the DGAT2 rs3060 polymorphism determines the lipid response to niacin in patients with high triglyceride levels.
In the 2 studies, we found a consistent association between baseline LDL-C level and percentage in LDL-C in response to ER niacin. This is in line with the observation in the HPS2-THRIVE study where the percentage reduction in LDL-C was dependent on the baseline values divided into 3 groups and this influenced the cardiovascular outcome with those with baseline LDL-C > 2 mmol/L having the best outcome and those with LDL-C < 1.5 mmol/L the worst outcome.6 This is a distinguishing feature from the LDL-C-lowering effects of statins for which the percentage changes in LDL-C are largely independent of baseline values although that is not the case for the percentage changes in HDL-C and triglycerides with statins.34
Unexpectedly, despite having lower LDL-C levels before niacin therapy, patients receiving statins had a greater LDL-C response to ER niacin than those not on statins suggesting there may be an interaction between statins and ER niacin, which may be a pharmacokinetic or a pharmacodynamic effect. A recent multiple-dose pharmacokinetic interaction study in 18 healthy volunteers showed that coadministration of ER niacin and ezetimibe/simvastatin resulted in a small increase in drug exposure for all the 3 drugs (22% for ER niacin, 9% for ezetimibe, 20% for simvastatin, and 35% for simvastatin acid).35 Other recent studies have also shown increases in systemic exposure, particularly to simvastatin acid when given with ER niacin, although a single dose study in Chinese healthy volunteers found no pharmacokinetic interaction between niacin and simvastatin.36,37 This may suggest repeated doses are needed for the interaction to occur. Lower body weight was associated with a greater reduction in LDL-C in response to niacin in the primary study, suggesting drug exposure might play a role in determining the lipid-lowering effect of niacin. It has been suggested that the side effects of niacin including rash are dose dependent.38 We previously also reported that an exanthematous eruption with ER niacin/laropiprant was dose dependent and appeared to be associated with lower body size in Chinese patients with dyslipidemia.17
Of interest, there was only a small increase in apoAI (4.4% ± 9.6%) in response to ER niacin/laropiprant compared with the HDL-C changes (23.3% ± 22.7%), which was only measured in the primary study. This is consistent with the published data from large randomized clinical trials with ER niacin.39,40 In the AIM-HIGH trial, the addition of ER niacin to simvastatin therapy during a 3-year mean follow-up period was associated with a 25% increase in HDL-C, but was only associated with a 4.1% increase in apoAI.39 However, it has been shown that with statins the percentage increase in apoA-I was almost identical to that of HDL-C.34 ApoAI is the major surface apoprotein on HDL particles and is considered an indicator of total HDL mass or particle number although the number of apoAI molecules per HDL particle may vary (2, 3, or 4). Recent studies suggested that apoAI may play a crucial role in protection against atherosclerosis.41–43 A recent study on the effect of niacin on HDL apoAI kinetics reported by Pang et al44 demonstrated that ER niacin increased HDL apoAI concentration by lowering apoAI fractional catabolic rate and this was associated with increased with HDL particle size as suggested by the higher HDL-C:apoAI ratio. It has been proposed that the failure of the 2 large outcome studies with ER niacin may be related to the changes in structural and functional complexity of HDL particles not adequately captured by changes in total HDL-C concentration45 in groups of patients with very low baseline level of LDL-C, which as discussed above can predict a poor LDL-C response to niacin.18
In recent years, genetic variants, particularly in drug transporters, have been identified to play an important role in determining the safety and lipid-lowering effect of some statins.27,46,47 To the best of our knowledge, the present study is the first to explore the contribution of genetic factors to the wide variation in the lipid responses to niacin. This study has several limitations. Firstly, we assessed 2 different formulations of niacin (ER niacin/laropiprant and ER niacin alone) at different doses so that we could not combine all data together to increase the study power. Laropiprant is thought to have no effect on lipids and thus polymorphisms in DGAT may have similar impact (if any) on the lipid response to these 2 formulations of niacin. Secondly, the primary study with ER niacin/laropiprant and the 2 small studies with ER niacin included in the replication study had different study designs (baseline characteristics, dose and duration of therapy, etc) and this might influence the results and contribute to the discrepancies between the studies. In addition, many patients were receiving statins or other lipid-lowering therapy before and throughout the study; and thus, we cannot obtain the lipid-lowering effect of niacin in relation to a baseline without treatment in all subjects, but this may reflect the real-world lipid-lowering effect of niacin in the statin era. The association between the DGAT2 rs3060 T>C polymorphism and the pretreatment LDL-C levels in the primary study might be confounded by the various statin regimens among patients and this cannot be thoroughly evaluated. Furthermore, we did not examine the effect of genetic polymorphisms on the pretreatment lipid profiles and the lipid response to niacin in patients with or without statins separately due to the small number of subjects in the subgroups. In this study, we only assessed rs3060 in DGAT2 based on the previous findings but the functionality of this SNP or any of the other SNPs reported in DGAT2 are still unclear and thus may not be the best DGAT2 variant for assessment. Future studies with larger sample size are needed to verify the findings observed in this study.
In conclusion, this study showed that the DGAT1 rs7003945 polymorphism had no significant effects on the lipid responses to niacin in Chinese dyslipidemic patients. Subjects with the DGAT2 rs3060 T>C variant showed reduced responses for some lipid fractions only in the replication study which varied according to the baseline phenotype and which largely became nonsignificant after adjusting for the pretreatment lipid values. These findings need to be verified in larger pharmacogenetic studies. The major determinant of LDL-C reduction was the pretreatment level, which may suggest that niacin would be less effective in subjects with lower pretreatment LDL-C. Patients receiving simvastatin also appeared to have greater reductions in triglycerides and LDL-C with niacin, which may suggest a pharmacokinetic or a pharmacodynamic interaction.
ACKNOWLEDGEMENT
The authors thank Evelyn Chau for helping in patient recruitment and Emily Poon for helping in genotyping and all patients for their participation in the study. No writing assistance was utilized in the production of this manuscript. Manuscript presented in part at the 82nd European Atherosclerosis Society Congress, Madrid, Spain, May 31, 2014 to June 3, 2014.
Footnotes
Abbreviations: apo = apolipoprotein, DGAT = acyl-CoA,diacylglycerol acyltransferase, ER = extended release, HDL-C = high-density lipoprotein cholesterol, LDL-C = low-density lipoprotein cholesterol, SNP = single nucleotide polymorphism.
The clinical study with ER niacin/laropiprant (Tredaptive) was an investigator-initiated study supported by a grant from Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Whitehouse Station, NJ. The funding source has no role in the conduct of the study or interpretation of the data. The clinical study with ER niacin (Niaspan) was supported in part by an unrestricted grant from Abbott Laboratories Ltd. This funding source had no role in the conduct of the study or interpretation of the data.
BT received research funding and lecture fees from Merck Sharp & Dohme Corp. and from Abbott Laboratories Ltd. The remaining authors have no conflicts of interest to disclose.
SY received research funding and lecturer fees from Merck Sharp & Dohme (MSD) Corp.
REFERENCES
- 1.Carlson LA. Nicotinic acid: the broad-spectrum lipid drug. A 50th anniversary review. J Intern Med 2005; 258:94–114. [DOI] [PubMed] [Google Scholar]
- 2.Chapman MJ, Le Goff W, Guerin M, et al. Cholesteryl ester transfer protein: at the heart of the action of lipid-modulating therapy with statins, fibrates, niacin, and cholesteryl ester transfer protein inhibitors. Eur Heart J 2010; 31:149–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coronary Drug Project Research Group. Clofibrate and niacin in coronary heart disease. JAMA 1975; 231:360–381. [PubMed] [Google Scholar]
- 4.Canner PL, Berge KG, Wenger NK, et al. Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin. J Am Coll Cardiol 1986; 8:1245–1255. [DOI] [PubMed] [Google Scholar]
- 5.Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 2011; 365:2255–2267. [DOI] [PubMed] [Google Scholar]
- 6.Landray MJ, Haynes R, Hopewell JC, et al. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med 2014; 371:203–212. [DOI] [PubMed] [Google Scholar]
- 7.Kang I, Kim SW, Youn JH. Effects of nicotinic acid on gene expression: potential mechanisms and implications for wanted and unwanted effects of the lipid-lowering drug. J Clin Endocrinol Metab 2011; 96:3048–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lauring B, Taggart AK, Tata JR, et al. Niacin lipid efficacy is independent of both the niacin receptor GPR109A and free fatty acid suppression. Sci Transl Med 2012; 148:ra115. [DOI] [PubMed] [Google Scholar]
- 9.Cases S, Smith SJ, Zheng YW, et al. Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc Natl Acad Sci U S A 1998; 95:13018–13023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cases S, Stone SJ, Zhou P, et al. Cloning of DGAT2, a second mammalian diacylglycerol acyltransferase, and related family members. J Biol Chem 2001; 276:38870–38876. [DOI] [PubMed] [Google Scholar]
- 11.Ganji SH, Tavintharan S, Zhu D, et al. Niacin noncompetitively inhibits DGAT2 but not DGAT1 activity in HepG2 cells. J Lipid Res 2004; 45:1835–1845. [DOI] [PubMed] [Google Scholar]
- 12.Le Bloc’h J, Leray V, Chetiveaux M, et al. Nicotinic acid decreases apolipoprotein B100-containing lipoprotein levels by reducing hepatic very low density lipoprotein secretion through a possible diacylglycerol acyltransferase 2 inhibition in obese dogs. J Pharmacol Exp Ther 2010; 334:583–589. [DOI] [PubMed] [Google Scholar]
- 13.Ludwig EH, Mahley RW, Palaoglu E, et al. DGAT1 promoter polymorphism associated with alterations in body mass index, high density lipoprotein levels and blood pressure in Turkish women. Clin Genet 2002; 62:68–73. [DOI] [PubMed] [Google Scholar]
- 14.Coudreau SK, Tounian P, Bonhomme G, et al. Role of the DGAT gene C79T single-nucleotide polymorphism in French obese subjects. Obes Res 2003; 11:1163–1167. [DOI] [PubMed] [Google Scholar]
- 15.Kantartzis K, Machicao F, Machann J, et al. The DGAT2 gene is a candidate for the dissociation between fatty liver and insulin resistance in humans. Clin Sci (Lond) 2009; 116:531–537. [DOI] [PubMed] [Google Scholar]
- 16.Hu M, Chu WC, Yamashita S, et al. Liver fat reduction with niacin is influenced by DGAT-2 polymorphisms in hypertriglyceridemic patients. J Lipid Res 2012; 53:802–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang YL, Hu M, Chang M, et al. A high incidence of exanthematous eruption associated with niacin/laropiprant combination in Hong Kong Chinese patients. J Clin Pharm Ther 2013; 38:528–532. [DOI] [PubMed] [Google Scholar]
- 18.Hu M, Tomlinson B. Niacin for reduction of cardiovascular risk. N Engl J Med 2014; 371:1941–1942. [DOI] [PubMed] [Google Scholar]
- 19.Ng CF, Lee CP, Ho AL, et al. Effect of niacin on erectile function in men suffering erectile dysfunction and dyslipidemia. J Sex Med 2011; 8:2883–2893. [DOI] [PubMed] [Google Scholar]
- 20.Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem 1972; 18:499–502. [PubMed] [Google Scholar]
- 21.Sakurabayashi I, Saito Y, Kita T, et al. Reference intervals for serum apolipoproteins A-I, A-II, B, C-II, C-III, and E in healthy Japanese determined with a commercial immunoturbidimetric assay and effects of sex, age, smoking, drinking, and Lp(a) level. Clin Chim Acta 2001; 312:87–95. [DOI] [PubMed] [Google Scholar]
- 22.Yamazaki T, Sasaki E, Kakinuma C, et al. Increased very low density lipoprotein secretion and gonadal fat mass in mice overexpressing liver DGAT1. J Biol Chem 2005; 280:21506–21514. [DOI] [PubMed] [Google Scholar]
- 23.Millar JS, Stone SJ, Tietge UJ, et al. Short-term overexpression of DGAT1 or DGAT2 increases hepatic triglyceride but not VLDL triglyceride or apoB production. J Lipid Res 2006; 47:2297–2305. [DOI] [PubMed] [Google Scholar]
- 24.Willer CJ, Sanna S, Jackson AU, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet 2008; 40:161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kathiresan S, Willer CJ, Peloso GM, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet 2009; 41:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teslovich TM, Musunuru K, Smith AV, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010; 466:707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu M, Tomlinson B. Pharmacogenomics of lipid-lowering therapies. Pharmacogenomics 2013; 14:981–995. [DOI] [PubMed] [Google Scholar]
- 28.Hu M, Lui SS, Mak VW, et al. Pharmacogenetic analysis of lipid responses to rosuvastatin in Chinese patients. Pharmacogenet Genomics 2010; 20:634–637. [DOI] [PubMed] [Google Scholar]
- 29.Chasman DI, Giulianini F, MacFadyen J, et al. Genetic determinants of statin-induced low-density lipoprotein cholesterol reduction: the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Circ Cardiovasc Genet 2012; 5:257–264. [DOI] [PubMed] [Google Scholar]
- 30.Deshmukh HA, Colhoun HM, Johnson T, et al. Genome-wide association study of genetic determinants of LDL-c response to atorvastatin therapy: importance of Lp(a). J Lipid Res 2012; 53:1000–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hopewell JC, Parish S, Offer A, et al. Impact of common genetic variation on response to simvastatin therapy among 18 705 participants in the Heart Protection Study. Eur Heart J 2012; 24: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Monetti M, Levin MC, Watt MJ, et al. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab 2007; 6:69–78. [DOI] [PubMed] [Google Scholar]
- 33.Yu XX, Murray SF, Pandey SK, et al. Antisense oligonucleotide reduction of DGAT2 expression improves hepatic steatosis and hyperlipidemia in obese mice. Hepatology 2005; 42:362–371. [DOI] [PubMed] [Google Scholar]
- 34.Barter PJ, Brandrup-Wognsen G, Palmer MK, et al. Effect of statins on HDL-C: a complex process unrelated to changes in LDL-C: analysis of the VOYAGER Database. J Lipid Res 2010; 51:1546–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kosoglou T, Zhu Y, Statkevich P, et al. Assessment of potential pharmacokinetic interactions of ezetimibe/simvastatin and extended-release niacin tablets in healthy subjects. Eur J Clin Pharmacol 2011; 67:483–492. [DOI] [PubMed] [Google Scholar]
- 36.Lauring B, Dishy V, De Kam PJ, et al. Effects of extended-release niacin and extended-release niacin/laropiprant on the pharmacokinetics of simvastatin in healthy subjects. Am J Ther 2014; 14: [DOI] [PubMed] [Google Scholar]
- 37.Liu M, Wang XL, Zhang D, et al. Pharmacokinetics of niacin, simvastatin and their metabolites in healthy Chinese subjects after single and multiple doses of a fixed dose combination tablet of niacin extended release/simvastatin. Drug Res (Stuttg) 2013; 23: [DOI] [PubMed] [Google Scholar]
- 38.Piepho RW. The pharmacokinetics and pharmacodynamics of agents proven to raise high-density lipoprotein cholesterol. Am J Cardiol 2000; 86:35L–40L. [DOI] [PubMed] [Google Scholar]
- 39.Albers JJ, Slee A, O’Brien KD, et al. Relationship of apolipoproteins A-1 and B, and lipoprotein(a) to cardiovascular outcomes: the AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes). J Am Coll Cardiol 2013; 62:1575–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bays HE, Shah A, Lin J, et al. Consistency of extended-release niacin/laropiprant effects on Lp(a), ApoB, non-HDL-C, Apo A1, and ApoB/ApoA1 ratio across patient subgroups. Am J Cardiovasc Drugs 2012; 12:197–206. [DOI] [PubMed] [Google Scholar]
- 41.deGoma EM, deGoma RL, Rader DJ. Beyond high-density lipoprotein cholesterol levels evaluating high-density lipoprotein function as influenced by novel therapeutic approaches. J Am Coll Cardiol 2008; 51:2199–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moore RE, Navab M, Millar JS, et al. Increased atherosclerosis in mice lacking apolipoprotein A-I attributable to both impaired reverse cholesterol transport and increased inflammation. Circ Res 2005; 97:763–771. [DOI] [PubMed] [Google Scholar]
- 43.Lee JY, Parks JS. ATP-binding cassette transporter AI and its role in HDL formation. Curr Opin Lipidol 2005; 16:19–25. [DOI] [PubMed] [Google Scholar]
- 44.Pang J, Chan DC, Hamilton SJ, et al. Effect of niacin on high-density lipoprotein apolipoprotein A-I kinetics in statin-treated patients with type 2 diabetes mellitus. Arterioscler Thromb Vasc Biol 2014; 34:427–432. [DOI] [PubMed] [Google Scholar]
- 45.Al-Hijji M, Martin SS, Joshi PH, et al. Effect of equivalent on-treatment apolipoprotein levels on outcomes (from the AIM-HIGH and HPS2-THRIVE). Am J Cardiol 2013; 112:1697–1700. [DOI] [PubMed] [Google Scholar]
- 46.Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy – a genomewide study. N Engl J Med 2008; 359:789–799. [DOI] [PubMed] [Google Scholar]
- 47.Tomlinson B, Hu M, Lee VW, et al. ABCG2 polymorphism is associated with the low-density lipoprotein cholesterol response to rosuvastatin. Clin Pharmacol Ther 2010; 87:558–562. [DOI] [PubMed] [Google Scholar]