

Abstract
Statins have a variety of myotoxic effects and can trigger the development of inflammatory myopathies or myasthenia gravis (MG) mediated by immunomodulatory properties. Autoantibodies to 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) have been identified in patients with statin-associated myopathy. The purpose of the present study is to develop an enzyme-linked immunosorbent assay (ELISA) of anti-HMGCR antibodies and to elucidate the clinical significance of anti-HMGCR antibodies in Japanese patients with inflammatory myopathies or MG.
We enrolled 75 patients with inflammatory myopathies, who were all negative for anti-signal recognition particle and anti-aminoacyl transfer RNA synthetase antibodies. They were referred to Keio University and National Center of Neurology and Psychiatry between October 2010 and September 2012. We also studied 251 patients with MG who were followed at the MG Clinic at Keio University Hospital. Anti-HMGCR antibodies were detected by ELISA. We investigated demographic, clinical, radiological, and histological findings associated with anti-HMGCR antibodies.
We established the anti-HMGCR ELISA with the recombinant protein. Protein immunoprecipitation detected autoantigens corresponding to HMGCR. Immunohistochemistry using muscle biopsy specimens revealed regenerating muscle fibers clearly stained by polyclonal anti-HMGCR antibodies and patients’ serum. Anti-HMGCR autoantibodies were specifically detected in 8 patients with necrotizing myopathy. The seropositivity rate in the necrotizing myopathy patients was significantly higher than those in the patients with other histological diagnoses of inflammatory myopathies (31% vs 2%, P = 0.001). Statins were administered in only 3 of the 8 anti-HMGCR-positive patients. Myopathy associated with anti-HMGCR antibodies showed mild limb weakness and favorable response to immunotherapy. All 8 patients exhibited increased signal intensities on short T1 inversion recovery of muscle MRI. Of the 251 patients with MG, 23 were administered statins at the onset of MG. One late-onset MG patient experienced MG worsening after 4-wk treatment with atorvastatin. However, anti-HMGCR antibodies were not detected in the 251 MG patients except for one early-onset MG patient with no history of statin therapy.
Anti-HMGCR antibodies are a relevant clinical marker of necrotizing myopathy with or without statin exposure, but they are not associated with the onset or deterioration of MG.
Support was received in the form of a grant from the Japanese Ministry of Education, Science, Sports and Culture (no. 26461298); a Health and Labor Sciences Research Grant on Intractable Diseases (Neuroimmunological Diseases) from the Ministry of Health, Labor and Welfare of Japan; Intramural Research Grants (Nos. 23–5 and 23–4) for Neurological and Psychiatric Disorders of NCNP; Grants for Research on Intractable Diseases and Comprehensive Research on Disability Health and Welfare from the Ministry of Health, Labor and Welfare of Japan; a Grant-in-Aid for Scientific Research (B) from MEXT (No. 24390227), and a Grant-in-Aid for Challenging Exploratory Research (24659437).
INTRODUCTION
Statins lower cholesterol levels by specifically inhibiting 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), a key enzyme in the cholesterol biosynthesis pathway. Statins are associated with ≥1 myotoxic effects including myalgia, elevation of creatine kinase and transaminases, and weakness. Muscle problems occurred in 10% to 25% of patients treated with statins in clinical practice and in approximately 13% of participants of published clinical trials.1 Statins have immunomodulatory properties, and they may unmask or worsen certain neuromuscular disorders including myasthenia gravis (MG), myotonic dystrophy, McArdle disease, and mitochondrial myopathy.2 The most severe problem is the development of inflammatory myopathy requiring immunosuppressive therapy; this is now known to be mediated by anti-HMGCR antibodies.3,4
Anti-HMGCR antibodies were first found as 200- and 100-kDa proteins using protein immunoprecipitation.5 In 2011, the 100-kDa autoantigen was identified as HMGCR by Mammen et al.4 Anti-HMGCR antibodies were detectable using different methods, including enzyme-linked immunosorbent assay (ELISA), addressable laser beam assay, and chemiluminscent immunoassay.4,6–8 These assays showed qualitative agreement and level of anti-HMGCR antibodies showed significant correlation.8 Clinical features associated with anti-HMGCR antibodies detected by these methods in United States and Europe; however, there were no detection methods in Japan.
MG is the most common autoimmune neuromuscular disorder mediated by autoantibodies to acetylcholine receptor (AChR) or muscle-specific tyrosine kinase.9 MG can be exacerbated by a variety of medications, which increase weakness by interrupting neuromuscular junction transmission. Some drugs potentially induce a new onset of MG. In 2002, Parmer et al10 first reported the case of a 67-year-old woman who suffered from the aggregation of generalized MG 12 weeks after being treated with atorvastatin. Special attention has been paid to the association between MG and statins. Some investigators suggested that MG occurs after therapy with statins, and others suggested that statins worsen previously diagnosed MG.10–15 The myotoxic effects of statins on MG symptoms were speculated to be immune-mediated, but the pathogenesis has not been investigated.
The purpose of the present study was to develop an ELISA of anti-HMGCR antibodies and to elucidate the clinical significance of anti-HMGCR antibodies in Japanese patients with inflammatory myopathies and MG.
METHODS
Patients
We examined 75 adult patients with inflammatory myopathies, who were referred to Keio University or National Center of Neurology and Psychiatry between October 2010 and September 2012. We included patients with the definite diagnosis of idiopathic inflammatory myopathies by a comprehensive histological examination.16 In addition, the patients were all negative for anti-signal recognition particle (SRP) and anti-aminoacyl transfer RNA synthetase antibodies using RNA immunoprecipitation assay. The 75 patients’ mean age at the examination was 61 ± 15 years (range 20–82), and the sex breakdown (M:F) was 30:45.
Histological diagnoses were based on the established criteria.17,18 Briefly, sporadic inclusion body myositis was diagnosed by the identification of rimmed vacuoles with non-necrotic fibers invaded by mononuclear cells or increased major histocompatibility complex (MHC) class I expression. Polymyositis was diagnosed based on exclusively endomysial inflammation cell infiltrate surrounding or invading non-necrotic muscle fibers, accompanied by ubiquitous MHC class I expression. Dermatomyositis was diagnosed by clinical criteria including a rash typical of dermatomyositis and the identification of perifascicular atrophy. Necrotizing myopathy was diagnosed based on the observation of necrotic fibers with diffuse distribution without or with minimal inflammatory cell infiltration.
We also studied 251 patients with MG, who were followed at the MG Clinic at Keio University Hospital. The diagnosis of MG was based on clinical, electrophysiologic, and immunologic criteria.9 The clinical classification and quantitative MG score were graded based on the recommendation issued by the Task Force of the Medical Advisory Board of the Myasthenia Gravis Foundation of America.19 Disease subtypes were divided into early-onset, late-onset, and thymoma-associated MG. As disease controls and normal controls, we used serum samples from 25 patients with Duchenne muscular dystrophy and 30 healthy volunteers.
Clinical information was retrospectively obtained for all patients by reviewing their clinical charts. All clinical samples and information were collected after the patients and controls gave their written informed consent as approved by the Institutional Review Boards of both the National Center of Neurology and Psychiatry and Keio University. All analyses were performed using statistical analysis software (IBM/SPSS version 20).
Anti-HMGCR ELISA
Our anti-HMGCR ELISA was developed based on the original method with some modifications.4 First, 96-well polyvinyl plates (Smilon multiwell plate H type; Sumitomo Bakelite) were coated with C-terminal recombinant HMGCR protein (Sigma, St. Louis, MO) at 0.1 μg/mL diluted in phosphate buffered. The remaining blocking sites were blocked with 3% bovine serum albumin. The wells were incubated with serum samples diluted at 1:400 and subsequently with peroxidase-conjugated anti-human IgG (Jackson Immuno Research, Westgrove, PA) diluted 1:100000. The antibody binding was visualized by incubation with tetramethylbenzidine (1 mg/mL) in phosphate-citrate buffer. The reaction was stopped by 1 mol/L sulfuric acid. The optical density at 450 nm (OD450) was read with an automatic plate reader (Biorad, Hercules, CA). Samples were tested in duplicate. The antibody index was calculated from the OD450 of the samples divided by the OD450 of the referential serum (patient 1 in Table 1). The cut-off value was set as the mean + 5SD of 30 healthy control sera.20
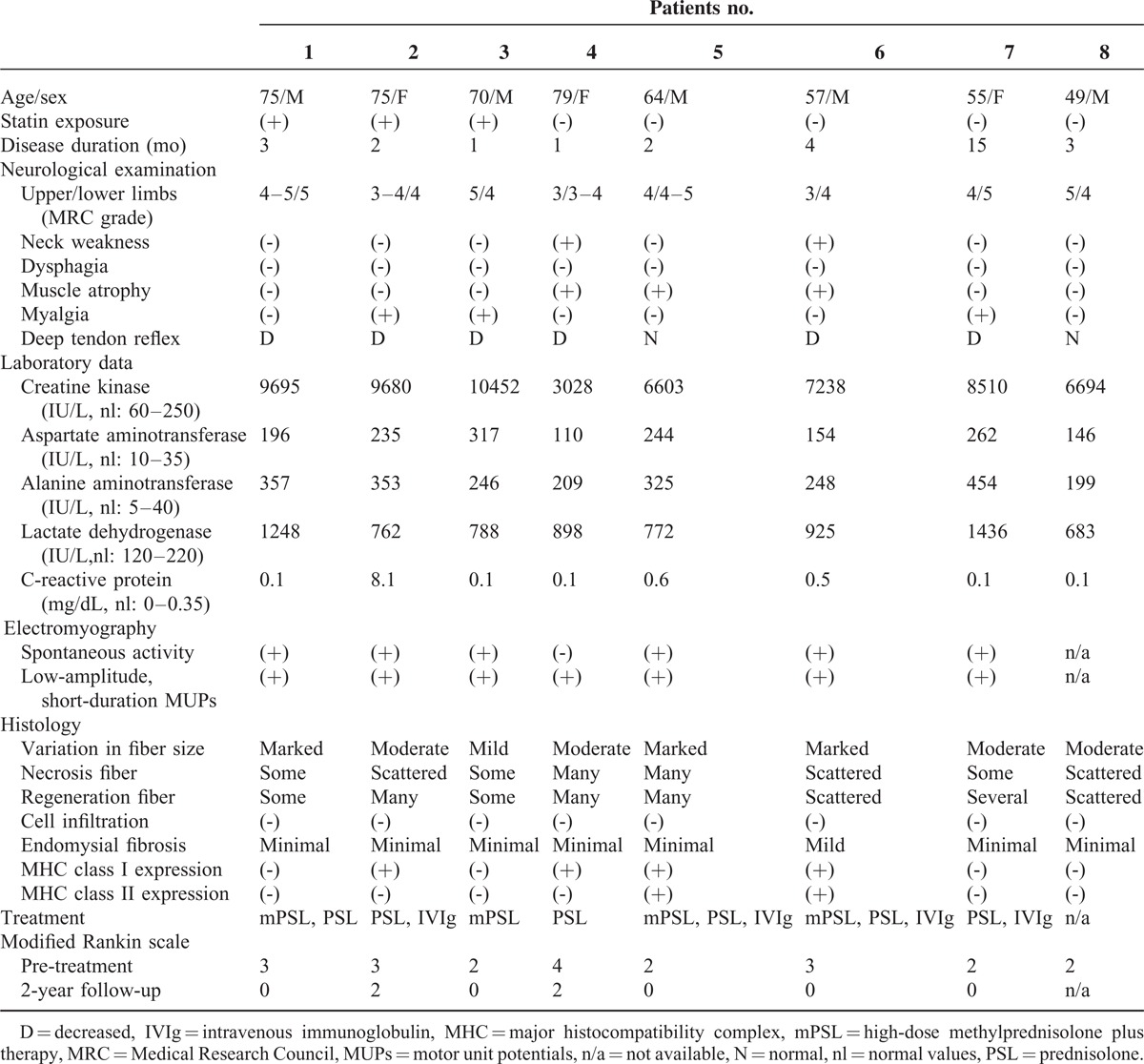
TABLE 1.
Characteristics of the 8 Patients With anti-HMGCR Antibodies
Protein Immunoprecipitation Assay
Autoantigens were analyzed by protein immunoprecipitation assay using 35S-labeled RD cellular extracts.21 RD cells (5 × 106 per sample) were cultured in methionine-free DMEM (Sigma) containing 3% heat-inactivated fetal bovine serum in the presence of 20 μCi/mL 35S-methionine for 14 h. The 35S-labeled cells were suspended in an ice-cold buffer containing 500 mmol/L NaCl, 0.1% Nonidet P-40, 10 mmol/L Tris-HCl, and a cocktail of protease inhibitors (Complete; Roche, Indianapolis, IN), and sonicated intermittently on ice for a total of 90 s. The supernatant (containing 35S-labeled soluble proteins originating from the nuclei, cytoplasm, and cellular membrane) was recovered by centrifugation (13,000g for 15 min) and used as the antigen source. Two milligrams of protein A-Sepharose CL-4B (Pharmacia Biotech, Little Chalfont) was incubated with 10 μL of a human serum sample. The immunoglobulins that were bound to protein A-Sepharose beads were then incubated with the 35S-labeled cellular extracts for 2 h. The immunoprecipitated material was resolved by electrophoresis on SDS-7.5% polyacrylamide gels, which were subsequently treated with 0.5 mol/L sodium salicylate to enhance the radioactivity, and evaluated by autoradiography using a BAS-5000 system (Fuji Film, Tokyo).
Immunohistochemistry
Six micrometer sections of frozen muscle tissue from biopsies were prepared. The sections were incubated with monoclonal mouse anti-neural cell adhesion molecule (NCAM) antibodies (Leica, Wetzlar) diluted 1:25, polyclonal rabbit anti-HMGCR antibodies (Sigma) diluted 1:125 and serum samples diluted 1:40. After incubation with the primary antibodies for 16 h, the sections were incubated for 2 h with a fluorescein isothiocyanate-conjugated anti-mouse, anti-rabbit or anti-human IgG antibody (Jackson Immuno-Research), and the sections were examined with a fluorescence microscope (Eclipse E-800, Nikon, Tokyo).
RESULTS
Anti-HMGCR ELISA
Since the cut-off value was set as the mean + 5 × SD of 30 healthy control sera, the cut-off of anti-HMGCR index was 0.48. Positivity for the anti-HMGCR antibody was observed in 8 of 26 the patients with necrotizing myopathy (Figure 1). However, only one of the 24 patients with sporadic inclusion body myositis had a slight elevation of anti-HMGCR index. There was no positivity of anti-HMGCR antibodies in the 25 patients with polymyositis or dermatomyositis, or in the 25 patients with Duchenne muscular dystrophy. The seropositivity rate in the 26 necrotizing myopathy patients was significantly higher compared with those of the 49 patients with other inflammatory myopathies (31% vs 2%, P = 0.001).
FIGURE 1.
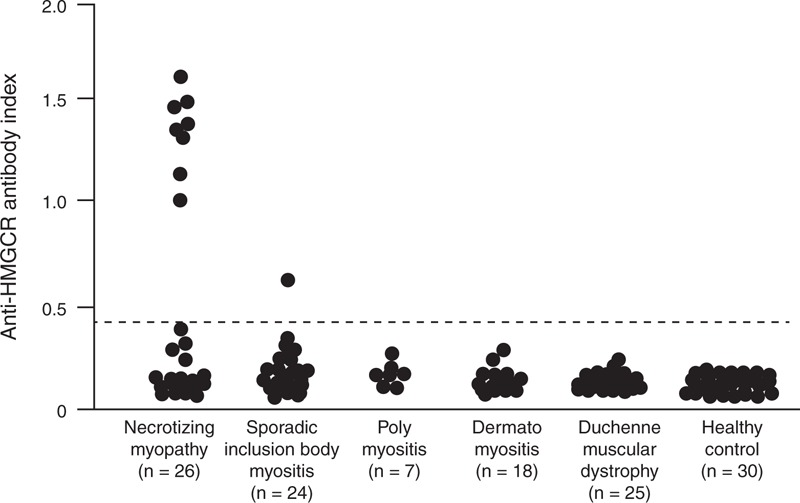
Anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) ELISA. Antibodies reactive with recombinant HMGCR protein by ELISA in sera from inflammatory myopathy patients, Duchenne muscular dystrophy patients, and healthy controls. The cut-off level for positivity is indicated by the broken line (anti-HMGCR index: 0.48).
Protein Immunoprecipitation Assay
We analyzed autoantigens immunoprecipitated by anti-HMGCR-positive sera using the protein immunoprecipitation assay. Representative results obtained from 6 patients with anti-HMGCR antibodies detected by anti-HMGCR ELISA are shown in Figure 2A. Anti-HMGCR-positive sera immunoprecipitated the doublet autoantigens located around 50 kDa (lanes 1–6). However, no immunoprecipitates were found in sera without anti-HMGCR antibodies (lanes 7 and 8). Moreover, we added an excess of the recombinant HMGCR protein (50 ng) to the patient 1’ serum at the incubation with protein A-Sepharose. The 50-kDa doublet autoantigens were clearly absorbed (lane 2 in Figure 2B).
FIGURE 2.
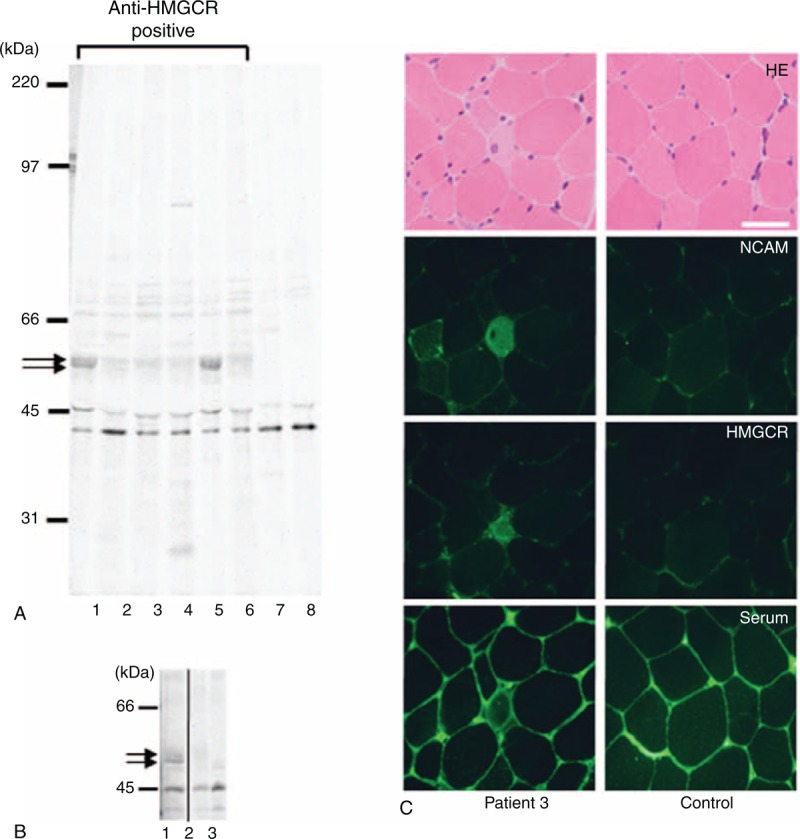
Confirmation of the HMGCR immunoreactivity. (A) Autoradiograms of immunoprecipitated 35S-labeled RD extracts from serum samples are shown. Immunoprecipitated materials were analyzed on SDS-7.5% polyacrylamide gels. The positions of the molecular weight standards are at the left. Arrows indicate the 50-kDa doublet precipitates detected in the serum sample containing anti-HMGCR antibodies (lanes 1–6). (B) Arrows indicate the 50-kDa doublet precipitates detected in the patient 1 sera (lane 1), but not in a healthy control (lane 3). The autoantigens were absorbed in the presence of recombinant HMGCR protein (lane 2). The panel has been cropped between lanes 2 and 3 to exclude immunoprecipitations that are irrelevant to the current study. (C) Muscle sections were obtained from patient 3 (left panels) and control (right panels). Sections were stained with hematoxylin-eosin (HE), polyclonal anti-neural cell adhesion molecules (NCAM) antibody, polyclonal anti-HMGCR antibody, and anti-HMGCR-positive sera. Scale bar = 50 μm.
We determine the sensitivity and specificity of the ELISA using this cutoff relative to the protein immunoprecipitation.4,5 Among 75 patients with inflammatory myopathies, 8 sera immunoprecipitated HMGCR protein and all of them were positive by anti-HMGCR ELISA. Conversely, among 9 sera positivity by anti-HMGCR ELISA, 8 were positive by protein immunoprecipitation. Therefore, the sensitivity and specificity of the anti-HMGCR ELISA are 100% and 98.5%, respectively.”
Immunohistochemistry
We next performed immunohistochemistry using the muscle tissues obtained from patient 3 (Figure 2C). Regenerated muscle fibers were clearly detected by anti-NCAM antibody. In addition, HMGCR was expressed in the regenerated fibers. Anti-HMGCR-positive sera produced similar staining on the regenerated muscle fibers. Thus, the immunoreactivity was clearly co-localized with staining by the polyclonal anti-HMGCR antibody and patient's sera (left panels, Fig. 2C). In contrast, the anti-HMGCR antibody and patient's sera did not show any staining on the muscle fibers of control muscle.
Clinical Features of Patients with Anti-HMGCR Antibodies
The clinical features of eight patients (five men and three women) with anti-HMGCR-positive necrotizing myopathy are summarized in Table 1. Their mean age was 66 years, ranging from 49 to 79 years. All but patient 7 deteriorated within two months with a markedly increase level of creatine kinase. The clinical course suggested the initial diagnosis of rhabdomyolysis. Atrovastatins were administered in 3 patients who were over 70 years’ old. The diseases did not recover after the cessation of the statin treatment. Neurological examinations showed symmetrical and proximal limb weakness. Arms and legs were equally affected. The severe limb weakness with the grade ≤2/5 assessed by manual muscle strength (Medical Research Council scale grade) was not observed. No patients had dysphagia. Electromyography also indicated myopathic motor unit potentials (MUPs) in all patients.
With regard to the histology, all patients showed necrotic and regenerating muscle fibers without inflammatory cell infiltration. Endomysial fibrosis was minimal. MHC class I and class II expression were detected in 50% and 25% of the 8 anti-HMGCR-positive patients, respectively. The 2-year follow-up was available in 7 patients. All patients required immunotherapy and responded well. The recovery of muscle weakness was observed several weeks after the therapy. Since the creatine kinase persisted in higher levels, intravenous immunoglobulin therapy was added in 4 patients. None of the 7 patients experienced disease relapse. Neurological outcome evaluated using the modified Rankin scale showed that 5 of the 7 patients were able to return to their normal daily lives.
Muscle magnetic resonance images (MRIs) were useful for evaluating the distribution of inflammation. Short T1 inversion recovery images in particular showed high signal intensities in all 8 patients. Focal or diffuse abnormal signals were seen in trunk and limb muscles (Figure 3). In contrast to the neurological examination, asymmetry was found on muscle MRI.
FIGURE 3.
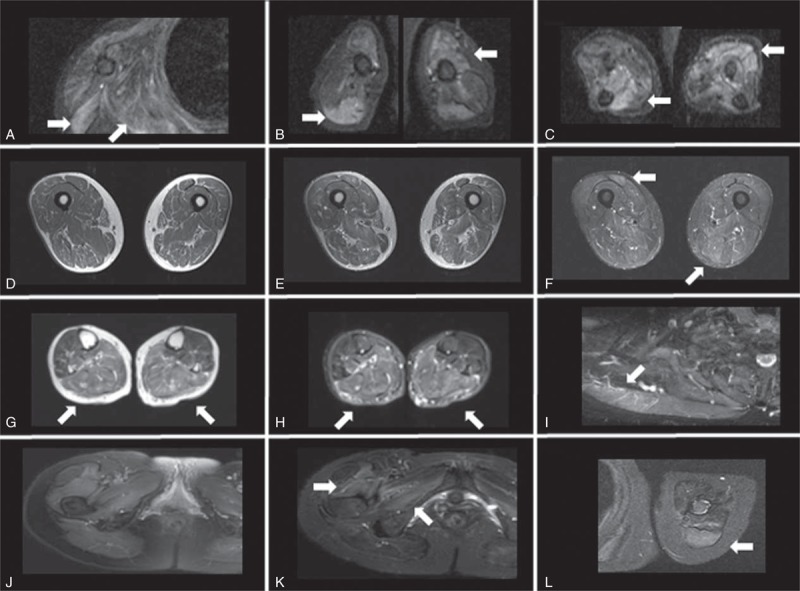
Muscle MRI of patients with anti-HMGCR antibodies. (A–C) Patient 2. Increased short T1 inversion recovery (STIR) signal abnormalities involving deltoid, infraspinatus muscles (A), biceps brachii and triceps brachii (B), and forearm muscles (C). (D–F) Patient 3. Images of thighs on T1 images (D), T2 images (E), and STIR images (F). High intensity in biceps femoris and semitendinousus muscles on STIR images. (G, H) Patient 4. Increased T2/STIR signal abnormalities in posterior calves. (I) Patient 6. Increased STIR signal in trapezius muscle. (J–L) Patient 7. STIR images of pelvis (J) and enhancement in vastus lateralis and obturator internus muscles (K). Increased signal with enhancement of triceps brachii on STIR images (L).
Statin Exposure and Anti-HMGCR Antibodies in MG Patients
The profiles of 251 patients with MG are indicated in Table 2. Of the 251 patients with MG, 23 (9%) including the 5 early-onset, 10 late-onset, and 8 thymoma-associated MG received statins at the disease onset. Statin brands were atorvastatin in 9 patients, pravastatin in 4, fluvastatin in 4, simvastatin in 3, pivastatin in 2, and rosuvastatin in 1 patient. In contrast, only 1 late-onset MG patient experienced MG worsening after statin exposure (Figure 4). Briefly, this 68-year-old woman had a diagnosis of ocular myasthenia at the age of 62 years. Her diplopia and ptosis were well controlled by pyridostigmine and achieved 18-month remission. However, she developed diplopia and ptosis after a 4-week treatment with atorvastatin at the age of 66 years. Her quantitative MG score was increased to 8 with an elevation titer of anti-AChR antibody. Since pyridostigmine was not fully effective, she required prednisolone at a daily dose of 10 mg.
TABLE 2.
Profiles of 251 Patients with Myasthenia Gravis

FIGURE 4.
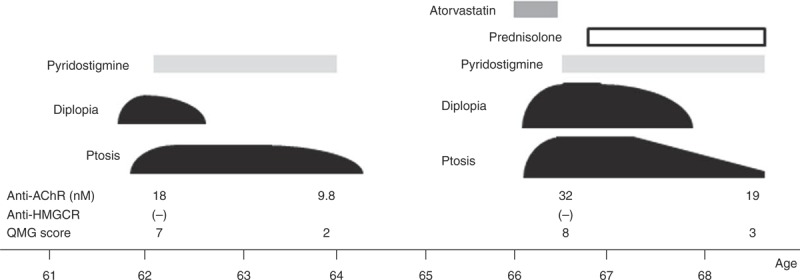
Clinical course of a 68-year-old woman with MG worsening after statin treatment. Anti-AChR = anti-acetylcholine receptor antibody, QMG = quantitative myasthenia gravis.
We examined the presence of anti-HMGCR antibodies by conducting our ELISA, using 251 serum samples from the patients with MG. Only 1 (0.4%) early-onset female patient was found to be seropositive for anti-HMGCR antibodies (anti-HMGCR index: 0.6). She had mild generalized MG (MG Foundation of America class 2A) with no history of statin treatment. She achieved pharmacological remission after treatment with prednisolone and extended thymectomy. In contrast, anti-HMGCR antibodies were not found in the 23 MG patients who had received statins at disease onset. Moreover, the patient with statin-associated MG exacerbation was also negative for anti-HMGCR antibodies at both the onset and the worsening of MG.
DISCUSSION
We established an anti-HMGCR ELISA and showed the following clinical relevance of its use:
anti-HMGCR autoantibodies were specifically detected in necrotizing myopathy,
myopathy associated with anti-HMGCR antibodies was characterized by mild limb weakness and favorable response to immunotherapy with or without statin exposure;
of 251 MG patients, 1 woman (0.4%) with no history of statin therapy had anti-HMGCR antibody.
The molecular weight of HMGCR is 97-kDa. Original reports indicated that a 100-kDa autoantigen in protein immunoprecipitation was successfully identified as HMGCR.4,5 In contrast, our studies showed the doublet autoantigens at around 50-kDa. It is likely that we detected 2 different cleaved forms of the HMGCR molecule. The discrepancy between the previous and present studies may be due to differences in the experimental protocols such as the culture cell lines used as the antigen source. Moreover, the HMGCR expression was upregulated predominantly in the regenerated muscle fibers of anti-HGMCR-positive patients.4 Our findings demonstrated that the HMGCR expression in the muscle fibers was clearly co-localized with patients’ serum. Taken together, the reactivity of our anti-HMGCR ELISA was successfully supported by other, different methods.
The most important pathogenesis of necrotizing myopathy is believed to be autoantibody-mediated. It is necessary to identify autoantibodies in sera of patients with necrotizing myopathy, since there is little or no evidence suggesting autoimmune mechanisms in muscle histology. Anti-SRP antibodies were found in 34 (53%) of 64 patients with necrotizing myopathy.16 In the present study, we demonstrated that 31% of the 26 anti-SRP-negative necrotizing myopathy patients had anti-HMGCR antibodies. Our results showed that only 3 (38%) of the 8 anti-HMGCR-positive patients had undergone statin exposure. This prevalence was consistent with that in a European cohort (44%).7 Taken these findings together, we emphasize that anti-HMGCR antibodies can be regarded as the second serological marker of necrotizing myopathy as well as a marker of statin-induced myopathy.
Importantly, there were clear differences in the clinical characteristics associated with autoantibodies to SRP and HMGCR in our investugation.22 The neurological manifestations of the anti-HMGCR-positive patients were mild limb weakness with good response to immunotherapy. Older patients tended to have anti-HMGCR antibodies.3 In contrast, anti-SRP myopathy was characterized by severe limbs weakness and atrophy as well as bulbar and trunk muscle involvement. Younger patients showed severe clinical deficits.22
Previous reports revealed the clinical features of 15 patients with statin-associated MG.10–15 The ages were ranged as 41 to 71 years (average 58 years) and the sex ratio was 11:4 (M:F). Newly-onset MG was observed in 8 patients and worsening of MG in 7 patients. The patients experienced MG symptoms after 1 to 16 weeks after the statin exposure (average, 4 weeks and within 2 weeks in 8 patients). Ocular MG was observed in 3 patients and generalized MG in 12 patients. The antibodies status was AChR-positive in 10 patients, muscle-specific tyrosine kinase-positive in 4, and seronegative in 1 patient. Discontinuance of statins and initiation of pyridostigmine were effective, but immunotherapy was necessary in 8 patients.
In our case series, it is likely that statins were less involved in the new onset of MG in 23 patients because myasthenic symptoms did not develop within several weeks after the start of the statin therapy. However, 1 patient's MG worsened after 4 weeks of statin use, after an 18-month MG remission. Her symptoms were limited to ocular myasthenia, but prednisolone was necessary to control her disease.
Oh et al13 reported that MG worsening occurred in 6 (11%) of their 54 MG patients with statin treatment. However, the actual incidence of statin-associated MG exacerbation seems to be lower. In clinical practice, we also feel that statins should be used in patients with MG for the same indications as in individuals without MG.23 Statins should be withdrawn if exacerbation of MG occurs, or if the anti-AChR antibody concentration increases markedly.
A limitation of the present study is that anti-HMGCR antibodies were evaluated in only 1 statin-associated MG patient, although the cases of 251 MG patients were examined. However, we think that anti-HMGCR antibodies do not affect the function of neuromuscular transmission. We regard seropositivity with a low titer of anti-HMGCR antibodies in 1 MG patient as a non-specific phenomenon. In a previous report, although organ-specific autoantibodies targeted to thyroid, gastric parietal cells, adrenal cortex, and islet cells were found in up to 30% of 283 MG patients, they had no clinical or pathogenic relevance.24
Our anti-HMGCR ELISA showed an extremely low frequency (0.4%) of seropositivity in MG patients. In this regard, a community-based study also showed that statin users without myopathy were all negative for anti-HMGCR antibodies.25 Based on its high specificity of antibody detection, we contend that the anti-HMGCR ELISA is a useful tool for the diagnosis of necrotizing myopathy, discriminating from other various muscle problems linked to statin exposure.
In conclusion, anti-HMGCR antibodies are a relevant clinical marker of necrotizing myopathy with or without statin-exposure, but they are not associated with the onset or deterioration of MG.
Acknowledgments
None.
Footnotes
Abbreviations: ELISA = enzyme-linked immunosorbent assay, HMGCR = 3-hydroxyl-3-methylglutatyl-coenzyme A reductase, MG = myasthenia gravis, MHC = major histocompatibility complex, MRI = magnetic resonance image, NCAM = neural cell adhesion molecules, OD450 = optical density at 450 nm, SRP = signal recognition particle.
Conflict of interest of interest disclosures: None reported.
REFERENCES
- 1.Ganga HV, Slim HB, Thompson PD. A systematic review of statin-induced muscle problems in clinical trials. Am Heart J 2014; 168:6–15. [DOI] [PubMed] [Google Scholar]
- 2.de Sousa E, Howard J. More evidence for the association between statins and myasthenia gravis. Muscle Nerve 2008; 38:1085–1086. [DOI] [PubMed] [Google Scholar]
- 3.Mohassel P, Mammen AL. Statin-associated autoimmune myopathy and anti-HMGCR autoantibodies. Muscle Nerve 2013; 48:477–483. [DOI] [PubMed] [Google Scholar]
- 4.Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum 2011; 63:713–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christopher-Stine L, Casciola-Rosen LA, Hong G, et al. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum 2010; 62:2757–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drouot L, Allenbach Y, Jouen F. et al: Exploring necrotizing autoimmune myopathies with a novel immunoassay for anti-3-hydroxy-3-methyl-glutaryl-CoA reductase autoantibodies. Arthritis Res Ther 2014; 16:R39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allenbach Y, Drouot L, Rigolet A, et al. Anti-HMGCR autoantibodies in European patients with autoimmune necrotizing myopathies: inconstant exposure to statin. Medicine (Baltimore) 2014; 93:150–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Musset L, Miyara M, Benveniste O, et al. Analysis of autoantibodies to 3-hydroxy-3-methylglutaryl-coenzyme A reductase using different technologies. J Immunol Res 2014; 405956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meriggioli MN, Sanders DB. Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurol 2009; 8:475–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parmar B, Francis PJ, Ragge NK. Statins, fibrates, and ocular myasthenia. Lancet 2002; 360:717. [DOI] [PubMed] [Google Scholar]
- 11.Cartwright MS, Jeffery DR, Nuss GR, et al. Statin-associated exacerbation of myasthenia gravis. Neurology 2004; 63:2188. [DOI] [PubMed] [Google Scholar]
- 12.Purvin V, Kawasaki A, Smith KH, et al. Statin-associated myasthenia gravis: report of 4 cases and review of the literature. Medicine (Baltimore) 2006; 85:82–85. [DOI] [PubMed] [Google Scholar]
- 13.Oh SJ, Dhall R, Young A, et al. Statins may aggravate myasthenia gravis. Muscle Nerve 2008; 38:1101–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elsais A, Lund C, Kerty E. Ptosis, diplopia and statins: an association? Eur J Neurol 2008; 15:e92–e93. [DOI] [PubMed] [Google Scholar]
- 15.Gale J, Danesh-Meyer HV. Statins can induce myasthenia gravis. J Clin Neurosci 2014; 21:195–197. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki S, Yonekawa T, Kuwana M, et al. Clinical and histological findings associated with autoantibodies detected by RNA immunoprecipitation in inflammatory myopathies. J Neuroimmunol 2014; 274:202–208. [DOI] [PubMed] [Google Scholar]
- 17.Hoogendijk JE, Amato AA, Lecky BR, et al. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10-12 October 2003, Naarden, The Netherlands. Neuromuscul Disord 2004; 14:337–345. [DOI] [PubMed] [Google Scholar]
- 18.Hilton-Jones D, Miller A, Parton M, et al. Inclusion body myositis: MRC Centre for Neuromuscular Diseases, IBM workshop, London 2008. Neuromuscul Disord 2010; 20:142–147. [DOI] [PubMed] [Google Scholar]
- 19.Jaretzki A, 3rd, Barohn RJ, Ernstoff RM, et al. Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America. Ann Thorac Surg 2000; 70:327–334. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki S, Utsugisawa K, Iwasa K, et al. Autoimmunity to endoplasmic reticulum chaperone GRP94 in myasthenia gravis. J Neuroimmunol 2011; 237:87–92. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki S, Satoh T, Yasuoka H, et al. Novel autoantibodies to a voltage-gated potassium channel Kv1.4 in a severe form of myasthenia gravis. J Neuroimmunol 2005; 170:141–149. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki S, Hayashi YK, Kuwana M, et al. Myopathy associated with antibodies to signal recognition particle: disease progression and neurological outcome. Arch Neurol 2012; 69:728–732. [DOI] [PubMed] [Google Scholar]
- 23.Gilhus NE. Is it safe to use statins in patients with myasthenia gravis? Nat Clin Pract Neurol 2009; 5:8–9. [DOI] [PubMed] [Google Scholar]
- 24.Klein R, Marx A, Strobel P, et al. Autoimmune associations and autoantibody screening show focused recognition in patient subgroups with generalized myasthenia gravis. Hum Immunol 2013; 74:1184–1193. [DOI] [PubMed] [Google Scholar]
- 25.Mammen AL, Pak K, Williams EK, et al. Rarity of anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibodies in statin users, including those with self-limited musculoskeletal side effects. Arthritis Care Res (Hoboken) 2012; 64:269–272. [DOI] [PMC free article] [PubMed] [Google Scholar]