

Abstract
Atypical teratoid/rhabdoid tumors (AT/RTs) are rare, highly malignant central nervous system tumors that predominantly occur in young children.
A 22-year-old woman presented with a 4-year history of relapsing tinnitus and gradual hearing loss. Neuroimaging revealed an enhanced intrinsic left internal auditory canal mass. The patient underwent radiotherapy treatment. Three years later, the tumor size continued to increase, as observed by imaging, and ultimately evolved into the left cerebellopontine angle. As a consequence, a total tumor resection was performed, and a pathological diagnosis of AT/RT was made. Aggressive radiotherapy and chemotherapy treatment continued; however, the tumor recurred within 11 months after the total tumor resection. The patient died within 4 months of the second operation.
Histopathologically, the tumor contained characteristic rhabdoid cells with areas that resembled a classical primitive neuroectodermal tumor. Immunostaining showed loss of INI1 protein expression in tumor cells, and fluorescence in situ hybridization showed a hemizygous deletion of the hSNF5/INI1 gene region on 22q11.2.
This is the first report of an AT/RT that arised from the acoustic nerve in a young adult. Despite manifold diagnostic and therapeutic advances, the prognosis of patients with AT/RT remains poor.
INTRODUCTION
Atypical teratoid/rhabdoid tumors (AT/RTs) are highly malignant central nervous system (CNS) tumors that primarily affect young children, and characterized by varying numbers of distinctive rhabdoid cells. In 1995, Rorke et al first reported this tumor in the brain and termed it AT/RT.1 AT/RTs are often misdiagnosed as other types of brain tumors due to the lack of specific clinical and neuroimaging features. This study reports a case of AT/RT that originated in the auditory nerves; in addition, a review of the current literature is provided. Informed consent was obtained from the patient's father.
CASE REPORT
A 22-year-old woman presented with a 4-year history of progressive hearing loss on the left side. Left tinnitus and hearing loss prompted admission at a different hospital. A magnetic resonance imaging (MRI) scan of the brain revealed a heterogeneous contrast-enhancing mass that measured 0.5 × 0.3 × 0.3 cm in the left internal auditory canal, which resulted in the diagnosis of an acoustic neuroma (Figure 1A). The patient recovered from tinnitus, but her hearing loss deteriorated after symptomatic treatment. One year later, the patient was readmitted to the hospital due to complete loss of hearing on the left side, hypophasis of the left eye, absence of corneal reflex, presence of shallow left nasolabial folds, and skewing of the mouth angle to the right. A computed tomography (CT) scan indicated the presence of a mass that measured 0.5 × 0.5 × 0.4 cm in the left internal auditory canal (Figure 1B). The patient received a 60-Gy dose of fractioned irradiation and partially recovered.
FIGURE 1.

MRI of the brain revealed a heterogeneous contrast-enhancing mass in the left internal auditory canal (A) in Dec 2008. One year later, the mass had increased in size (B). In July 2011, the mass had further increased in size, and had evolved into the left CPA with heterogeneous enhancement (C). CPA = cerebellopontine angle, MRI = magnetic resonance imaging.
The patient was admitted to the hospital for a third time in July 2011 complaining of a constant headache that had persisted for 4 weeks. An MRI scan of the brain showed an increase in the original size of the mass and that the mass had evolved into the left cerebellopontine angle (CPA). The scan also revealed a heterogeneous enhancing signal and cyst formation in the mass (Figure 1C). The tumor was completely removed and sent for pathological evaluation. Microscopically, the tumor was composed of sheets of compact cells. The majority of the tumor cells demonstrated a rhabdoid phenotype, characterized by an eccentrically placed vesicular nucleus, a prominent nucleolus, and an eosinophilic inclusion-like cytoplasm (Figure 2A). Areas of the tumor showed a primitive neuroectodermal tumor (PNET)-like, blue-colored small-cell appearance. Mitoses were frequently observed. Immunostaining revealed a lack of integrase interactor 1 (INI1) protein expression in the tumor cells, and fluorescence in situ hybridization (FISH) revealed a hemizygous deletion of the INI1 gene region on 22q11.2 (Figure 2B and C). The tumor cells were either entirely or partially positive for glial fibrillary acidic protein, epithelial membrane antigen (EMA), vimentin, oligodendrocyte transcription factor 2, nestin, and actin, but were negative for desmin, S100 protein, and SRY-related HMG-box gene 10 (SOX-10). The mindbomb homolog 1 labeling index was 45%. The patient received a 5-cycle chemotherapeutic regimen that included vincristine, etoposide, cyclophosphamide, methotrexate, and cisplatinum, with the eventual goal of hematopoietic stem cell transplantation. Additionally, a 54-Gy dose of fractioned irradiation at the site of the resected tumor was given 5 months after the first surgery.
FIGURE 2.
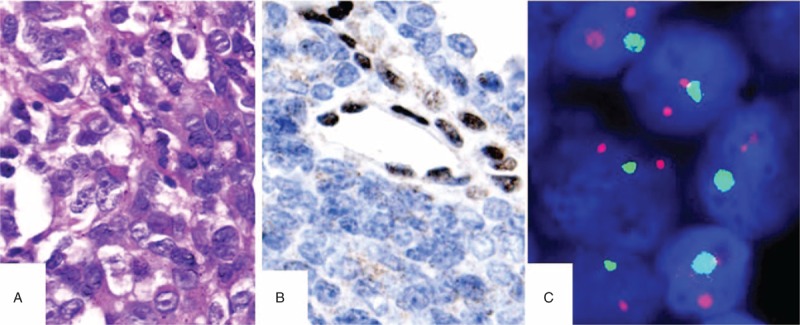
The tumor was completely removed in July 2011. The majority of the tumor cells was of the rhabdoid phenotype and displayed a vesicular nucleus, prominent nucleolus, and an eosinophilic cytoplasm (A). Immunostaining showed a lack of INI1 protein expression in the tumor cells (B), and FISH showed a deletion of the INI1 gene (C). FISH = fluorescence in situ hybridization.
The patient was then admitted to the hospital for a fourth time because she experienced neck aches and weakness of the lower limbs for a period of 10 consecutive days. An MRI scan of the brain revealed a well-demarcated intraspinal mass at the C6-T7 level that was hypointense on T1-weighted images, slightly hyperintense on T2-weighted images, and homogeneously contrast enhancing (Figure 3A and B). The tumor was completely removed and subjected to a pathological examination. Microscopically, the tumor contained characteristic rhabdoid cells with areas that resembled a classical PNET (Figure 3C). Immunostaining showed that the tumor cells had decreased INI1 protein expression (Figure 3D), similar to that of the left cerebellopontine mass, which was described a year earlier. A final diagnosis of AT/RT was then made. In the postoperative period, 2 additional cycles of high-dose chemotherapy that consisted of carboplatin, thiotepa, etoposide, and methotrexate were given to the patient. Although the patient received chemotherapy, she died 4 months later.
FIGURE 3.

In June 2012, MRI revealed a well-demarcated intraspinal mass at the C6-T7 level with homogeneous contrast enhancement (A and B). Microscopically, the tumor was primarily composed of characteristic rhabdoid cells (C) and lacked INI1 immunoreactivity (D). MRI = magnetic resonance imaging.
DISCUSSION
AT/RTs occur most frequently in young children under the age of 3 years (median age 2 years).2,3 However, approximately 45 cases of AT/RT in adults have been reported in the literature.4 AT/RTs account for approximately 1% to 2% of all pediatric CNS tumors.5 Men appear to be affected slightly more frequently than women, with a reported ratio of 3:2 to 2:1 (M:F).3,6 This tumor may arise in either the supratentorial or infratentorial areas, but is predominantly observed in the supratentorial region.6,7 Other sites of origin include the brainstem and the spinal cord in rare cases. In other rare cases, the tumor originates from the cranial nerves.8–10 The present report details a case of AT/RT that originated in the auditory nerves in a young adult, and until now, no such cases have been reported.
Difficulty in the diagnosis of patients with AT/RT can arise as a result of the nonspecific neuroimaging features of AT/RT. The neuroimaging features of AT/RT often mimic those of other CNS embryonal tumors, in particular, medulloblastomas, where the cerebellum is involved. Often, AT/RT is misdiagnosed as another type of brain tumor particularly when an unusual site of the brain is affected. For example, the present case of AT/RT that is discussed in this report was misdiagnosed as an acoustic neuroma. The CT scans of patients show that the tumors are most often slightly hyperattenuated or iso-attenuated compared with the adjacent brain parenchyma, with calcification in only a few cases.11 AT/RTs show predominantly iso signal intensity on T1- and T2-weighted MRI images with heterogeneous contrast enhancement. In addition, AT/RTs contain few areas of peritumoral edema11 and more areas of apparent necrosis or cystic change compared with typical medulloblastoma.12 Yong et al13 indicated that a supratentorial tumor with a thick, wavy (irregular) heterogeneous enhanced wall that surrounds a central cystic region is suggestive of AT/RT in the appropriate clinical setting, particularly in a child of preschool age.
Grossly, AT/RTs are soft, fleshy, pink-gray tumors that often contain areas of necrosis or hemorrhage. Histopathologically, the primary diagnostic component is the presence of rhabdoid cells, which consist of an eccentrically placed vesicular nucleus, a prominent nucleolus, and homogeneous pink cytoplasm that may appear to contain inclusions. Indeed, AT/RTs often feature several histopathological patterns similar to those of other CNS embryonal tumors, including the presence of “small blue cells”.2,3 Cytopathology is also an important method for the diagnosis of AT/RT. Typical rhabdoid cells are found in most AT/RT cases with dissemination into the cerebrospinal fluid.14 Immunophenotypic heterogeneity is a key indicator for the diagnosis of AT/RT. The majority of tumors show focal immunoreactivity for epithelial membrane antigen, vimentin, glial fibrillary acidic protein, neurofilament protein, synaptophysin, and cytokeratins.3 Almost all AT/RTs demonstrate at least focal positivity for smooth muscle actin (SMA), but are typically negative for desmin and germ cell antigens. AT/RTs demonstrate a characteristic loss of INI1 protein expression.15 Another useful diagnostic tool is FISH; however, only approximately 70% of cases show a deletion of the INI1 gene region on 22q11.2.16
The most important differential diagnosis of AT/RT is medulloblastoma of the posterior fossa in a young child.2 Other important differential diagnoses are other embryonal tumors, choroid plexus carcinoma and anaplastic ependymoma. Indeed, in a child <3 years of age, any tumor with a high proliferation index is potentially an AT/RT. Ancillary diagnostic studies are critical in the determination of the distinction of these embryonal tumors. Approximately 5% of AT/RTs that contain ependymoblastic rosettes result in a considerable number of cases with a differential diagnosis of embryonal tumor with abundant neuropils and true rosettes; this tumor type can be distinguished based on a high level of focal amplification of chromosome 19q13.41.3,17 The loss of INI1 expression may play a key role in the ability to distinguish AT/RT from the aforementioned tumors. In addition, in the present case, the AT/RT originated from the acoustic nerve and involved the CPA. This should distinguish this type of tumor from an epithelial malignant peripheral nerve sheath tumor in which the loss of INI1 expression occurs in approximately 50% cases.18,19 The present case demonstrated a more pure AT/RT pattern and contained rhabdoid cells and PNET-like small blue cells, but lacked features of peripheral neural differentiation. Immunohistochemically, the combination of S100 protein and SOX-10 positivity and EMA and cytokeratin 7 (CK7) negativity has a high predictive value for the diagnosis of MPSNT19–21 and distinguishes it from the present case of AT/RT, which was focally positive for EMA and CK7 and negative for S100 protein and SOX-10.
A defining molecular feature of AT/RT is the loss of the putative tumor suppressor gene hSNF5/INI1 located on chromosome 22q11.2.22 Loss of the INI1 protein can be seen in almost all cases of AT/RTs, and deletions or other mutations of the INI1 gene are detected in approximately 70% of all cases. Although the mechanism of tumorigenesis is unknown, the loss of INI1 leads to a reduction of p16 and an increase in cell proliferation via the retinoblastoma-signaling pathway.23
Surgical removal of the AT/RT is the primary treatment. Adjuvant therapy includes chemotherapy, radiotherapy, autologous bone marrow transplantation, and stem cell support that may yield some effects in a subset of patients.7,24–29 Radiation is an effective therapy, but is avoided in patients who are <3 years of age due to long-term neurocognitive sequelae. Most long-term survivors undergo radiation therapy as a part of their upfront or salvage therapy, and it has been suggested that outcomes may improve if patients receive radiation therapy earlier in their treatment regimen. There is not an accepted standard chemotherapy, but intensive alkylator-based chemotherapy regimens, regimens with high-dose methotrexate, and regimens that include high-dose chemotherapy with stem cell rescue may be more effective in these patients.29 AT/RTs are highly aggressive malignancies, and their biological invasiveness contributes to a poor prognosis with a median survival of 17 months.7 Disease progression is the major cause of death.7 Negative contributing factors include young age, presence of metastatic disease, infratentorial location, and efficacy of chemotherapy, particularly when young age and metastatic disease are identified as independent risk factors.30
Footnotes
Abbreviations: AT/RT = atypical teratoid/rhabdoid tumor, CK7 = cytokeratin 7, CNS = central nervous system, CPA = cerebellopontine angle, CT = computed tomography, EMA = epithelial membrane antigen, FISH = fluorescence in situ hybridization, GFAP = glial fibrillary acidic protein, INI1 = integrase interactor 1, MIB-1 = mindbomb homolog 1, MPNST = malignant peripheral nerve sheath tumor, MRI = magnetic resonance imaging, Olig-2 = oligodendrocyte transcription factor 2, PNET = primitive neuroectodermal tumor, Rb = retinoblastoma, SI = signal intensity, SOX-10 = SRY-related HMG-box gene 10.
This work was supported by the Natural Science Foundation of Fujian Province, P.R.C. (Grant No. 2014J01413).
The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Rorke LB, Packer R, Biegel J. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood. J Neurooncol 1995; 24:21–28. [DOI] [PubMed] [Google Scholar]
- 2.Rorke LB, Packer RJ, Biegel JA. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg 1996; 85:56–65. [DOI] [PubMed] [Google Scholar]
- 3.Burger PC, Yu IT, Tihan T, et al. Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study. Am J Surg Pathol 1998; 22:1083–1092. [DOI] [PubMed] [Google Scholar]
- 4.Souki C, Abdel-Rhaman M, Qasem A, et al. Atypical teratoid rhabdoid tumor in adulthood. Clin Neuropathol 2014; 33:245–250. [DOI] [PubMed] [Google Scholar]
- 5.Reddy AT. Atypical teratoid/rhabdoid tumors of the central nervous system. J Neurooncol 2005; 75:309–313. [DOI] [PubMed] [Google Scholar]
- 6.Hilden JM, Meerbaum S, Burger P, et al. Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol 2004; 22:2877–2884. [DOI] [PubMed] [Google Scholar]
- 7.Athale UH, Duckworth J, Odame I, et al. Childhood atypical teratoid rhabdoid tumor of the central nervous system: a meta-analysis of observational studies. J Pediatr Hematol Oncol 2009; 31:651–663. [DOI] [PubMed] [Google Scholar]
- 8.Wykoff CC, Lam BL, Brathwaite CD, et al. Atypical teratoid/rhabdoid tumor arising from the third cranial nerve. J Neuroophthalmol 2008; 28:207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verma A, Morriss C. Atypical teratoid/rhabdoid tumor of the optic nerve. Pediatr Radiol 2008; 38:1117–1121. [DOI] [PubMed] [Google Scholar]
- 10.Beschorner R, Mittelbronn M, Koerbel A, et al. Atypical teratoid-rhabdoid tumor spreading along the trigeminal nerve. Pediatr Neurosurg 2006; 42:258–263. [DOI] [PubMed] [Google Scholar]
- 11.Lee IH, Yoo SY, Kim JH, et al. Atypical teratoid/rhabdoid tumors of the central nervous system: imaging and clinical findings in 16 children. Clin Radiol 2009; 64:256–264. [DOI] [PubMed] [Google Scholar]
- 12.Meyers SP, Khademian ZP, Biegel JA, et al. Primary intracranial atypical teratoid/rhabdoid tumors of infancy and childhood: MRI features and patient outcomes. AJNR Am J Neuroradiol 2006; 27:962–971. [PMC free article] [PubMed] [Google Scholar]
- 13.Au Yong KJ, Jaremko JL, Jans L, et al. How specific is the MRI appearance of supratentorial atypical teratoid rhabdoid tumors? Pediatr Radiol 2013; 43:347–354. [DOI] [PubMed] [Google Scholar]
- 14.Choi J, Kim H, Kim SH. Atypical teratoid/rhabdoid tumor: analysis of cytomorphologic features in CSF, focused on the differential diagnosis from mimickers. Diagn Cytopathol 2012; 40:592–596. [DOI] [PubMed] [Google Scholar]
- 15.Judkins AR. Immunohistochemistry of INI1 expression: a new tool for old challenges in CNS and soft tissue pathology. Adv Anat Pathol 2007; 14:335–339. [DOI] [PubMed] [Google Scholar]
- 16.Bruch LA, Hill DA, Cai DX, et al. A role for fluorescence in situ hybridization detection of chromosome 22q dosage in distinguishing atypical teratoid/rhabdoid tumors from medulloblastoma/central primitive neuroectodermal tumors. Hum Pathol 2001; 32:156–162. [DOI] [PubMed] [Google Scholar]
- 17.Gessi M, Pfister S, Hans VH, et al. Absence of chromosome 19q13.41 amplification in a case of atypical teratoid/rhabdoid tumor with ependymoblastic differentiation. Acta Neuropathol 2011; 121:283–285. [DOI] [PubMed] [Google Scholar]
- 18.Hornick JL, Dal Cin P, Fletcher CD. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol 2009; 33:542–550. [DOI] [PubMed] [Google Scholar]
- 19.Carter JM, O’Hara C, Dundas G, et al. Epithelioid malignant peripheral nerve sheath tumor arising in a schwannoma, in a patient with “neuroblastoma-like” schwannomatosis and a novel germline SMARCB1 mutation. Am J Surg Pathol 2012; 36:154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olsen SH, Thomas DG, Lucas DR. Cluster analysis of immunohistochemical profiles in synovial sarcoma, malignant peripheral nerve sheath tumor, and Ewing sarcoma. Mod Pathol 2006; 19:659–668. [DOI] [PubMed] [Google Scholar]
- 21.Thway K, Fisher C. Malignant peripheral nerve sheath tumor: pathology and genetics. Ann Diagn Pathol 2014; 18:109–116. [DOI] [PubMed] [Google Scholar]
- 22.Biegel JA. Molecular genetics of atypical teratoid/rhabdoid tumor. Neurosurg Focus 2006; 20:E11. [DOI] [PubMed] [Google Scholar]
- 23.Imbalzano AN, Jones SN. Snf5 tumor suppressor couples chromatin remodeling, checkpoint control, and chromosomal stability. Cancer Cell 2005; 7:294–295. [DOI] [PubMed] [Google Scholar]
- 24.Tekautz TM, Fuller CE, Blaney S, et al. Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol 2005; 23:1491–1499. [DOI] [PubMed] [Google Scholar]
- 25.Strother D. Atypical teratoid rhabdoid tumors of childhood: diagnosis, treatment and challenges. Expert Rev Anticancer Ther 2005; 5:907–915. [DOI] [PubMed] [Google Scholar]
- 26.Nicolaides T, Tihan T, Horn B, et al. High-dose chemotherapy and autologous stem cell rescue for atypical teratoid/rhabdoid tumor of the central nervous system. J Neurooncol 2010; 98:117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lafay-Cousin L, Hawkins C, Carret AS, et al. Central nervous system atypical teratoid rhabdoid tumours: the Canadian Paediatric Brain Tumour Consortium experience. Eur J Cancer 2012; 48:353–359. [DOI] [PubMed] [Google Scholar]
- 28.Park ES, Sung KW, Baek HJ, et al. Tandem high-dose chemotherapy and autologous stem cell transplantation in young children with atypical teratoid/rhabdoid tumor of the central nervous system. J Korean Med Sci 2012; 27:135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ginn KF, Gajjar A. Atypical teratoid rhabdoid tumor: current therapy and future directions. Front Oncol 2012; 2:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.von Hoff K, Hinkes B, Dannenmann-Stern E, et al. Frequency, risk-factors and survival of children with atypical teratoid rhabdoid tumors (AT/RT) of the CNS diagnosed between 1988 and 2004, and registered to the German HIT database. Pediatr Blood Cancer 2011; 57:978–985. [DOI] [PubMed] [Google Scholar]