

Abstract
Hemophagocytic syndrome (HPS) is a potentially life-threatening complication of systemic inflammatory disorders. Adult-onset Still disease (AOSD) is one of the systemic autoimmune diseases associated with reactive hemophagocytic syndrome (RHS). This study aimed to evaluate the characteristic findings, predictive factors, and prognosis of RHS in patients with AOSD.
We retrospectively evaluated 109 patients diagnosed with AOSD and reviewed their clinical data and laboratory findings, including the biopsy results of 21 AOSD patients with RHS. Moreover, data from 17 hemophagocytic lymphohistiocytosis (HLH) patients evaluated during the same period were compared with those from the RHS patients.
Twenty-one patients (19.3%) developed RHS during the course of AOSD, and only 7 patients (6.4%) were confirmed by bone marrow, liver, or lymph node biopsy. AOSD patients with RHS showed significantly higher frequencies of splenomegaly, hepatomegaly, and lymphadenopathy than did those without RHS. Moreover, patients with RHS showed significantly higher relapse rates than those without RHS (61.9% vs 18.2%, P < 0.001). Possible triggering factors inducing hemophagocytosis were detected in 16 of 21 RHS patients (76.2%): disease flare in 12 patients (75%), infection in 3 patients (18.8%), and drug use in 1 patient (6.3%). AOSD patients with RHS showed higher frequencies of leukopenia, anemia, thrombocytopenia, hypoalbuminemia, hypofibrinogenemia, hypertriglyceridemia, hyperferritinemia, and elevated lactate dehydrogenase levels than did those without RHS. Multivariate logistic regression with forward selection procedure showed that low platelet count (<121,000/mm3), anemia, and hepatomegaly were independent predictors of RHS. Patients with definite RHS and those with probable RHS showed comparable results. Although RHS is a life-threatening complication of AOSD, long-term prognosis was observed to be similar in patients with and those without RHS. Compared to RHS patients, HLH patients had poor prognosis, such as higher death rates (52.9% vs 9.5%, P = 0.005).
RHS can be considered when an AOSD patient shows at least 2 of the following 3 findings: low platelet count, anemia, and hepatomegaly. Diagnostic confirmation by biopsy may not be essential if typical clinical findings of RHS are present. Moreover, prognosis of RHS was better than that of HLH diagnosed by the presence of trilineage cytopenia at admission.
INTRODUCTION
Hemophagocytic syndrome (HPS) is a severe and potentially life-threatening complication of systemic inflammatory disorders and is also known as macrophage activation syndrome.1–3 Clinically, it is characterized by acute fever, cytopenia, lymphadenopathy, hepatosplenomegaly, intravascular coagulation, and raised serum levels of ferritin, triglycerides, and liver enzymes. The pathogenesis of this syndrome is excessive activation and proliferation of T lymphocytes and macrophages, which lead to phagocytosis of hematopoietic cells in the bone marrow or other organs of the reticuloendothelial system with hypersecretion of proinflammatory cytokines, including interferon-γ, interleukin (IL)-1, IL-6, IL-18, and tumor necrosis factor (TNF)-α.4–6
HPS may be primary (familial) or secondary (reactive) to autoimmune systemic diseases, malignancies, infections, and drug use.2,7 The primary type is also called familial hemophagocytic lymphohistiocytosis (HLH) and is developed especially in childhood. It is revealed that mutations in the perforin gene related to T-cell cytotoxicity often play a role in the development of familial HLH.8 Reactive hemophagocytic syndrome (RHS) is triggered by various infections, hematologic malignancies, and systemic autoimmune diseases.9,10
Systemic lupus erythematosus (SLE) and adult-onset Still disease (AOSD) are the main systemic autoimmune diseases related to RHS.1,11–14 In these autoimmune diseases, RHS can occur with either a disease flare or an active infection from a complication of immunosuppressive treatment. RHS is known to be the most severe complication of systemic-onset juvenile idiopathic arthritis or Still disease, but this complication has been poorly studied in AOSD.15–17 RHS and AOSD share several clinical characteristics and laboratory findings including fever, lymphadenopathy, hepatosplenomegaly, elevated liver enzyme levels, and hyperferritinemia, and these conditions cannot be easily distinguished from each other.18–20 However, most of the previous studies on AOSD related to RHS have been case reports.21–24 Therefore, the inducing factors, course, treatment, and prognosis of RHS in AOSD are not yet clearly known. It is suggested that RHS in AOSD is more common than has been previously recognized in the literature and has probably been underdiagnosed in several cases.13,18,20
In our study, we reviewed clinical data including laboratory findings, course, treatments, and prognosis in 21 AOSD patients with RHS and compared these data with those from 88 AOSD patients without RHS and 17 HLH patients without AOSD, who were evaluated during the same period. Moreover, we compared 7 definite RHS patients with biopsy-proven hemophagocytosis with 14 probable RHS patients without that. This study aimed to evaluate the characteristic findings, predictive factors, and prognosis of RHS in AOSD patients.
MATERIALS AND METHODS
We retrospectively evaluated 109 patients who had AOSD and were treated at the Department of Rheumatology, Ajou University Hospital, between 1996 and 2013. The patients were diagnosed with AOSD according to the Yamaguchi criteria, after exclusion of infectious, hematologic, and autoimmune diseases.25 This study was approved by the institutional review board of our hospital.
All patients underwent laboratory tests including complete blood count, erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), ferritin, lactate dehydrogenase (LDH), fibrinogen, and triglyceride levels, liver function tests, and urinalysis. Moreover, patients with cytopenia or fever of unknown origin underwent bone marrow, liver, or lymph node biopsy. Clinical characteristics including age, gender, length of follow-up, clinical features, treatments, outcomes, potential triggering factors for RHS, and complications were evaluated using a standardized form. Furthermore, systemic disease score was evaluated using the method described by Pouchot et al,26 in which a score from 0 to 12 is assigned, and 1 point is added for each of the following manifestations: fever, typical rash, pleuritis, pneumonia, pericarditis, hepatomegaly, or abnormal liver function tests, splenomegaly, lymphadenopathy, leukocytosis ≥15,000/mm2, sore throat, myalgia, and abdominal pain.
Of the AOSD patients, 21 (19.3%) were diagnosed with RHS. Seven RHS patients (6.4%) were confirmed by bone marrow, liver, or lymph node biopsy; however, 14 patients (12.8%) without biopsy-proven hemophagocytosis were considered to have probable RHS. Probable RHS was characterized by the following 3 criteria: high fever with unexplained progressive cytopenia affecting at least 2 cell lineages, occurrence of RHS during the course of AOSD in spite of proper treatment, and exclusion of malignancy-associated RHS.18 Three patients with infection at the time of RHS were not excluded because infection can trigger RHS in AOSD patients. Furthermore, we examined 17 HLH patients without autoimmune disease during the same period and compared these with the RHS patients.
All data were expressed as mean ± standard deviation, and a value of P < 0.05 was considered statistically significant. Prognostic factors and responses to medication were assessed using a χ2 test or Fisher exact test and independent sample t test or Mann–Whitney U test wherever appropriate. The cutoff values, sensitivity, specificity, and area under the receiver-operating characteristic (ROC) curve for significantly different laboratory and clinical variables were calculated by ROC analysis, and odds ratios (ORs) for RHS were evaluated by univariate analysis. Finally, multivariate logistic regression with forward selection procedure was performed for significant variables identified by univariate analysis. The Kaplan–Meier method was used to report survival curves and estimate the mean survival and the 95% confidence intervals (CIs). To assess the differences in survival between the 2 groups (AOSD with RHS vs AOSD without RHS and AOSD with RHS vs HLH), log rank test, and Cox proportional hazard model was applied. The statistic analyses were performed using IBM SPSS 20.0 (SPSS Inc, Chicago, IL), and the Kaplan–Meier graph was created using R version 2.13.0 (R Foundation for Statistical Computing, Vienna, Austria).
RESULTS
Clinical Findings in AOSD Patients With RHS
In this study, 130 patients were reviewed retrospectively, of which 21 patients were excluded because they did not meet the Yamaguchi criteria or were confirmed to have other autoimmune diseases such as SLE. Finally, 109 AOSD patients were evaluated.
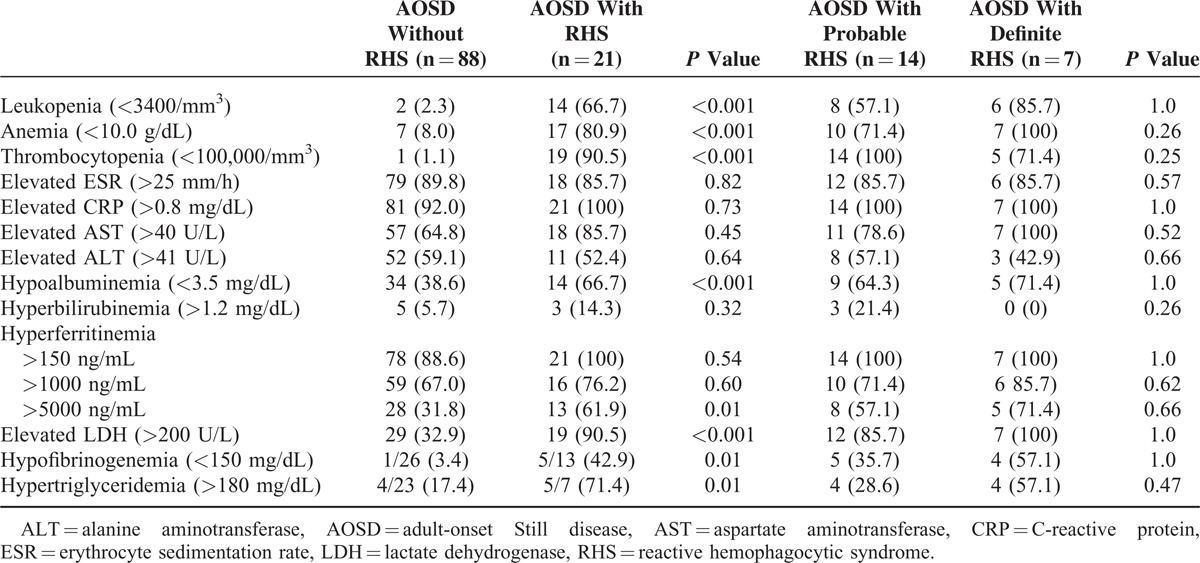
Table 1 shows the clinical characteristics and frequency of clinical features in AOSD patients according to RHS diagnosis. Twenty-one patients (19.3%) developed RHS during the course of AOSD, and only 7 patients were confirmed by bone marrow, liver, or lymph node biopsy. There were no significant differences in age at diagnosis, gender, and follow-up period in AOSD patients, according to RHS diagnosis. Among RHS patients, 15 patients (71.4%) showed hemophagocytic features within 1 month after AOSD diagnosis. Compared with AOSD patients without RHS, patients with RHS showed significantly higher frequencies of splenomegaly, hepatomegaly, and lymphadenopathy, but lower incidences of sore throat. No difference in Pouchot score was observed between patients with and without RHS (5.0 ± 1.6 vs 5.2 ± 1.7).
TABLE 1.
Clinical Characteristics in AOSD Patients According to RHS
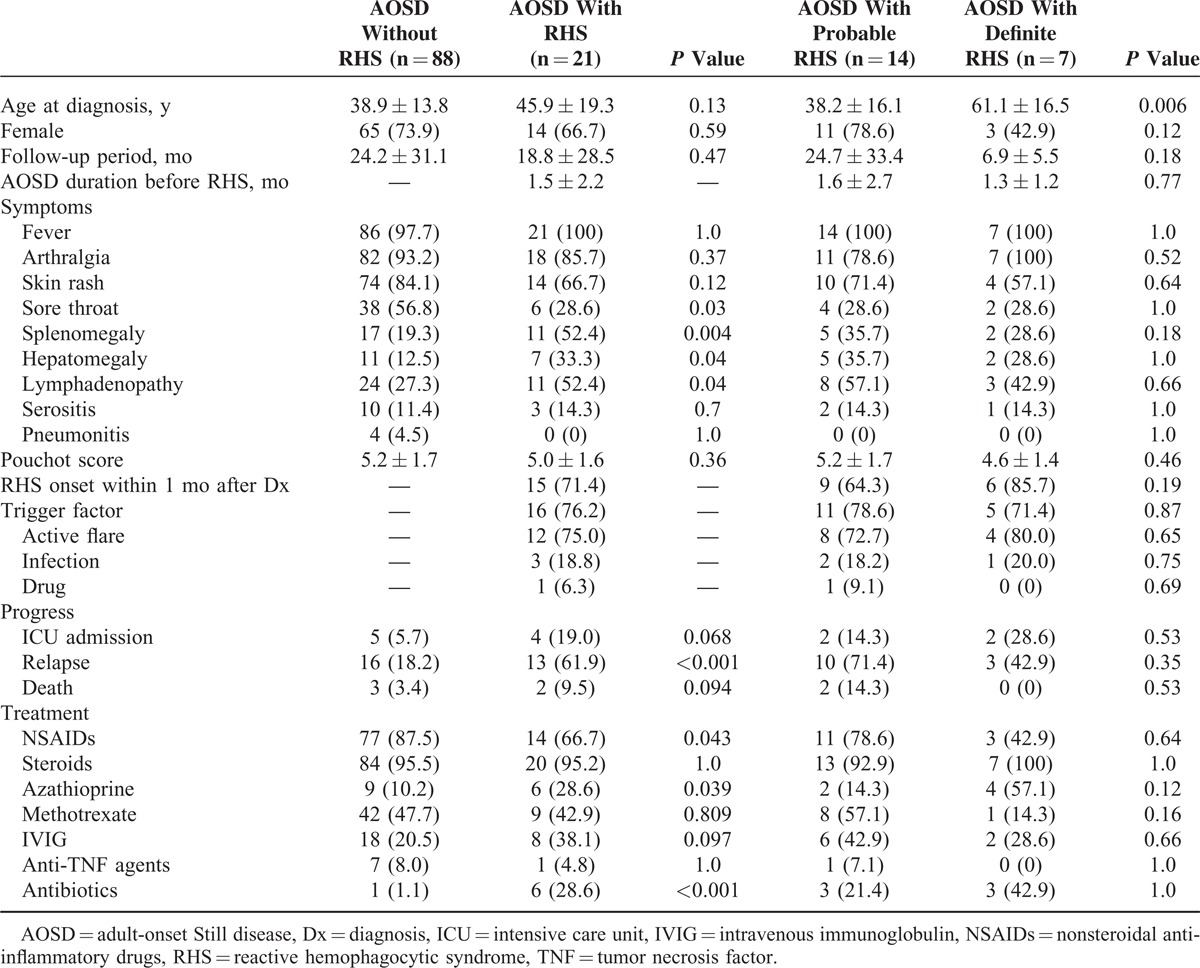
Overall, patients with RHS showed higher frequencies of intensive care unit (ICU) admission and relapse than did those without RHS, but only relapse frequency was significantly higher (61.9% vs 18.2%, P < 0.001). The survival of AOSD patients with RHS was not different with those without RHS (hazard ratio [HR] 7.48, 95% CI [0.52–107.38], P = 0.1389) (Figure 1). Nearly all AOSD patients received corticosteroids, irrespective of RHS. Most RHS patients mainly received oral steroids, and only 2 patients received intravenous steroids because they could not receive oral steroids and pulse doses were necessary. Methotrexate was the most commonly used disease-modifying antirheumatic drug as a steroid-sparing agent, although other disease-modifying antirheumatic drugs including azathioprine, hydroxychloroquine, sulfasalazine, and leflunomide were used. In some patients, intravenous immunoglobulin and anti-TNF agents were used with systemic steroids. There was no difference in the use of immunosuppressive treatments in AOSD patients according to RHS, except for the more common use of azathioprine in those with RHS. Nonsteroidal anti-inflammatory drugs (NSAIDs) were less commonly used in AOSD patients with RHS, whereas antibiotics were more commonly used.
FIGURE 1.
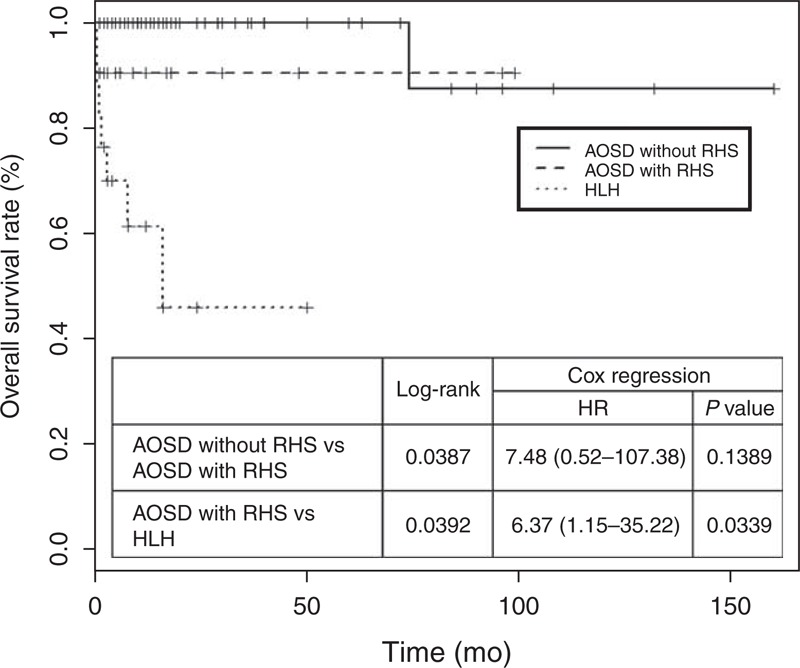
Kaplan–Meier estimates of overall survival in AOSD without RHS, and AOSD with RHS and HLH. The AOSD without RHS patients had a longer median overall survival than the AOSD with RHS patients (P value 0.0387 by log-rank test), and AOSD with RHS patients had a longer median overall survival than HLH patients (P value 0.0392 by log-rank test). The HR of RHS was 7.48 (95% CI, 0.52–107.38; P = 0.1389) and the HR of HLH compared with AOSD with RHS was 6.37 (95% CI, 1.15–35.22; P = 0.0339). AOSD = adult-onset Still disease, CI = confidence interval, HLH = hemophagocytic lymphohistiocytosis, HR = hazard ratio, RHS = reactive hemophagocytic syndrome.
Possible triggering factors inducing hemophagocytosis were detected in 16 of 21 RHS patients (76.2%). Active disease flares were reported in 12 patients (75%), infection in 3 patients (18.8%), and suspicious drug use (antibiotics) in 1 patient (6.3%). Two patients developed acute pharyngitis and 1 patient developed acute pyelonephritis. Five patients (23.8%) with no suspected triggering factors showed bilineage or trilineage cytopenia without any flare symptoms, signs of infection, and suspicious drug use. However, 2 of 5 RHS patients with no triggering factors died despite ICU management, whereas all RHS patients with triggering factors recovered. AOSD patients with definite RHS and those with probable RHS showed no differences in clinical characteristics, except age at diagnosis, which was higher in patients with definite RHS.
Biopsy Findings in AOSD Patients With RHS
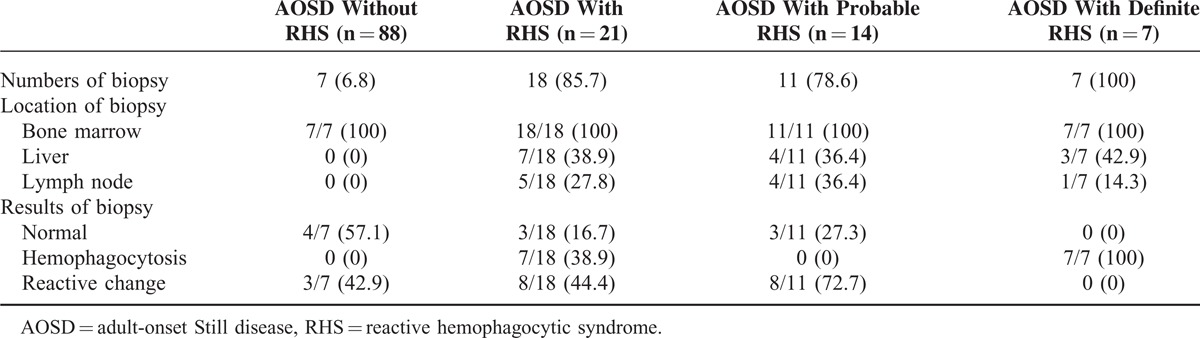
Biopsies were conducted to evaluate the cause of bilineage or trilineage cytopenia or fever of unknown origin. Eighteen patients with RHS (85.7%) and only 7 patients without RHS (6.8%) underwent biopsies (Table 2). Bone marrow biopsy (100%) was most commonly conducted, followed by liver biopsy (38.9%) and lymph node biopsy (27.8%) in RHS patients. All patients with definite RHS showed hemophagocytosis on biopsy, whereas patients with probable RHS showed normal findings (27.3%) or reactive changes (72.7%). Seven AOSD patients without RHS and with leukocytosis underwent biopsy to exclude hematologic malignancies such as myeloproliferative disease. The results were normal (57.1%) or showed reactive changes (42.9%). Reactive plasmacytosis was observed in most cases of reactive changes.
TABLE 2.
Biopsy Findings in AOSD Patients According to RHS
Laboratory Findings in AOSD Patients With RHS
White blood cell count and platelet count, hemoglobin, albumin, and fibrinogen levels were significantly lower in AOSD patients with RHS than in those without RHS (Table 3). However, ferritin, LDH, and triglyceride levels were significantly higher in patients with RHS. AOSD patients with definite RHS and those with probable RHS showed about the same laboratory findings. Frequencies of abnormal laboratory findings were higher in AOSD patients with RHS than in those without RHS (Table 4). In particular, RHS patients showed higher frequencies of leukopenia, anemia, thrombocytopenia, hypoalbuminemia, hypofibrinogenemia, hypertriglyceridemia, and elevated LDH levels. Patients with RHS showed significantly higher frequencies of ferritin levels >5000 ng/mL than did those without RHS (61.9% vs 31.8%, P = 0.01).
TABLE 3.
Laboratory Findings in AOSD Patients According to RHS
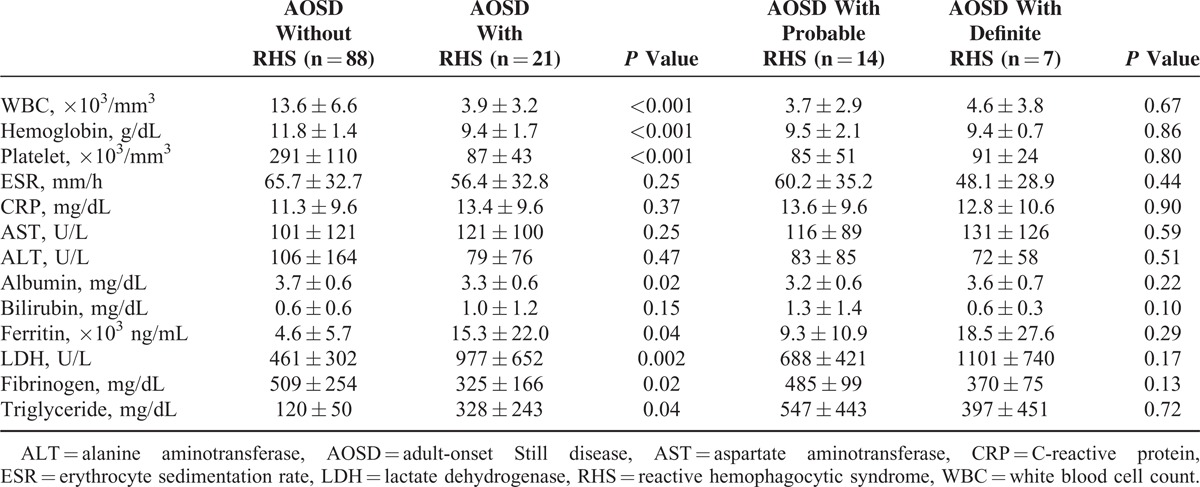
TABLE 4.
Frequencies of Abnormal Laboratory Findings in Patients According to RHS
ROC Curve Analysis in AOSD Patients With RHS
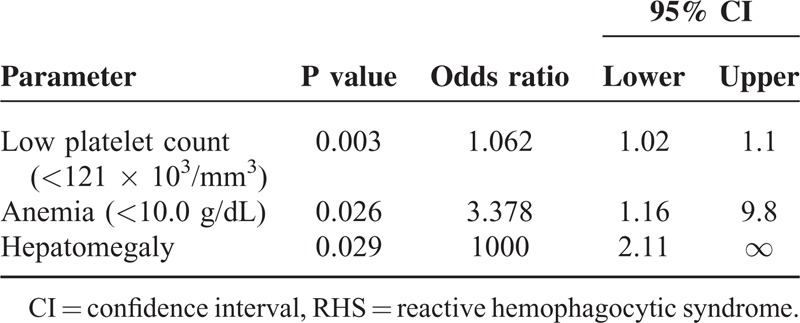
The cutoff values, sensitivity, specificity, and area under the ROC curve (AUC) for significantly different laboratory and clinical variables were calculated by ROC analysis, and ORs for RHS were evaluated by univariate analysis (Table 5). Among the variables, low platelet count with a cutoff value of <121,000/mm3 showed the best sensitivity (96.6%), specificity (95.2%), and AUC value (0.98) as an independent factor, and hypertriglyceridemia (>170 mg/dL) and hypofibrinogenemia (<493 mg/dL) had 100% specificity. To determine predictors of RHS, multivariate logistic regression with forward selection procedure was used for the 9 significant variables identified by univariate analysis. Low platelet count, anemia, and hepatomegaly were found to be significantly independent predictors of RHS (Table 6). AOSD patients showing at least 2 of the 3 above-mentioned findings had the best AUC value (0.87) for RHS diagnosis.
TABLE 5.
Sensitivity and Specificity of Clinical and Laboratory Findings for Diagnosis of RHS

TABLE 6.
Parameters for the Diagnosis of RHS by Multivariate Logistic Regression Analysis
Comparison Between AOSD Patients With RHS and HLH Patients
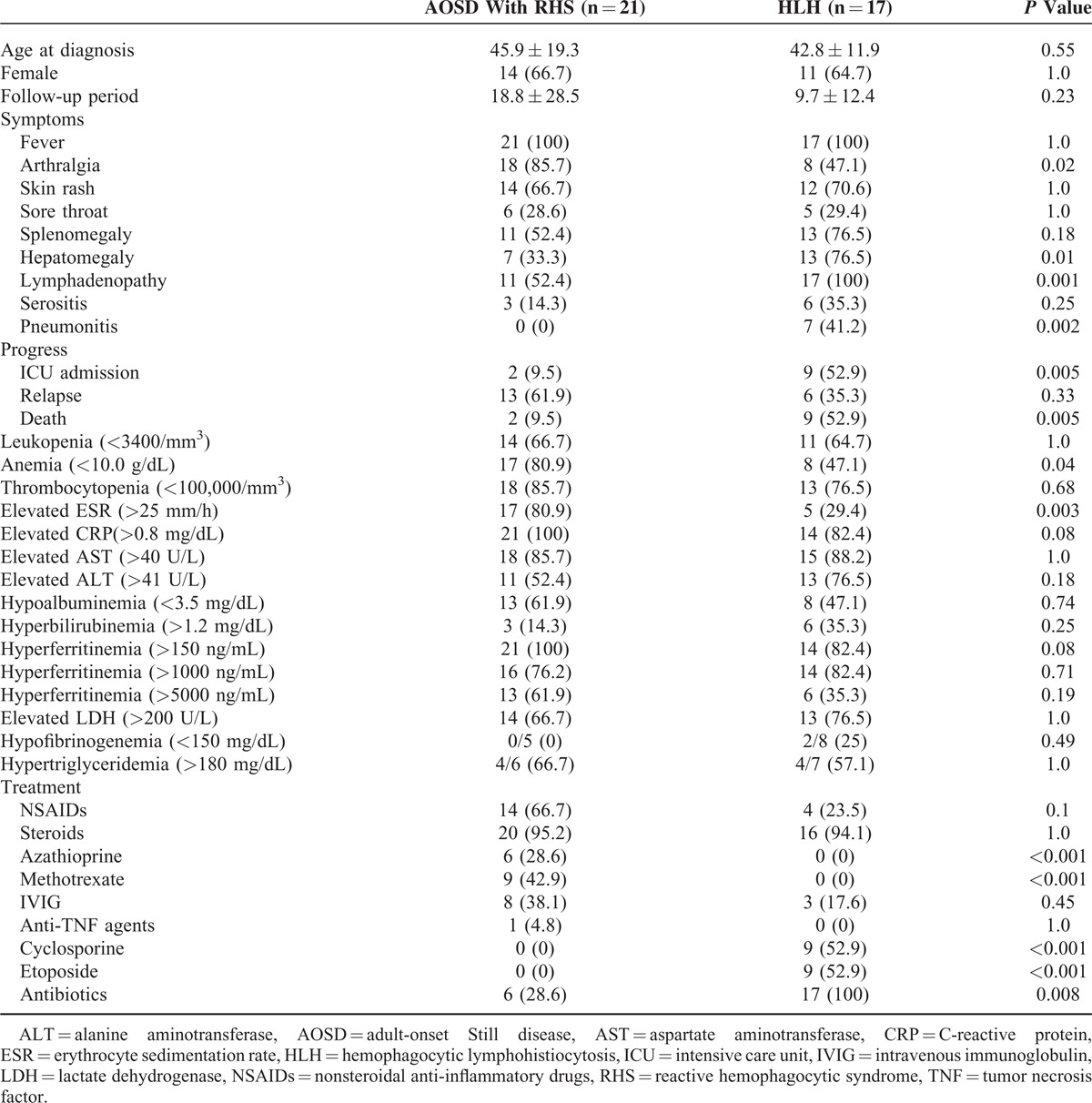
Twenty-one RHS patients were compared with 17 HLH patients evaluated during the same period (Table 7). The frequencies of hematologic manifestations including hepatomegaly (76.5% vs 33.3%, P = 0.01), lymphadenopathy (100% vs 52.4%, P = 0.001), and pneumonitis (41.2% vs 0%, P = 0.002) were significantly higher in HLH patients than in RHS patients. Overall, HLH patients had poor prognosis, such as higher ICU admission and death rates (52.9% vs 9.5%, P = 0.005). The mortality of HLH was significantly higher than AOSD patients with RHS (HR 6.37, 95% CI [1.15–35.22], P = 0.0339) (Figure 1). However, the frequencies of arthralgia, anemia, and elevated ESR were higher in RHS patients. Treatments were different in RHS and HLH patients. Higher frequency of methotrexate use was observed in RHS patients than in HLH patients (42.9% vs 0%). However, compared with RHS patients, 9 HLH patients had higher rates of etoposide and cyclosporine use (52.9% vs 0%), and 17 HLH patients had higher rates of antibiotic use (100% vs 28.6%). NSAID use was higher in RHS patients than in HLH patients (66.7% vs 23.5%), but this difference was not significant (P = 0.1). Steroids were used in almost all HLH (94.1%) and RHS (95.2%) patients.
TABLE 7.
Comparison of Clinical Characteristics Between AOSD Patients With RHS and HLH Patients
DISCUSSION
HPS may be primary (HLH) or secondary (RHS) to infection, malignancies, and drug use.1–3 Moreover, RHS may occur in autoimmune systemic inflammatory diseases such as juvenile idiopathic arthritis, SLE, and AOSD.11–17 Macrophage activation is one of the important clinicopathological findings in AOSD.27 RHS and AOSD manifest similar clinical features such as fever, hepatosplenomegaly, elevated liver enzyme levels, and hyperferritinemia.19 Above all, the presence of cytopenia in at least 2 cell lineages is a distinguished feature of RHS.18
The most important immunological abnormality in primary or secondary HPS is impaired cytotoxic function.4–6 Defective cytotoxicity may result in incomplete pathogen destruction and persistent macrophage activation.28–30 Persistent macrophage activation may result in high levels of various cytokines including interferon-γ, TNF-α, IL-1, IL-6, IL-8, and IL-18, and ferritin production. Persistent macrophage activation in AOSD may lead to RHS after disease flare up or exposure to triggering factors such as infection or use of suspicious drugs such as methotrexate, hydroxychloroquine, and anti-TNF agents.3,30,31
In the present study, the most common triggering factor was disease flare (57.1%), followed by infection (14.3%) and suspicious drug use (4.8%), and no specific triggering factor was noted in 23.8% of the cases. RHS patients without triggering factors showed higher death rates than those with triggering factors, and this may be explained that disease flare, infection, or suspicious drug use can be managed by immunosuppressive treatment, antibiotic therapy, or stopping the offending drug, respectively. Therefore, AOSD patients with bilineage or trilineage cytopenia and without a triggering factor require more careful attention.
Although HPS is characterized by macrophage activation with hemophagocytosis, very few studies have focused on macrophage activation in HPS patients. Phenotypic markers expressed by macrophages in a small number of primary HPS patients comprise complement receptors (CD11b, CD21, CD35, and CD36) and activation markers (CD25 and CD30), whereas those from secondary HPS patients and healthy controls do not have these.32 Therefore, study of macrophage subpopulations such as classic (M1) or type-2-activated macrophages is important to determine the cause of HPS in patients.33
In a retrospective study of 50 patients with AOSD, 6 patients (12%) were reported to have RHS, of which 1 patient died.20 Another retrospective study of 26 AOSD patients showed that 3 patients (11.5%) developed RHS and 1 patient died because of uncontrolled infection.14 A recent retrospective case–control study showed that 8 (15.3%) of 52 AOSD patients developed RHS, and RHS was the first manifestation of AOSD in 7 patients.18
In our study, 21 (19.3%) of 109 AOSD patients developed RHS, and 4 patients were treated in the ICU, of which 2 patients died. Among them, 15 patients (71.4%) were confirmed to have RHS within 1 month after AOSD diagnosis. These findings reveal that most RHS may occur at the very beginning during the course of AOSD, even if a simultaneous diagnosis of AOSD and RHS is common. Moreover, it was reported that 63.2% of SLE patients were diagnosed with macrophage activation within 1 month after SLE diagnosis.34
We compared the clinical and laboratory findings of definite RHS patients confirmed by biopsy and probable RHS patients. The probable RHS patients may have a milder form of the disease, which may not have been detected by biopsy, or these patients may not have true HPS. Our data show that definite RHS and probable RHS patients showed almost similar clinical characteristics and laboratory findings. It has been reported that histological confirmation of hemophagocytosis is not always required for HLH diagnosis.19,35,36 Our data also support that RHS in AOSD can be diagnosed without bone marrow, liver, or lymph node biopsy-proven hemophagocytosis if there is sufficient clinical proof of hemophagocytosis.
Laboratory findings helped to differentiate RHS patients from patients without RHS, whereas clinical findings were rather nonspecific. The clinical findings showed that frequencies of hepatomegaly, splenomegaly, and lymphadenopathy were higher in RHS patients. Moreover, the frequency of abnormal complete blood count results including leukopenia, anemia, and thrombocytopenia was higher in RHS patients. Above all, low platelet count (<121,000/mm3) produced the best AUC (0.98) with the highest combined sensitivity (96.6%) and specificity (95.2%) as an independent predictor of RHS. Multiple logistic regression analysis showed that only low platelet count, anemia, and hepatomegaly were significant predictors of RHS. In particular, AOSD patients showing at least 2 of the 3 above-mentioned findings could be diagnosed as RHS with the best AUC value (0.87). Of 21 RHS patients, 16 (76.2%) presented with leukocytosis (>10,000/mm3) at admission, and within 1 month, they developed cytopenia in at least 2 lineages including leukopenia. Only 5 RHS patients (23.8%) presented with leukopenia and thrombocytopenia at admission, and they showed worse prognosis than the RHS patients presenting with leukocytosis at admission. However, 13 of 17 HLH patients (76.5%) presented with pancytopenia at admission, whereas no HLH patient presented with leukocytosis. It has been suggested that a low ESR is an important sign for distinguishing between RHS and other inflammatory disorders.16 However, in the present study, neither ESR nor CRP showed significant differences according to the presence of RHS in AOSD patients.
The treatment guidelines for RHS in AOSD have not been established.1 For juvenile idiopathic arthritis-associated RHS, the main treatment is the administration of high-dose steroids.1–3 However, refractory cases of RHS have been reported despite the application of the high-dose steroid treatment.17,20–22 Conflicting results have been reported with the administration of high-dose intravenous immunoglobulins, cyclosporin, cyclophosphamide, plasma exchange, etoposide, and anti-TNF agents.1–3,37–39 In Japanese patients with systemic autoimmune disease and RHS, high-dose steroid monotherapy was given in 26 cases, being effective in 12 (46%), and 10 out of 15 patients resistant to steroids were administered cyclosporin, cyclophosphamide, or tacrolimus, leading to remission in 80%.14
In our study, cyclosporin, cyclophosphamide, and etoposide were not used because steroid monotherapy was effective in 16 of 20 RHS patients (80%). Methotrexate (42.9%) and azathioprine (28.6%) were used as steroid-sparing agents, and intravenous immunoglobulin (38.1%) was administered for the rapid correction of severe thrombocytopenia (<50,000/mm3). On the basis of the increase in serum TNF-α levels in RHS patients, anti-TNF agents can be used for the treatment of AOSD patients with RHS.38,39 In the present study, etanercept was used in 1 patient because of steroid resistance, and the patient recovered without any side effects. However, cyclosporine, etoposide, and high-dose steroids were used as the main chemotherapy agents in HLH patients (47.1%). Furthermore, more than half of the HLH patients (52.9%) died although aggressive chemotherapy and prophylactic antibiotics were used.
Our study has several potential limitations. Data were collected retrospectively; therefore, some data may be missing and some laboratory findings were not available in few patients. Moreover, a potential bias may exist because all the clinical and laboratory data were collected from a single center. However, our study has the largest sample size to date and included 2 control groups such as AOSD patients without RHS and HLH patients without systemic autoimmune diseases.
In conclusion, RHS can be considered when an AOSD patient shows at least 2 of the following 3 findings: low platelet count (<121,000/mm3), anemia, and hepatomegaly. Diagnostic confirmation by biopsy may not be essential if typical clinical and laboratory findings of RHS are reported. Although RHS is a life-threatening complication of AOSD, long-term prognosis is found to be similar in AOSD patients with and without RHS. Moreover, prognosis of HLH diagnosed by the presence of trilineage cytopenia at admission is worse than that of RHS. Further studies in larger populations are required to confirm our results on the diagnosis of RHS in AOSD patients.
Acknowledgment
The authors would gratefully acknowledge Dr Dae-Ryong Kang (Department of Medical Humanities & Social Medicine, Office of Biostatistics, Ajou University School of Medicine, Suwon, Korea) for his statistical help.
Footnotes
Abbreviations: AOSD = adult-onset Still disease, AUC = area under the ROC curve, ESR = erythrocyte sedimentation rate, HLH = hemophagocytic lymphohistiocytosis, HPS = hemophagocytic syndrome, ICU = intensive care unit, IL = interleukin, LDH = lactate dehydrogenase, RHS = reactive hemophagocytic syndrome, ROC = receiver-operating characteristic, SLE = systemic lupus erythematosus, TNF = tumor necrosis factor.
The authors have no funding and conflicts of interest to disclose.
REFERENCES
- 1.Ramos-Casals M, Brito-Zeron P, Lopez-Guillermo A, et al. Adult haemophagocytic syndrome. Lancet 2014; 383:1503–1516. [DOI] [PubMed] [Google Scholar]
- 2.Janka GE. Hemophagocytic syndrome. Blood Rev 2007; 21:235–253. [DOI] [PubMed] [Google Scholar]
- 3.Ravelli A. Macrophage activation syndrome. Curr Opin Rheumatol 2002; 14:548–552. [DOI] [PubMed] [Google Scholar]
- 4.Larroche G, Mouthon L. Pathogenesis of hemopahgocytic syndrome. Autoimmun Rev 2004; 3:69–75. [DOI] [PubMed] [Google Scholar]
- 5.Arceci RJ. Whem T cells and macrophage do not talk: the hemophagocytic syndrome. Curr Opin Hematol 2008; 15:359–367. [DOI] [PubMed] [Google Scholar]
- 6.Canna SW, Behrens EM. Not all hemophagocytes are created equally: appreciating the heterogeneity of the hemophagocytic syndrome. Curr Opin Rheumatol 2012; 24:113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Imashuku S. Differential diagnosis of hemophagocytic syndrome: underlying disorders and selection of the most effective treatment. Int J Hematol 1997; 66:135–151. [DOI] [PubMed] [Google Scholar]
- 8.Henter JI, Horne A, Arico M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007; 48:124–131. [DOI] [PubMed] [Google Scholar]
- 9.Grom AA. Macrophage activation syndrome and reactive hemophagocytic lymphohistiocytosis: the same entities? Curr Opin Rheumatol 2003; 15:587–590. [DOI] [PubMed] [Google Scholar]
- 10.Castillo L, Carcillo J. Secondary hemophagocytic lymphohistiocytosis and severe sepsis/systemic inflammatory response syndrome/multiorgan dysfunction syndrome/macrophage activation syndrome share common intermediate phenotypes on a spectrum of inflammation. Pediatr Crit Care Med 2009; 10:387–392. [DOI] [PubMed] [Google Scholar]
- 11.Dhote R, Simon J, Papo T, et al. Reactive hemophagocytic syndrome in adult systemic disease: report of twenty-six cases and literature review. Arthritis Rheum 2003; 49:633–639. [DOI] [PubMed] [Google Scholar]
- 12.Kumakura S, Ishikura H, Kondo M, et al. Autoimmune-associated hemophagocytic syndrome. Mod Rheumatol 2004; 14:205–215. [DOI] [PubMed] [Google Scholar]
- 13.Tristano AG. Macrophage activation syndrome: a frequent but under-diagnosed complication associated with rheumatic diseases. Med Sci Monit 2008; 14:RA27–36. [PubMed] [Google Scholar]
- 14.Fukaya S, Yasuda S, Hashimoto T, et al. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: analysis of 30 cases. Rheumatology (Oxford) 2008; 47:1686–1691. [DOI] [PubMed] [Google Scholar]
- 15.Lin CI, Yu HH, Lee JH, et al. Clinical analysis of macrophage activation syndrome in pediatric patients with autoimmune diseases. Clin Rheumatol 2012; 31:1223–1230. [DOI] [PubMed] [Google Scholar]
- 16.Stephan JL, Kone-Paut I, Gamambrun C, et al. Reactive haemophagocytic syndrome in children with inflammatory disorders. A retrospective study of 24 patients. Rheumatology (Oxford) 2001; 40:1285–1292. [DOI] [PubMed] [Google Scholar]
- 17.Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child 2001; 85:421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hot A, Toh ML, Cope B, et al. Reactive hemophagocytic syndrome in adult-onset Still disease: clinical feature and long-term outcome: a case-conrol study of 8 patients. Medicine (Baltimore) 2010; 89:37–46. [DOI] [PubMed] [Google Scholar]
- 19.Felix FH, Leal LK, Fontenele JB. Cloak and dagger: the case for adult onset Still disease and hemophagocytic lymphohistiocytosis. Rheumatol Int 2009; 29:973–974. [DOI] [PubMed] [Google Scholar]
- 20.Arlet JB, Le TH, Marinho A, et al. Reactive haemophagocytic syndrome in adult-onset Still's disease: a report of six patients and a review of the literature. Ann Rheum Dis 2006; 65:1596–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burgi U, Mendez A, Hasler P, et al. Hemophagocytic syndrome in adult-onset Still's disease (AOSD): a must for biologics? Case report and brief review of the literature. Rheumatol Int 2012; 32:3269–3272. [DOI] [PubMed] [Google Scholar]
- 22.Fujii K, Kitamura Y, Osugi Y, et al. A case of adult-onset Still's disease complicated by hemophagocyti syndrome and interstitial pneumonia with pneumomediastium/recurrent pneumonia. Int J Rheum Dis 2012; 15:e60–e62. [DOI] [PubMed] [Google Scholar]
- 23.Pamuk ON, Pamuk GE, Usta U, et al. Hemophagocytic syndrome in one patient with adult onset Still's disease: presentation with febrile neutropenia. Clin Rheumatol 2007; 26:797–800. [DOI] [PubMed] [Google Scholar]
- 24.Kim HS, Park KW, Kim SK, et al. Two cases of adult onset Still's disease with comcomitant hemophagocytic syndrome. J Rheum Dis 2007; 14:160–165. [Google Scholar]
- 25.Yamaguchi M, Ohta A, Tsunematu T, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol 1992; 19:424–430. [PubMed] [Google Scholar]
- 26.Pouchot J, Sampalis JS, Beaudet F, et al. Adult Still's disease: manifestations, disease course, and outcome in 62 patients. Medicine (Baltimore) 1991; 70:118–136. [PubMed] [Google Scholar]
- 27.Lambotte O, Cacoub P, Costedoat N, et al. High ferritin and low glycosylated ferritin may also be a marker of excessive macrophage activation. J Rheumatol 2003; 30:1027–1028. [PubMed] [Google Scholar]
- 28.Wulffraat NM, Rijkers GT, Elst E, et al. Reduced perforin expression in systemic juvenile idiopathic arthritis is restored by autologous stem-cell transplantation. Rheumatology (Oxford) 2003; 42:375–379. [DOI] [PubMed] [Google Scholar]
- 29.Villanueva J, Lee S, Giannini EH, et al. Natural killer cell dysfunction is a distinguishing feature of systemic onset juvenile rheumatoid arthritis and macrophage activation syndrome. Arthritis Res Ther 2005; 7:R30–R37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grom AA. Natural killer cell dysfunction: A common pathway in systemic-onset juvenile rheumatoid arthritis, macrophage activation syndrome, and hemophagocytic lymphohistiocytosis? Arthritis Rheum 2004; 50:689–698. [DOI] [PubMed] [Google Scholar]
- 31.Amenomori M, Migita K, Miyashita T, et al. Cytomegalovirus-associated hemophagocytic syndrome in a patient with adult onset Still's disease. Clin Exp Rheumatol 2005; 23:100–102. [PubMed] [Google Scholar]
- 32.Buckley PJ, O’Laughlin S, Komp DM. Histiocytes in familial and infection-induced/idiopathic hemophagocytic syndromes may exhibit phenotypic differences. Pediatr Pathol 1992; 12:51–66. [DOI] [PubMed] [Google Scholar]
- 33.Mosser DM. The many faces of macrophage activation. J Leukoc Biol 2003; 73:209–212. [DOI] [PubMed] [Google Scholar]
- 34.Parodi A, Davi S, Pringe AB, et al. Macrophage activation syndrome in juvenile systemic lupus erythematosus: a multinational multicenter study of thirty-eight patients. Arthritis Rheum 2009; 60:3388–3399. [DOI] [PubMed] [Google Scholar]
- 35.Gupta A, Weitzman S, Abdelhaleem M. The role of hemophagocytosis in bone marrow aspirates in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2008; 50:192–194. [DOI] [PubMed] [Google Scholar]
- 36.Janka GE, Schneider EM. Modern management of children with haemophagocytic lymphohistiocytosis. Br J Haematol 2004; 124:4–14. [DOI] [PubMed] [Google Scholar]
- 37.Emmenegger U, Frey U, Reimers A, et al. Hyperferritinemia as indicator for intravenous immunoglobulin treatment in reactive macrophage activation syndromes. Am J Hematol 2001; 68:4–10. [DOI] [PubMed] [Google Scholar]
- 38.Takahashi N, Naniwa T, Banno S. Successful use of etanercept in the treatment of acute lupus hemophagocytic syndrome. Mod Rheumatol 2008; 18:72–75. [DOI] [PubMed] [Google Scholar]
- 39.Henzan T, Nagafuji K, Tsukamoto H, et al. Success with infl iximab in treating refractory hemophagocytic lymphohistiocytosis. Am J Hematol 2006; 81:59–61. [DOI] [PubMed] [Google Scholar]