

Abstract
The connection between Helicobacter pylori (Hp) infection and eye diseases has been increasingly reported in the literature and in active research. The implication of this bacterium in chronic eye diseases, such as blepharitis, glaucoma, central serous chorioretinopathy and others, has been hypothesized. Although the mechanisms by which this association occurs are currently unknown, this review describes shared pathogenetic mechanisms in an attempt to identify a lowest common denominator between eye diseases and Hp infection.
The aim of this review is to assess whether different studies could be compared and to establish whether or not Hp infection and Eye diseases share common pathogenetic aspects. In particular, it has been focused on oxidative damage as a possible link between these pathologies.
Text word search in Medline from 1998 to July 2014.
152 studies were included in our review.
Were taken into considerations only studies that related eye diseases more frequent and/or known.
Likely oxidative stress plays a key role. All of the diseases studied seem to follow a common pattern that implicates a cellular response correlated with a sublethal dose of oxidative stress. These alterations seem to be shared by both Hp infections and ocular diseases and include the following: decline in mitochondrial function, increases in the rate of reactive oxygen species production, accumulation of mitochondrial DNA mutations, increases in the levels of oxidative damage to DNA, proteins and lipids, and decreases in the capacity to degrade oxidatively damaged proteins and other macromolecules. This cascade of events appears to repeat itself in different diseases, regardless of the identity of the affected tissue. The trabecular meshwork, conjunctiva, and retina can each show how oxidative stress may acts as a common disease effector as the Helicobacter infection spreads, supported by the increased oxidative damage and other inflammation.
INTRODUCTION
Helicobacter pylori (Hp) is a major pathogen that is etiologically associated with gastritis, peptic ulcer disease, gastric cancer and primary gastric lymphoma. Worldwide, it is one of the most common chronic infections, although the exact mode of Hp transmission remains controversial. Transmission during transit disorders of the gastrointestinal tract has been suggested, although there is no evidence to date of its transmission during outbreaks of gastroenteritis.1,2 The host response to Hp precipitates the induction of damage to the gastric epithelium and therefore plays an integral role in Hp pathogenesis. Therefore, the Hp infection is acquired by oral ingestion of the bacterium and is mainly transmitted within families in early childhood.3 However, it is very difficult to identify the initial site of the Hp infection. Some authors have considered the role of Hp infections with eye diseases, including some afflictions as widespread as glaucoma and others as uncommon as mucosa-associated lymphoid tissue (MALT) lymphoma. Despite the repeated implication of Hp in the etiology of eye diseases, we still do not know the real of how Hp begins to target the eye. Mindel and Rosenberg, in 1997, for the first time, described Hp and ocular pathology.4 There are common risk factors that can be evident in several diseases. For instance, genome stability is essential for maintaining cellular and organism homeostasis, but it is subject to many discussions. One ubiquitous threat is from a class of compounds known as reactive oxygen species (ROS), which can indiscriminately react with many cellular biomolecules including proteins, lipids, and DNA to produce a variety of oxidative lesions. The human eye is constantly exposed to sunlight and artificial lighting. Exogenous sources of ROS such as UV light, visible light, ionizing radiation, chemotherapeutics, and environmental toxins may contribute to the oxidative damage in the tissues.5 The aging eye also appears to be at considerable risk from oxidative stress. Mitochondria are the major target of ROS, and alterations in the efficiency of mitochondrial respiration resulting in superoxide production. This precedes subsequent reactions that form potentially dangerous ROS species such as the hydroxyl radical, hydrogen peroxide and peroxynitrite. Therefore, during the life ROS-induced damage in the eye may consist of oxidation of proteins and DNA damage occurs in the various tissues of the eye. The ROS may be the trait d’union between eye diseases and infection by HP. The purpose of this review is to investigate the relationships between the eye and Hp infections and emphasize the common elements among the various diseases of the eye and the Hp infection and emphasize the elements that are shared between the different diseases of the eye and the Hp infection which are the possible common denominator of their pathogenesis.
METHODS
We performed a literature search consisting of a text word search in Medline from 1998 to July 2014. Articles dealing with pathogenetic aspects of HP infection, eye disease, and oxidative damage and stress were selected and reviewed. The papers were sought in two databases Pub Med and Science Direct. The combinations of words that we used were: “Helicobacter AND eye” papers found: 82 (37); “Helicobacter AND pathogenesis AND eye” papers found: 76 (46); Helicobacter pylori infection AND pathogenesis AND oxidative stress“ papers found: 131 (77); “Eye AND pathogenesis AND oxidative stress“ papers found: 1557 (157). We reviewed only papers written in English. The number of papers actually reviewed is shown in brackets: all abstracts were read and, if subject was compatible with our article, the paper was reviewed in detail.
This article is a review, so it was not necessary to ask for the approval of this study to the ethics committee of the IRCCS San Martino University Hospital - IST.
Helicobacter and Blepharitis
Blepharitis is a common condition where the eyelids are inflamed, with oily particles and bacteria coating the eyelid margin near the base of the eyelashes. The underlying causes of blepharitis are not completely understood, it can be associated with a bacterial eye infection, dry eyes symptoms and certain types of skin conditions such as acne rosacea. From a dermatological aspect, a diagnosis of blepharitis is done if one or more lesions is observed on the eyelid margin in association with a non-granulomatous inflammatory reaction. This lesion is classified as rosacea blepharitis if the following were present: flushing; persistent erythema with scattered telangiectasia, papules and pustule; sprays of vessels, especially on the nose or cheeks; or phymas. Seborrheic blepharitis is diagnosed in the presence of the following signs: greasy-looking scales and/or crusts; increased redness and cutaneous color variability; or clinical patterns on the trunk, scalp, and face (including lids). Mixed type of dermatitis or blepharitis is diagnosed if one or more lesions fulfilling the criteria of both of the classification systems previously described were observed.6 (Figure 1) Rosacea is considered in conjunction with diseases of the anterior segment, such as blepharitis, nodular conjunctivitis, episcleritis and painful marginal infiltrates keratitis. It is interesting to underline that digestive troubles are more connected to rosacea than to seborrhea,7 although the two pathological conditions are closely related. According to Boni R., “Diseases of Seborrheic origin include rosacea, acne, gram-negative folliculitis, demodex-folliculorum, perioral dermatitis as well as Seborrheic dermatitis.”8
FIGURE 1.
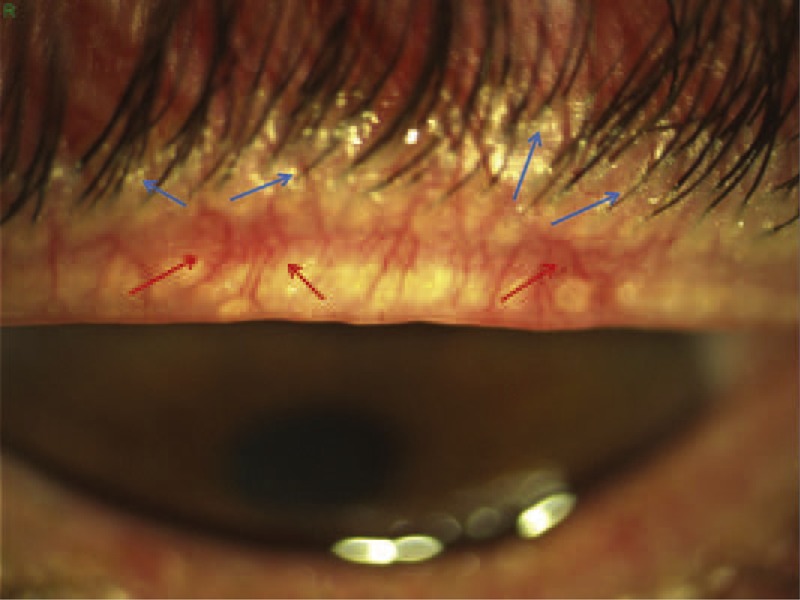
Anterior blepharitis of a mixed type of the upper eyelid. This shows a squamous blepharitis-hyperemic. Very evident the presence of collarettes, meibomian secretion around eyelashes (blue arrows) and telangiectasia and very hyperemic vessels of the lid margin (red arrows). Although it is unknown the role of Hp infection in this type of pathology, its eradication leads to an improvement in both subjective and objective symptoms and can, especially in the young, alleviate blepharitis.
Hp is the primary cause of gastritis and a major contributor to peptic ulcer disease. There have been several investigations suggesting a possible etiologic role of Hp in rosacea.9 The link between seborrhea and Hp appears to be more uncertain, although it has been reported by other authors.10 Moreover, Seborrheic dermatitis may be observed in conjunction with other skin diseases, such as rosacea, blepharitis and ocular rosacea, and with acne vulgaris.11 Thus, it can therefore be claimed, although with some uncertainty, that digestive troubles may be correlated, or at least associated, with the presence of blepharitis. According to this hypothesis, another study was investigate the relationship between blepharitis and Hp.6 Although, possible sources of error must be considered when defining the association of two highly prevalent conditions, the data seem to validate an association between Hp infection and blepharitis. However, this association may still not be indicative of a causal association.12 Therefore, given that blepharitis and Hp infection are wide spread, it remains difficult to know whether this prevalence is real or rather random. Furthermore, several investigations have suggested a possible etiological role for Hp in rosacea,13 as the prevalence of Hp infection in patients with rosacea is higher than that in control subjects13,14 and Hp eradication treatment reduces the severity of rosacea.14,15 Overall, the relationship between blepharitis and Hp infection is not influenced by clinical appearance or degree16; it seems that their only common factor is chronic inflammation of the eyelid and gastrointestinal tract. Indeed, gastric epithelial cells release cytokines, such as interleukins, which act as proinflammatory stimuli, promoting the release of the other cytokines and contributing to the inflammatory state, in combination with the histamine from mast cell degranulation.17 Cross-mimicry mechanisms between bacterial and extradigestive antigens could affect extradigestive organs.18,19 Free radical and lipid peroxide generations are crucial to the attained events in inflammation; thus, Hp can increase the serum or tissue levels of nitric oxide,20 inducing vasodilatation, inflammation, and immune modulation. The effectiveness of the therapy is certainly connected with the lid inflammation, but it is not possible to know whether rosacea plays a greater role than the other blepharitis types.13,15 Chronic blepharitis is one of the most difficult ocular diseases to treat, and we do not know whether the antibiotics that treat the digestive disturbance have a secondary effect on the blepharitis. Thus, the Hp eradication therapy treats the infection at the same time as acts on the state and the flora of the eyelids. This is indeed plausible, as antibiotics may have local effects, in addition to systemic effects. Thus, these drugs may act on the conjunctival and lid bacteria, as well as on the Hp. In some cases, it has been demonstrated that the associations among chronic blepharitis, eyelid meibomian gland lipids and the microflora reveal important relationships between these lipids and chronic blepharitis disease states. Antibiotics, such as tetracycline, inhibit lipase activity, thereby decreasing the release of noxious free fatty acids.21 Dougherty et al22 have shown that tetracycline results in decreased bacteriological lipase activity in vitro.23 Therefore, it is reasonable to suppose that antibiotics act upon blepharitis. The cause of rosacea remains unknown, even if the associations between rosacea and certain digestive diseases, such as gastritis, hypochlorhydria, or a number of jejunal mucosal abnormalities, are well established.16 Among many theories, the role of Hp has often been a subject of investigation.24 Different studies have presented conflicting results. The mainstay treatments of ocular rosacea are topical metronidazole and oral tetracyclines, administered over several months. Furthermore, topical metronidazole, which is effective for stage I and stage II rosacea and avoids the toxicity of systemic treatment, is regarded as a first-line therapy.25 Rosacea responds well to oral antibiotics,26 and its systemic treatment includes metronidazole, which was used in this clinical study. Therefore, is difficult to understand how and whether there can be a direct infection of the eyelid with the Hp. Although Hp has been found in the mouth,27 the presence of Hp in a human oral cavity should be considered transient and independent of its oral status,28 but there is as of yet no research showing a direct causal relationship between Hp infection and diseases. Finally, there are currently no other studies seeking to understand the relationship between blepharitis and Hp.
Helicobacter and Glaucoma
Glaucoma affects more than 70 million people worldwide.29 Glaucoma is a progressive optic neuropathy characterized by a modification of optic nerve head and visual field damage, which result from the loss of retinal ganglion cells by apoptosis.30 Moreover, in high tension glaucoma oxidative stress is the “primum movens" that affects trabecular meshwork (TM),31 particularly its endothelial cells.32 In these develops a real mitochondriopaty. Indeed, mitochondria produce up to 90% of required cellular energy and play a crucial role in mediated cell death through apoptotic pathways.33 Damage accrued by the mitochondrial genome (mtgenome) is associated with increased cellular stress and organelle dysfunction.34 TM cells of patients with primary open angle glaucoma (POAG) have lower levels of ATP, as their functionality is endangered by an intrinsic mitochondrial complex I defect that leaves these cells mitochondrial respiratory chain-deficient.35 Mitochondrial DNA (mtDNA) deletion is dramatically increased in the TM of POAG patients, compared to controls, and the ratio between mtDNA and nuclear DNA is decreased; additionally, the amount of nuclear DNA per mg wet tissue is decreased,36 confirming that mitochondrial damage is severe in the TM of POAG patients. The trabecular meshwork altering both motility and cytoarchitecture, inducing cells die by apoptosis, losing barrier functions and altering the aqueous humor outflow. This is the reason intraocular pressure (IOP) increase occurs during glaucoma.32 Under normal physiological conditions, approximately 1%–5% of the oxygen consumed by mitochondria is converted to ROS, including superoxide anions, hydrogen peroxide, and hydroxyl radicals.37 Mitochondrial respiratory function declines with age,38,39 which increases the production of ROS and free radicals in mitochondria. Further, ROS and oxidants can function as intracellular signaling molecules, conditioning cell death or survival.40 The wide spectrum of alterations in aged individuals and senescent cells is correlated with the cellular response to a sublethal dose of oxidative stress. These alterations and responses include the following: (1) decline in mitochondrial respiratory function; (2) increase in the rate of production of ROS; (3) accumulation of mitochondrial DNA (mtDNA) mutations; (4) increase in the levels of oxidative damage to DNA, proteins and lipids; and (5) decrease in the capacities of degradation in oxidatively damaged proteins and other macromolecules.41 Responses to oxidative stress and their subsequent interactions in tissues result in deleterious effects on cellular functions, which culminate in aging and degenerative diseases. Oxidative modification and mutation of mtDNA occur with great ease, and the extent of such alterations of mitochondrial DNA increases exponentially with age. Oxidative modification in mtDNA is much more extensive than in nuclear DNA.42,43 Age-related alterations in the respiratory enzymes not only decrease ATP synthesis but also enhance the production of ROS by increasing electron leakage in the respiratory chain. With the accumulation of genetic defects in mechanisms of mitochondrial energy production, the issue of neuronal susceptibility to damage as a function of aging becomes important.44 Damage to mtDNA induces alterations to the polypeptides encoded by the mtDNA in the respiratory complexes, resulting in consequent decreases in electron transfer, further production of ROS and a vicious circle of oxidative stress and energetic decline. This deficiency in mitochondrial capacity is considered the cause of aging and age-related degenerative diseases.45 Markers of cellular senescence are found in the TM of patients with POAG to a much greater degree than in age-matched controls,46 besides, mitochondria provide a gene-environment interaction between environment and our genes.47 The delayed-onset and progressive course of age-related diseases are based on the accumulation of somatic mutations in the mtDNAs of postmitotic tissues. The variations in individual and regional predispositions to degenerative diseases and cancer may result from the interaction of modern dietary caloric intake and ancient mitochondrial genetic polymorphisms.47 As mentioned above ROS are most likely responsible for TM malfunctions, which lead to IOP increases.48 Indeed, during the course of POAG, the most severe TM alterations occur in the layers that are in closest contact with the aqueous humor of the anterior chamber,49 whose cells are exposed to relatively high hydrogen peroxide concentrations.50 Extensive and prolonged oxidative stress in vivo results in reduced TM cell adhesion, cell loss, and compromised TM integrity.51 The peculiar sensitivity of the TM to oxidative stress is consistent with the damage selectively induced, which triggers glaucoma pathogenic cascade.52 The oxidative damage detected in TM53 could not explain why patients with glaucoma exhibited low levels of circulating glutathione, suggesting a general compromise of the antioxidative defenses.54 Furthermore, the statistically significant correlations between TM oxidative damage, and IOP increases was observed in POAG55 cannot be explained by assuming this relationship as a result of drug administration. Moreover, the increased expression and activity of nitric oxide synthase in the TM of POAG patients are proportional to the visual field defect and could lead to increased nitrotyrosine levels, which in turn may serve as markers of oxidative stress in the progression of TM cell death in POAG.56 Antioxidant proteins are downregulated in the increased of nitric oxide synthase 2 and in the presence of other proteins that, under physiological conditions, are segregated inside cells into functional mitochondria.36 Under POAG pathological conditions, mitochondrial proteins can be detected in AH, thus demonstrating the occurrence of cell and mitochondrial damage and destruction.57 These data support the hypothesis that, as in many neurodegenerative diseases, there is mitochondrial dysfunction in glaucoma. Mitochondrial damage triggers intracellular calcium release and the activation of apoptosis through the intrinsic activation pathways.
Apoptosis occurring in ocular tissues during POAG is induced by a variety of mechanisms, primarily including mitochondrial damage but also inflammation, vascular dysregulation, and hypoxia.58 Overall, several proteome alterations confirm the occurrence of oxidative stress in the anterior chambers of POAG patients.57 In particular, the antioxidant enzymes superoxide dismutases 1/2 and glutathione S transferees 1 were significantly lower in POAG patients than in controls, while the pro-oxidant enzymes, nitric oxide synthesize 2 and glutamate ammonia ligase, were significantly higher in POAG patients than in controls59 (Figure 2).
FIGURE 2.
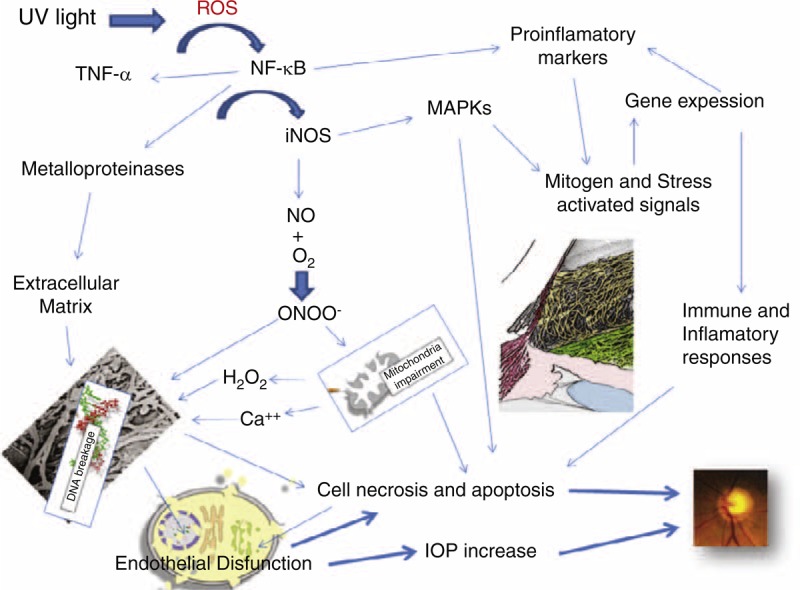
Glaucoma in oxidative stress plays a key role in mitochondrial dysfunction occurs and consequently the endothelial cells of the trabecular meshwork are not working as they should. There is an alteration of the extracellular matrix is accompanied by the activation of several metabolic pathways that result in an alteration of gene expression, an activation of inflammation and the immune response. This determines the malfunction of the trabecular meshwork and consequently the intraocular pressure increase. All this triggers the apoptosis of retinal ganglion cells.
Hp infection has been associated with glaucoma.60–62 A positive association between Hp infection and ROS production was first demonstrated in 1994.63 Recently, Hp bacteria were identified in the trabeculum and iris specimens from patients who underwent trabeculectomy for POAG.64 Furthermore, Hp infection locally induces a chronic inflammatory status consisting of polymorphonuclear neutrophil and lymphocyte recruitment at the infection site65 (Figure 3).
FIGURE 3.
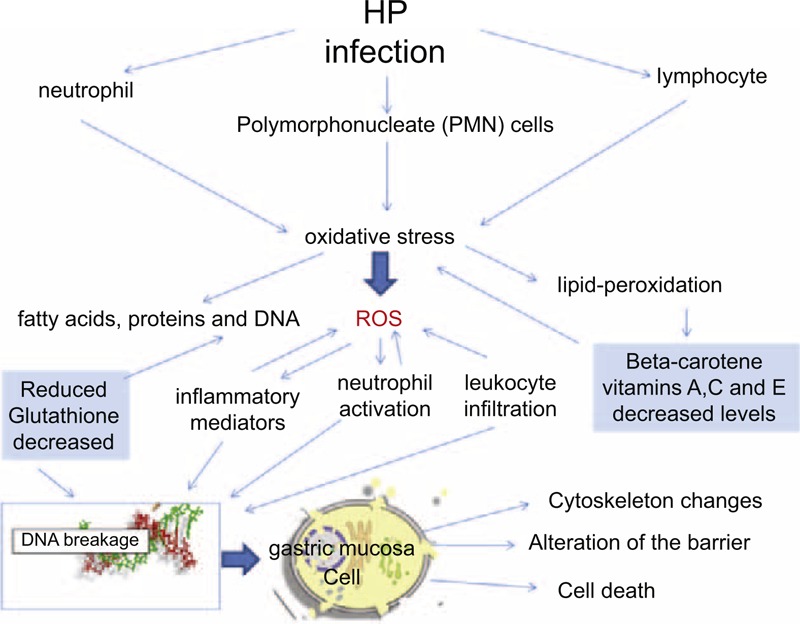
Sequence of the pathogenesis of Hp infection that increases the production of ROS and inflammatory cytokines, sees the recruitment of white blood cells in the presence of a decrease of antioxidant defenses leads to the alteration of gastric mucosa cells and thus of the gastric mucosal barrier.
Oxidative stress is exacerbated by both relative deficiencies of glutathione and of vitamins A, C, and E.66,67 In addition, the recruitment of neutrophils and the release of a variety chemoattractants/inflammatory mediators triggers an intense leukocyte infiltration of the gastric mucosa, which can cause tissue damage in the absence of antioxidants.68 This Gram-negative bacterium activates multiple oncogenic pathways in epithelial cells, including NF-kappaB, and induces epigenetic alterations, such as DNA methylation and histone modification, which play critical roles in oncogenic transformation.69
Hp can be classified into two different classes based on its ability to produce cytotoxins, such as CagA (cytotoxin-associated gene A) and VacA (Vacuolating-associated gene A).70 Inside the cells, VacA can target mitochondria, leading, at least in some cases, to the release of cytochrome c and apoptosis.71,72 Even if the actual sequence of events is unknown, a potential mechanism is that Hp infection reaches the mitochondrion from the endosomal compartment after accumulation, allowing a specific and direct interaction between the endosomal membranes and the mitochondrial outer membranes.73 The chain of events leading to the apoptosis of endothelial cells (in glaucoma) and of gastric mucosa cells (in gastric Hp infection) are oxidative stress with mitochondrial involvement; this is similar in both diseases. Another similarity between the two diseases is given by the alteration of the barrier function mediated by the signaling of the Rho family of GTPases.57 The rho-kinase pathway appears to mediate TM cell responses to cyclic mechanical stress.74 During the course of glaucoma, TM endothelial cell alterations arise, leading to increased pressure to other molecular events that then translate into the clinical apoptosis of ganglion cells and, thus, to the visual field detriment.58 Hp changes based on epithelial cell signaling and polarity, which can explain the pathogenesis of the carcinoma.75 Finally, it is worth noting the potential contributions of an Hp infection to vascular injury. Indeed, certain pathogens, such as Hp, can increase the synthesis of tissue factors, cell-surface thrombin expression, platelet adherence, and the expression of adhesion molecules, cytokines, and growth factors, even while decreasing prostacyclin release76,77 and causing endothelial cell injury.78 Endothelial dysfunction may be one of the underlying mechanisms by which multiple intracellular pathogens actually contribute to these early processes that lead to the development of atherosclerosis and to its progression to multi-vessel disease.79 Hp infection stimulates the production of the proinflammatory cytokines associated with the development of atherosclerosis,80 which arise cellular oxidative stress and endothelial dysfunction81; furthermore, Hp eradication can improve endothelial dysfunction.82 The high-tension glaucoma depends of the endothelial cell dysfunction in the TM.58 The anterior chamber of the eye is a real vessel83 that behaves as if it were a vase, expressing all proteins that act as early markers of plaque atherosclerosis.84 Still, we do not really know whether the prevalence of glaucoma is significantly different in Hp-infected patients from that in non-infected subjects. There are no epidemiological studies that have demonstrated the possible ethnic similarities and/or diversities regarding the associations between Hp and glaucoma among different countries.85 Therefore, Hp infection was associated with risk for normal tension glaucoma (NTG). Indeed, the retinal ganglion cells may be damaged in eyes within normal intraocular pressure in because the site of injury related with Hp infection may be not only trabecular meshwork but also the retinal ganglion cell itself. Probably, It due by decreasing ocular blood flow, secreting toxic materials, and causing antibody-induced apoptosis attributed to inflammation in the retrobulbar area, although the exact pathophysiology is still unclear.86
Helicobacter pylori and Central Serous Chorioretinopathy
Central serous chorioretinopathy (CSCR) is characterized by an acute, serous detachment of the sensory retina in the macular region by idiopathic breakdown of the outer blood-retina barrier formed by the retinal pigment epithelium.87 OCT images of such areas demonstrate elevation of neurosensory retina by the presence of subretinal fluid (Figure 4). Sometimes this involves a visual acuity decrease that is correlated with the amount of liquid present in the layers of the sensory retina. Although, CSCR typically affects young men and has been described as a benign and self-limiting disease, it has a tendency to re-occur.88 Some eyes with CSCR may, however, have a poor visual outcome due to retinal pigment epithelium atrophy, persistent pigment epithelial detachment, subretinal fluid, recurrences, and submacular choroidal neovascularization.89 The precise pathophysiology of central serous chorioretinopathy is uncertain. It was originally thought to be a disorder of the RPE. Choroidal microcirculation abnormalities, possibly caused by atherosclerotic lesions, could play a role.90 Indeed, the disease could originate from choroidal hyperperfusion.91–93 Hp has been associated with the development of atherosclerosis94 and with the enhanced instability of atherosclerotic plaques.95 Moreover, cross-mimicking mechanisms between antibodies against Cag-A and vascular wall antigens could be involved.95 In addition, a cross-reaction between antibodies against anti-heat shock proteins and homologous host proteins has been proposed.96,97 It should be noted that even this theory of “molecular mimicry” is not completely supported. CSCR patients often have higher levels of serum and urinary cortisol and catecholamines than healthy subjects.98,99 Hypercoagulability and the enhanced platelet aggregation have been described in patients with CSCR100 and could somehow affect the choroidal circulation and increase its permeability. Furthermore, even if its role is unclear, the platelet-derived growth factor contributes to the pathogenesis of CSCR.101 Usually, the main risk factors for the onset of CSCR are glucocorticoids.102 These hormones increase platelet aggregation and blood viscosity103 and are capable of producing a vasoconstrictive response.104 These factors together would reduce the vascular bed, but on the other hand, they could lead to hypoperfusion and an increase in endoluminal perfusion pressure, leading to serum leakage and the presence of small molecules in the retina.105 Nevertheless, although several potentially associated risk factors for this disease have been reported (Table 1) and numerous studies have been conducted on this disease over the years, many aspects of CSCR remain unclear. Moreover, a correlation between CSCR and Hp infection has been suggested.128–130 A possible point of contact between these 2 diseases may resemble the interaction between Hp infection and atherosclerosis. Indeed, chronic infection with Hp may be involved in the development of the atherosclerosis via endothelial dysfunction and systemic and vascular inflammation,131 even if more recent studies exclude this association.132,133 It is still interesting to note that anti-Hp treatment can produce the faster reabsorption of the subretinal fluid.134 Dang et al135 suggest that Hp eradication could increase central retinal sensitivity. It must be remembered, however, that the metronidazole used in Hp eradication therapy improves intestinal microcirculation in septic rats independently of the bacterial burden, causing a significant improvement in their functional capillary density.136 Furthermore, antibiotic treatment significantly reduced adverse cardiac events in patients with acute coronary syndromes; however, this effect was independent of Hp seropositivity.137
FIGURE 4.
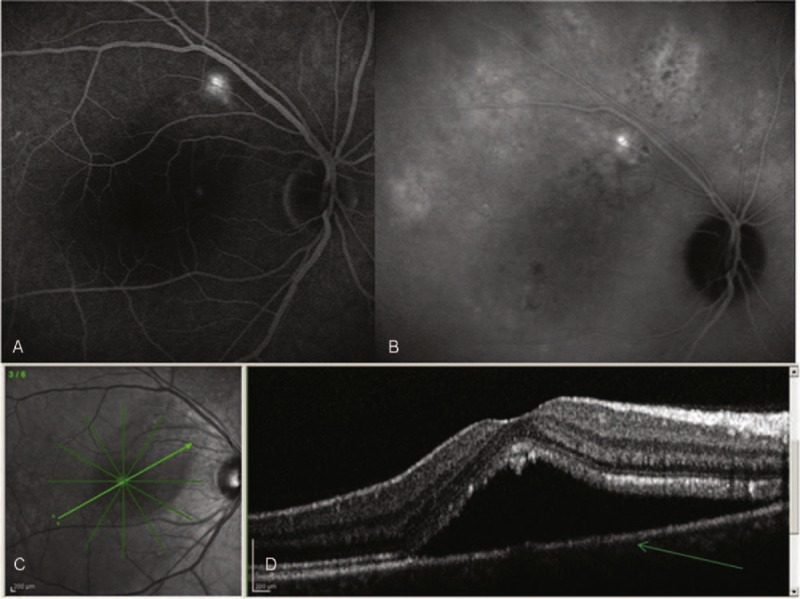
(A) Fluoroangiography of right eye; it shows a central serous chorioretinopathy which highlights the leakage (white) in the retinal pigment epithelium with pooling of the dye in the subretinal space. (B) Indocyanine green angiography shows an area with a late iperfluorescent likeage that suggests as an etiologic factor the choroidal hyperpermeability. (C) The optical coherence tomography (OCT) allows us a qualitative and quantitative assessment of the disease. (D) OCT cross-sectional image of a fovea with central serous chorioretinopathy: The detachment of the neurosensory retina is revealed as optically empty space within the retina. The retinal pigment epithelium remains attached to the choroid and it is evident by OCT as a thin layer (arrow). The retinal layers are intact but detached in the photoreceptors are thicker and a higher reflectivity.
TABLE 1.
Potentially Risk Factors in Central Serous Chorioretinopathy

Helicobacter pylori and Ocular Adnexal MALT Lymphoma
MALT lymphoma is a form of lymphoma involving the Mucosa-Associated Lymphoid Tissue (MALT) that has distinct features from all other forms of primary non-Hodgkin extranodal lymphoma (Figure 5). Ocular adnexal lymphoma is primarily found in older adults with a slight female preponderance. It occurs in the orbit, conjunctiva, and lacrimal gland, in decreasing order of frequency of involvement.138 From a histological point of view, this condition is characterized by a large prevalence of marginal zone B-cell histologic types and a varying degree of infiltrating reactive T-cells.139 The clinical manifestations depend on the identity of the compromised structures. For example, 25% of MALT lymphoma displays conjunctival involvement. Intra-orbital masses are present in 75% of cases, while bilateral involvement occurs in 10%–15% of cases.138 Intraorbital lymphoma is variably associated with exophthalmos, palpable mass or nodule, eyelid ptosis, diplopia, epiphora, and impaired ocular motility.140 Its clinical presentation usually consists of a single, slowly growing, painless mass that displaces the normal structures; however, its presentation can also be acute, with inflammatory-like signs and symptoms. Ocular infiltration is exceptional.141 Gastric MALT is known to be acquired in response to local infection by Hp, which is present in greater than 90% of these lymphomas.142 To colonize in the stomach, Hp must overcome the acidic environment of the stomach and then the gastric mucous layer. The hydrolysis of urea with the generation of ammonia may enable survival of this acid-sensitive organism in the gastric mucosa. Furthermore, ammonia generated by urea hydrolysis may also produce severe cytotoxic effects within the gastric epithelium.143 Due to the spiral shape and multiple polar flagella, which are used for motility,144 Hp stick out through the mucous layer and reach the gastric epithelium, where they sticks to its cells using the adhesins.145 Among infected individuals, approximately 10% develop peptic ulcer disease; 1% to 3% develop gastric adenocarcinoma; and <0.1% develop MALT lymphoma.146 The contact between Hp and the gastric epithelium generates an immune and inflammatory response.147 This activates the transcription factor NF-kB, inducing proinflammatory chemokines and recruiting neutrophils, monocytes, macrophages, and dendritic cells.148–151 Furthermore, this response regulates processes connected with B-cell development, growth, and survival by producing cytokines and growth factors and can also be responsible for activating cell apoptosis.152 Then, an adaptive immune response to the Hp infection emerges from the macrophages and dendritic cells located in the lamina propria of the gastric mucosa.150,153 Neutrophils, monocytes, and macrophages may phagocytose Hp, but they seem to be able to achieve this without intracellular killing,154 as Hp can survive within monocytes for up to 48 hours.155 The failure of the immune response to eliminate Hp results in chronic inflammation of the gastric mucosa. The sequential progression from chronic inflammation to mucosal atrophy, metaplasia, and dysplasia leads to carcinogenesis.156 In a small subset of individuals, chronic inflammation due to a persistent Hp infection can give rise to organized lymphoid tissue in the gastric mucosa, ultimately progressing to low-grade gastric B-cell lymphoma of the MALT type.157 Immunity to Hp and gastric immune-mediated damage is dependent on T cells.158 The prolonged interaction between the bacteria and the host immune mechanisms makes Hp a plausible infectious agent for triggering autoimmunity via molecular mimicry. The tumor cells of low-grade gastric MALT lymphoma (MALToma) are B cells that are still responsive to differentiation signals, such as cytokines produced by antigen-stimulated T cells, and that are dependent on stimulation by Hp-specific T cells for growth.157,158 The activity of these specific T cells, which are defective in both perforin- and Fas ligand-mediated cytotoxicity, consequently promotes both B-cell overgrowth and exhaustive B-cell proliferation.159 In the gastric mucosal cells, there are elevated levels of cytokines, including proliferation-inducing ligand (APRIL), which belongs to the tumor necrosis factor family. APRIL is produced by macrophages present in the gastric MALT infiltrate, close to the neoplastic cells,160 and may also induce B-cell transformation and their progression to diffuse large B-cell lymphoma. APRIL production by macrophages can be enhanced and maintained by activated T lymphocytes.161 The survival and transformation of B cells in malignant lymphoma require additional signals. They come either from T cells or directly from the antigenic autostimulation of lymphoma cells. Therefore, one or more neoplastic clones, derived from a gastric MALToma, are able to express molecules that powerfully stimulate B-cell activation and proliferation,162 colonize and replace the original follicles, eventually destroying the gastric glands to form lympho-epithelial lesions,163 and finally precipitate the onset of low-grade gastric MALT lymphoma. Although MALT lymphoma usually grows slowly and has a low propensity to spread, a small percentage of cases undergo high-grade transformations.
FIGURE 5.
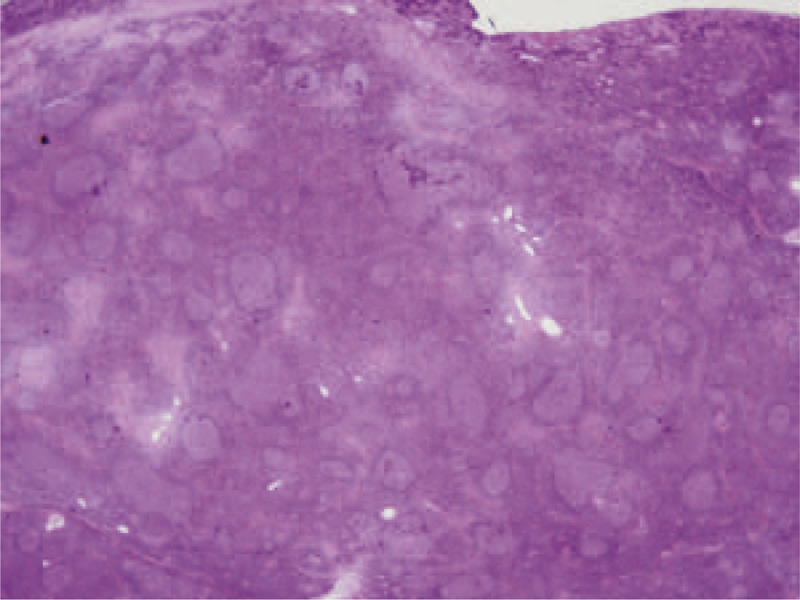
Extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma): the lymphoma cells infiltrate around reactive B-cell follicles, external to a preserved follicle mantle, in a marginal zone distribution. Marginal zone B cells have characteristics of small and medium size. The majority of patients present with stage I or II disease. The MALT lymphoma and its natural course is indolent and is slow to disseminate; recurrence may involve other extranodal sites.
Many MALT lymphomas at non-ocular sites are associated with an infectious etiology, supporting the model of antigen-driven lymphoma genesis.164 According to this model, an infection first triggers the chronic antigen stimulation of B cells and the production of antibodies. The proliferation of B cell clones becomes antigen-independent, and with uncontrolled proliferation, malignant transformations can occur.164 However, not all those with an Hp infection also have a MALT lymphoma. This indicates that the role played by genetic factors, is of great importance, as gastric MALT lymphoma presents with a series of recurrent genomic lesions, including chromosomal translocations and unbalanced aberrations. The accumulation of genetic abnormalities is associated with a loss of dependency from antigenic stimulation (with subsequent antibiotic resistance) and a possible histologic transformation.165 Moreover, 10%–20% of patients do not respond to Hp eradication treatments. This group often has a chromosome translocation, which suggests that there is another pathogenetic mechanism of MALT lymphoma that is thus far unknown.161
A higher incidence of MALT lymphoma has been reported in patients with chronic Hp infections, as well as in those with Sjögren syndrome.158 Similarly, patients with Sjögren syndrome have a much higher incidence of developing lymphoma, most of which are MALT type.166 Further studies are needed before Hp can be implicated as a significant contributor to the etiology of conjunctival MALT lymphoma.
Helicobacter pylori and Anterior Uveitis
Uveitis is a term used to describe different forms of intraocular inflammation involving the uveal tract of the eye; it is classified by anatomical location and time course of the disease.167 Acute anterior uveitis, also known as iridocyclitis or iritis, is an inflammatory disorder of the iris and/or pars plicata (anterior ciliary body) and anterior chamber that lasts no longer than 3 months. Intermediate uveitis, or pars planitis, consists of vitritis, defined as an inflammation of the cells in the vitreous, sometimes with snow banking, or the deposition of inflammatory material on the pars plana. Posterior uveitis indicates inflammation in the retina and/or choroid. Uveitis is a rare disease that is particularly prevalent in younger people.168 The etiological diagnosis of anterior uveitis can be established in approximately 60% of cases, while 75% of patients with intermediate uveitis remained without specific diagnosis. A specific diagnosis could be established in 78% of patients with posterior uveitis. Uveitis infections accounted for approximately 20% of the above cases.169 Otasevic et al170 have demonstrated that a high percentage of antibodies to Hp in the serum of a group of patients with acute anterior uveitis, some of whom were affected by spondyloarthropathies. Unfortunately, the sample examined was too small to allow for any concrete conclusions. Kim et al171 have detected on a sample of 165 subjects that Hp infection is associated with high IOP in anterior uveitis, but without finding a real causal connection between Hp infection and ocular hypertension. Thus, uveitis involved in a multitude of diseases; many chronic inflammatory diseases are associated with an elevated risk of uveitis, eg, rheumatoid arthritis, ankylosing spondylitis,172 Behcet disease,173 and Crohn disease.174 These diseases can induce other types of uveitis due to inflammation, even if they are not diagnosed as uveitis at the beginning. The breakdown of the blood-aqueous barrier in uveitis involves cellular infiltration, an increase in protein permeability, and the up-regulation of cytokines, such as TNF-α and IL-6, and chemokines, such as MCP-1, and MIP-1. In the aqueous humor and uveal regions,175 the exposure of the cells near the blood-aqueous barrier to inflammatory cytokines and chemokines could eventually cause cytotoxicity, leading to apoptosis or proliferation. Inflammation is associated with increased oxidative stress by elevated ROS, which could alter cellular and molecular targets and pathways crucial to normal tissue homeostasis.176,177 The generation of ROS in turn activates redox-sensitive transcription factors such as NF-κB, which controls the expression of a large number of genes involved in apoptosis, cell growth, survival, differentiation and immune response. Alterations in NF-κB activity are associated with a large number of diseases, including autoimmune, cancer and inflammatory diseases.178 NF-κB plays an important role in the regulation of immune and inflammatory responses. Pathogens, oxidants, cytokines, chemokines, and growth factors associated with oxidative stress trigger specific receptors and cause oxidative stress signaling cascades that lead to the activation of NF-κB. NF-κB activation is responsible for the expression of a wide variety of genes that encode cytokines (TNF, IL-1, IL-6), chemokines (MIP-1, MCP-1), adhesion molecules (ICAM, VCAM, E-Selectin), iNOS and Cox-2.175 ROS are important modulators of signaling pathways and can regulate both the apoptotic signaling and NF-kappaB transcription triggered by TNF. ROS can also cause redox modifications that inhibit NF-kappaB activation, leading to cell death triggered by TNF.179 The increased ROS levels during inflammation could be increased oxygen consumption in the uveitis or decreased antioxidative defense in the concerned tissue. The increased levels of ROS in the ocular cells cause a redox imbalance, leading to activation of redox signaling intermediates, which in turn activate transcription factors, including NF-κB, with the result of transcription of inflammatory marker genes.180,181 The reduced circulating levels of vitamin C in Hp infected subjects may contribute to the etiology of some diseases associated with antioxidant deficiency.182 The excessive ROS generation also weakens the tissue own antioxidant defense system, further aggravating the inflammation and ROS production and generating tissue damage in uveitis. This damage increases the level of metallo-proteases, which chew up intra-cellular and extracellular proteins, resulting in tissue injury.176 During Hp infection, activated macrophages produce the following pro-inflammatory cytokines: IL-1, IL-2, IL-6, IL-8, and TNF-α.183 Most of these cytokines are expressed in the aqueous humors of patients with idiopathic acute anterior uveitis.184 Therefore, the chance of an autoimmune reaction due to molecular mimicry may be possible.185,186
CONCLUSION
In conclusion, it is extremely difficult to compare the results of the studies that are currently available in the literature, as to do so would require a population-based study involving thousands of patients in order to effectively determine the prevalence of eye diseases in patients infected with Hp. Nevertheless, inadequate antioxidant protection or excess production of ROS creates conditions of oxidative stress, which is thought to play an important role in the aging of the eye and in many inflammatory eye diseases.5 In any case, it is difficult to understand how Hp infection can be linked to such varied pathologies. It is possible that this “link” might be the oxidative damage that recurs in circulatory disorders,187 inflammation188,189; and glaucoma.55,85 As we can also see in central serous chorioretinopathy and ocular adnexal MALT lymphoma, the effects of oxidative stress can be substantial. Inflammation in Hp infections and in eye diseases progresses through a series of common pathogenic aspects shared by the two entities, despite their differing clinical features. Indeed, adequate antioxidant defenses responsible for scavenging free radicals are essential for redox homeostasis and inhibition of inflammation. These are variables, eg, corneal epithelial cells have strong antioxidant defenses, conversely, other ocular tissues, such as the trabecular meshwork, are poorly equipped with antioxidant defenses and consequently less able to counteract injurious effects of ROS5 Therefore, therapy with antioxidants should prove beneficial for the clinical management of patients with Hp infection.190
Vitamin C appears to have a particularly important role, in fact within the cell, vitamin C helps to protect membrane lipids from peroxidation by recycling vitamin E.191 This could be relevant in all eye diseases that we talked about, especially in glaucoma where the Vitamin C has a direct effect on the trabecular meshwork where might improve their ability to degrade proteins within the lysosomal compartment and recover tissue function.192 Therefore Ascorbic acid supplementation may improve the effectiveness of Hp-eradication therapy.193 Besides, diets rich in naturally occurring ascorbic acid are associated with protection of the gastric corpus from atrophy and a reduction in the incidence of gastric cancer possibly through the ability of ascorbic acid to reduce oxidative damage to the gastric mucosa by scavenging carcinogenic N-nitroso compounds and free radicals and attenuating the Hp-induced inflammatory cascade.193 Further studies are required to prove that Ascorbate or other antioxidant supplementations have a significant impact on progress of the association between eye diseases and HP infection.
On the whole, the data reported in this review provide evidence that oxidative stress and inflammation represent common pathogenic mechanisms that play a major role in both Hp infection and several eye diseases.
Footnotes
Abbreviations: AH = aqueous humor, CSCR = central serous chorioretinopathy, Hp = Helicobacter pylori, IOP = intraocular pressure, MALT = mucosa-associated lymphoid tissue lymphoma, mtDNA = mitochondrial DNA, NTG = normal tension glaucoma, OCT = optical coherence tomography, POAG = primary open angle glaucoma, ROS = reactive oxygen species, TM = trabecular meshwork.
The authors declare that there are no conflicts of interest and to be in agreement with the Declaration of Helsinki.
REFERENCES
- 1.Parsonnet J, Shmuely H, Haggerty T. Fecal and oral shedding of Helicobacter pylori from healthy infected adults. JAMA 1999; 282:2240–2245. [DOI] [PubMed] [Google Scholar]
- 2.Leung WK, Siu KL, Kwok CK, et al. Isolation of Helicobacter pylori from vomitus in children and its implication in gastro-oral transmission. Am J Gastroenterol 1999; 94:2881–2884. [DOI] [PubMed] [Google Scholar]
- 3.Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med. 2002; 347:1175–1186. [DOI] [PubMed] [Google Scholar]
- 4.Mindel JS, Rosenberg EW. Is Helicobacter pylori of interest to ophthalmologists? Ophthalmology. 1997; 104:1729–1730. [DOI] [PubMed] [Google Scholar]
- 5.Saccà SC, Roszkowska AM, Izzotti A. Environmental light and endogenous antioxidants as the main determinants of non-cancer ocular diseases. Mutat Res. 2013; 752:153–171. [DOI] [PubMed] [Google Scholar]
- 6.Saccà SC, Pascotto A, Venturino GM, et al. Prevalence and treatment of Helicobacter pylori in patients with blepharitis. Invest Ophthalmol Vis Sci. 2006; 47:501–508. [DOI] [PubMed] [Google Scholar]
- 7.Kendall SN. Remission of rosacea induced by reduction of gut transit time. Clin Exp Dermatol. 2004; 29:297–299. [DOI] [PubMed] [Google Scholar]
- 8.Boni R, Rosacea acne and other diseases of the seborrheic spectrum. Schweiz Rundsch Med Prax. 2000; 89:566–570. [PubMed] [Google Scholar]
- 9.Diaz C, O’Callaghan CJ, Khan A, Ilchyshyn A. Rosacea: a cutaneous marker of Helicobacter pylori infection? Results of a pilot study. Acta Derm Venereol. 2003; 83:282–286. [DOI] [PubMed] [Google Scholar]
- 10.Charlett A, Dobbs RJ, Dobbs SM, et al. Parkinsonism: siblings share Helicobacter pylori seropositivity and facets of syndrome. Acta Neurol Scand. 1999; 99:26–35. [DOI] [PubMed] [Google Scholar]
- 11.Gupta AK, Bluhm R, Cooper EA, Summerbell RC, Batra R Seborrheic dermatitis. Dermatol Clin. 2003; 21:401–412. [DOI] [PubMed] [Google Scholar]
- 12.Fitzpatrick JE. Common inflammatory skin diseases of the elderly. Geriatrics. 1989; 44:40–46. [PubMed] [Google Scholar]
- 13.Utas S, Ozbakir O, Turasan A, Utas C. Helicobacter pylori eradication treatment reduces the severity of rosacea. J Am Acad Dermatol 1999; 40:433–435. [DOI] [PubMed] [Google Scholar]
- 14.Boixeda de Miquel D, Vázquez Romero M, Vázquez Sequeiros E, et al. Effect of Helicobacter pylori eradication therapy in rosacea patients. Rev Esp Enferm Dig 2006; 98:501–509. [DOI] [PubMed] [Google Scholar]
- 15.Szlachcic A, Sliwowski Z, Karczewska E, et al. Helicobacter pylori and its eradication in rosacea. J Physiol Pharmacol 1999; 50:777–786. [PubMed] [Google Scholar]
- 16.Michel JL, Cabibel F. Frequency, severity and treatment of ocular rosacea during cutaneous rosacea. Ann Dermatol Venereol. 2003; 130:20–24. [PubMed] [Google Scholar]
- 17.Gasbarrini A, Serricchio M, Tondi P, et al. Association of Helicobacter pylori infection with primary Raynaud phenomenon [letter]. Lancet 1996; 348:966–967. [DOI] [PubMed] [Google Scholar]
- 18.Gasbarrini A, Franceschi F. Autoimmune diseases and Helicobacter pylori infection. Biomed Pharmacother. 1999; 53:223–226. [DOI] [PubMed] [Google Scholar]
- 19.Amedei A, Bergman MP, Appelmelk BJ, et al. Molecular mimicry between Helicobacter pylori antigens and H, K-adenosine triphosphatase in human gastric autoimmunity. J Exp Med. 2003; 198:1147–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gurer MA, Erel A, Erbas D, et al. The seroprevalence of Helicobacter pylori and nitric oxide in acne rosacea. Int J Dermatol 2002; 41:768–770. [DOI] [PubMed] [Google Scholar]
- 21.Gutgesell VJ, Stern GA, Hood CI. Histopathology of meibomian gland dysfunction. Am J Ophthalmol. 1982; 94:383–387. [DOI] [PubMed] [Google Scholar]
- 22.Dougherty JM, Mc Culley JP, Silvany RE, Meyer DR. The role of tetracycline in chronic blepharitis: inhibition of lipase production in Staphylococci. Invest Ophthalmol Vis Sci. 1991; 32:2970–2975. [PubMed] [Google Scholar]
- 23.Gulbenkian A, Myers J, Fries D. Hamster flank organ hydrolase and lipase activity. J Invest Dermatol. 1980; 75:289–292. [DOI] [PubMed] [Google Scholar]
- 24.Crawford GH, Pelle MT, James WD. Rosacea: I. Etiology, pathogenesis, and subtype classification. J Am Acad Dermatol. 2004; 51:327–341. [DOI] [PubMed] [Google Scholar]
- 25.Cohen AF, Tiemstra JD. Diagnosis and treatment of rosacea. J Am Board Fam Pract. 2002; 15:214–217. [PubMed] [Google Scholar]
- 26.Rebora A. The management of rosacea. Am J Clin Dermatol. 2002; 3:489–496. [DOI] [PubMed] [Google Scholar]
- 27.Song Q, Lange T, Spahr A, et al. Characteristic distribution pattern of Helicobacter pylori in dental plaque and saliva detected with nested PCR. J Med Microbiol. 2000; 49:349–353. [DOI] [PubMed] [Google Scholar]
- 28.Al-Ahmad A, Kürschner A, Weckesser S, et al. Is Helicobacter pylori resident or transient in the human oral cavity? J Med Microbiol. 2012; 61:1146–1152. [DOI] [PubMed] [Google Scholar]
- 29.Leske MC. Open-angle glaucoma: an epidemiologic overview. Ophthalmic Epidemiol 2007; 14:166–172. [DOI] [PubMed] [Google Scholar]
- 30.Ritch R. Natural Compounds: evidence for a protective role in eye disease. Can J Ophthalmol 2007; 42:425–438. [PubMed] [Google Scholar]
- 31.Pescosolido N, Cavallotti C, Rusciano D, Nebbioso M. Trabecular meshwork in normal and pathological eyes. Ultrastruct Pathol. 2012; 36:102–107. [DOI] [PubMed] [Google Scholar]
- 32.Saccà SC, Izzotti A. Focus on molecular events in the anterior chamber leading to glaucoma. Cell Mol Life Sci. 2014; 71:2197–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brenner C, Kroemer G. Apoptosis: mitochondria; the death signal integrators. Science. 2000; 289:1150–1151. [DOI] [PubMed] [Google Scholar]
- 34.Van Houten B, Woshner V, Santos JH. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair (Amst). 2006; 5:145–152. [DOI] [PubMed] [Google Scholar]
- 35.He Y, Leung KW, Zhang YH, et al. Mitochondrial complex I defect induces ROS release and degeneration in trabecular meshwork cells of POAG patients: protection by antioxidants. Invest Ophthalmol Vis Sci. 2008; 49:1447–1458. [DOI] [PubMed] [Google Scholar]
- 36.Izzotti A, Saccà SC, Longobardi M, Cartiglia C. Mitochondrial damage in the trabecular meshwork of patients with glaucoma. Arch Ophthalmol. 2010; 128:724–730. [DOI] [PubMed] [Google Scholar]
- 37.Trounce I, Byrne E, Marzuki S. Decline in skeletal muscle mitochondrial chain function: possible factor in aging. Lancet 1989; 1:637–639. [DOI] [PubMed] [Google Scholar]
- 38.Yen TC, Chen YS, King KL, et al. Liver mitochondrial respiratory functions decline with age. Biochem Biophys Res Com 1989; 165:994–1003. [DOI] [PubMed] [Google Scholar]
- 39.Camougrand N, Rigoulet M. Aging and oxidative stress: studies of some genes involved both in aging and in response to oxidative stress. Resp Physiol 2001; 128:393–401. [DOI] [PubMed] [Google Scholar]
- 40.Tappia PS, Dent MR, Dhalla NS. Oxidative stress and redox regulation of phospholipase D in myocardial disease. Free Radic Biol Med. 2006; 41:349–361. [DOI] [PubMed] [Google Scholar]
- 41.Lee H-C, Wei Y-H. Mitochondrial alterations, cellular response to oxidative stress and defective degradation of proteins in aging. Biogerontology 2001; 2:231–244. [DOI] [PubMed] [Google Scholar]
- 42.Ames BN, Shigenaga MK, Hagen TM Oxidants, antioxidants, and the degenerative diseases of ageing. Proc Natl Acad Sci USA 1993; 90:7915–7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yakes FM, van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci USA 1997; 94:514–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parihar MS, Brewer GJ. Mitoenergetic failure in Alzheimer disease. Am J Physiol Cell Physiol. 2007; 292:C8–C23. [DOI] [PubMed] [Google Scholar]
- 45.Genova ML, Pich MM, Bernacchia A, et al. The mitochondrial production of reactive oxygen species in relation to aging and pathology. Ann N Y Acad Sci. 2004; 1011:86–100. [DOI] [PubMed] [Google Scholar]
- 46.Caprioli J. Invest Ophthalmol Vis Sci. 2013; 54: ORSF60-7. [DOI] [PubMed] [Google Scholar]
- 47.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005; 39:359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saccà SC, Izzotti A. Oxidative stress and glaucoma: injury in the anterior segment of the eye. Prog Brain Res. 2008; 173:385–407. [DOI] [PubMed] [Google Scholar]
- 49.Alvarado JA, Murphy C, Juster R. Trabecular meshwork cellularity in primary open angle glaucoma and nonglaucomatous normals. Ophthalmology 1984; 91:564–579. [DOI] [PubMed] [Google Scholar]
- 50.Kahn MG, Giblin FJ, Epstein DL. Glutathione in calf trabecular meshwork and its relation to aqueous humor outflow facility. Invest Ophthalmol Vis Sci. 1983; 24:1283–1287. [PubMed] [Google Scholar]
- 51.Zhou L, Li Y, Yue BY. Oxidative stress affects cytoskeletal structure and cell-matrix interactions in cells from an ocular tissue: the trabecular meshwork. J Cell Physiol. 1999; 180:182–189. [DOI] [PubMed] [Google Scholar]
- 52.Izzotti A, Saccà SC, Longobardi M, Cartiglia C. Sensitivity of ocular anterior chamber tissues to oxidative damage and its relevance to the pathogenesis of glaucoma. Invest Ophthalmol Vis Sci. 2009; 50:5251–5258. [DOI] [PubMed] [Google Scholar]
- 53.Izzotti A, Saccà SC, Cartiglia C, De Flora S. Oxidative deoxyribonucleic acid damage in the eyes of glaucoma patients. Am J Med. 2003; 114:638–646. [DOI] [PubMed] [Google Scholar]
- 54.Sacca SC, Bolognesi C, Battistella A, et al. Gene-environment interactions in ocular diseases. Mutat Res. 2009; 667:98–117. [DOI] [PubMed] [Google Scholar]
- 55.Saccà SC, Pascotto A, Camicione P, et al. Oxidative DNA damage in the human trabecular meshwork: clinical correlation in patients with primary open-angle glaucoma. Arch Ophthalmol. 2005; 123:458–463. [DOI] [PubMed] [Google Scholar]
- 56.Fernández-Durango R, Fernández-Martínez A, García-Feijoo J, et al. Expression of nitrotyrosine and oxidative consequences in the trabecular meshwork of patients with primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 2008; 49:2506–2511. [DOI] [PubMed] [Google Scholar]
- 57.Izzotti A, Longobardi M, Cartiglia C, Saccà SC. Proteome alterations in primary open angle glaucoma aqueous humor. J Proteome Res. 2010; 9:4831–4838. [DOI] [PubMed] [Google Scholar]
- 58.Sacc SC, Izzotti A. Oxidative stress in anterior segment of primary pen angle glaucoma, Glaucoma—Current Clinical and Research Aspects. Dr Gunvant Pinakin.(ed.); 2011; pp. 1–23. from: http://www.intechopen.com/books/glaucoma-current-clinical-and-researchaspects/oxidative-stress-in-anterior-segment-of-primary-open-angle-glaucoma [Google Scholar]
- 59.Bagnis A, Izzotti A, Centofanti M, Saccà SC. Aqueous humor oxidative stress proteomic levels in primary open angle glaucoma. Exp Eye Res. 2012; 103:55–62. [DOI] [PubMed] [Google Scholar]
- 60.Kountouras J, Mylopoulos N, Boura P, et al. Relationship between Helicobacter Pylori infection and glaucoma. Ophthalomology 2001; 108:599–604. [DOI] [PubMed] [Google Scholar]
- 61.Kontouras J, Mylopoulos N, Chatzopoulos D, et al. Eradication of Helicobacter pylori may be beneficial in the management of chronic open-angle glaucoma. Arch Intern Med 2002; 162:1237–1244. [DOI] [PubMed] [Google Scholar]
- 62.Kountouras J, Mylopoulos N, Konstas AG, et al. Increased levels of Helicobacter pylori IgG antibodies in aqueous humor of patients with primary open-angle and exfoliation glaucoma. Graefes Arch Clin Exp Ophthalmol 2003; 241:884–890. [DOI] [PubMed] [Google Scholar]
- 63.Davies GR, Simmonds NJ, Stevens TR, et al. Helicobacter pylori stimulates antral mucosal reactive oxygen metabolite production in vivo. Gut 1994; 35:179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zavos C, Kountouras J, Sakkias G, et al. Histological presence of Helicobacter pylori bacteria in the trabeculum and iris of patients with primary open-angle glaucoma. Ophthalmic Res. 2012; 47:150–156. [DOI] [PubMed] [Google Scholar]
- 65.Miedany Y, Baddour M, Ahmed I, Fahmy H. Sjogren's syndrome: concomitant Hp infection and possible correlation with clinical parameters. Joint Bone Spine 2005; 72:135–141. [DOI] [PubMed] [Google Scholar]
- 66.Halliwell B. Antioxidants in human health and diseases. Ann Rev Nutr 1996; 16:33–50. [DOI] [PubMed] [Google Scholar]
- 67.Correa P. The role of antioxidants in gastric carcinogenesis. Crit Rev Food Sci Nutr 1995; 53:59–64. [DOI] [PubMed] [Google Scholar]
- 68.Kountouras J, Chatzopoulos D, Zavos C. Reactive oxygen metabolites and upper gastrointestinal diseases. Hepatogastroenterology. 2001; 48:743–751. [PubMed] [Google Scholar]
- 69.Ding SZ, Goldberg JB, Hatakeyama M. Helicobacter pylori infection, oncogenic pathways and epigenetic mechanisms in gastric carcinogenesis. Future Oncol. 2010; 6:851–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Figura N, Tabaqchali S. Bacterial pathogenic factors. Curr Opin Gastroenterol 1996; 12 Suppl. 1:11–15. [Google Scholar]
- 71.Galmiche A, Rassow J, Doye A, et al. The N-terminal 34 kDa fragment of Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces cytochrome c release. EMBO J. 2000; 19:6361–6370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boquet P, Ricci V, Galmiche A, Gauthier NC. Gastric cell apoptosis and Hp: has the main function of VacA finally been identified? Trends Microbiol. 2003; 11:410–413. [DOI] [PubMed] [Google Scholar]
- 73.Galmiche A, Rassow J. Targeting of Helicobacter pylori VacA to mitochondria. Gut Microbes. 2010; 1:392–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ramos RF, Sumida GM, Stamer WD. Cyclic mechanical stress and trabecular meshwork cell contractility. Invest Ophthalmol Vis Sci. 2009; 50:3826–3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Osman MA, Bloom GS, Tagoe EA. Helicobacter pylori-induced alteration of epithelial cell signaling and polarity: a possible mechanism of gastric carcinoma etiology and disparity. Cytoskeleton (Hoboken). 2013; 70:349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Etingin OR, Silverstein RL, Friedman HM, Hajjar DP. Viral activation of the coagulation cascade: molecular interactions at the surface of infected endothelial cells. Cell. 1990; 61:657–662. [DOI] [PubMed] [Google Scholar]
- 77.Dechend R, Maass M, Gieffers J, et al. Chlamydia pneumoniae infection of vascular smooth muscle and endothelial cells activates NF-κB and induces tissue factor and PAI-1 expression: a potential link to accelerated arteriosclerosis. Circulation. 1999; 100:1369–1373. [DOI] [PubMed] [Google Scholar]
- 78.Zhu J, Quyyumi AA, Norman JE, et al. Effects of total pathogen burden on coronary artery disease risk and C-reactive protein levels. Am J Cardiol. 2000; 85:140–146. [DOI] [PubMed] [Google Scholar]
- 79.Prasad A, Zhu J, Halcox JP, et al. Predisposition to atherosclerosis by infections: role of endothelial dysfunction. Circulation. 2002; 106:184–190. [DOI] [PubMed] [Google Scholar]
- 80.Chen BF, Xu X, Deng Y, et al. Relationship between Helicobacter pylori infection and serum interleukin-18 in patients with carotid atherosclerosis. Helicobacter. 2013; 18:124–128. [DOI] [PubMed] [Google Scholar]
- 81.Bidault G, Garcia M, Vantyghem MC, et al. Lipodystrophy-linked LMNA p.R482W mutation induces clinical early atherosclerosis and in vitro endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2013; 33:2162–2171. [DOI] [PubMed] [Google Scholar]
- 82.Blum A, Tamir S, Mualem K, et al. Endothelial dysfunction is reversible in Helicobacter pylori-positive subjects. Am J Med. 2011; 124:1171–1174. [DOI] [PubMed] [Google Scholar]
- 83.Izzotti A, Saccà SC, Di Marco B, et al. Antioxidant activity of timolol on endothelial cells and its relevance for glaucoma course. Eye (Lond). 2008; 22:445–453. [DOI] [PubMed] [Google Scholar]
- 84.Saccà SC, Centofanti M, Izzotti A. New proteins as vascular biomarkers in primary open angle glaucomatous aqueous humor. Invest Ophthalmol Vis Sci. 2012; 53:4242–4253. [DOI] [PubMed] [Google Scholar]
- 85.Izzotti A, Saccà SC, Bagnis A, Recupero SM. Glaucoma and Helicobacter pylori infection: correlations and controversies. Br J Ophthalmol. 2009; 93:1420–1427. [DOI] [PubMed] [Google Scholar]
- 86.Kim JM, Park KH, Kim SH, et al. Investigation of the Association between Helicobacter pylori infection and normal tension glaucoma. Invest Ophthalmol Vis Sci. 2011; 52:665–668. [DOI] [PubMed] [Google Scholar]
- 87.Wang M, Munch IC, Hasler PW, et al. Central serous chorioretinopathy. Acta Ophthalmol. 2008; 86:126–145. [DOI] [PubMed] [Google Scholar]
- 88.Yap EY, Robertson DM. The long-term outcome of central serous chorioretinopathy. Arch Ophthalmol. 1996; 114:689–692. [DOI] [PubMed] [Google Scholar]
- 89.Loo RH, Scott IU, Flynn HW, Jr, et al. Factors associated with reduced visual acuity during long-term follow-up of patients with idiopathic central serous chorioretinopathy. Retina. 2002; 22:19–24. [DOI] [PubMed] [Google Scholar]
- 90.Kitaya N, Nagaoka T, Hikichi T, et al. Features of abnormal choroidal circulation in central serous chorioretinopathy. Br J Ophthalmol 2003; 87:709–712. 10.1136/bjo.87.6.709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Piccolino Cardillo F, Borgia L. Central serous chorioretinopathy and indocyanine green angiography. Retina. 1994; 14:231–242. [DOI] [PubMed] [Google Scholar]
- 92.Guyer DR, Yannuzzi LA, Slakter JS, et al. Digital indocyanine-green videoangiography of occult choroidal neovascularization. Ophthalmology. 1994; 101:1727–1735. [DOI] [PubMed] [Google Scholar]
- 93.Spaide RF, Hall L, Haas A, et al. Indocyanine green videoangiography of older patients with central serous chorioretinopathy. Retina. 1996; 16:203–213. [DOI] [PubMed] [Google Scholar]
- 94.Adiloglu AK, Nazli C, Cicioglu-Aridogan B, et al. Gastroduodenal Helicobacter pylori infection diagnosed by Helicobacter pylori stool antigen is related to atherosclerosis. Acta Cardiol. 2003; 58:335–339. [DOI] [PubMed] [Google Scholar]
- 95.Gabrielli M, Santoliquido A, Cremonini F, et al. CagA-positive cytotoxic Hp strains as a link between plaque instability and atherosclerotic stroke. Eur Heart J. 2004; 25:64–68. [DOI] [PubMed] [Google Scholar]
- 96.Franceschi F, Sepulveda AR, Gasbarrini A, et al. Cross-reactivity of anti-CagA antibodies with vascular wall antigens: possible pathogenic link between Helicobacter pylori infection and atherosclerosis. Circulation 2002; 106:430–434. [DOI] [PubMed] [Google Scholar]
- 97.Lamb DJ, El-Sankary W, Ferns GA. Molecular mimicry in atherosclerosis: a role for heat shock proteins in immunisation. Atherosclerosis 2003; 167:177–185. [DOI] [PubMed] [Google Scholar]
- 98.Bouzas EA, Scott MH, Mastorakos G, et al. Central serous chorioretinopathy in endogenous hypercortisolism. Arch Ophthalmol. 1993; 111:1229–1233. [DOI] [PubMed] [Google Scholar]
- 99.Tewari HK, Gadia R, Kumar D, et al. Sympathetic-parasympathetic activity and reactivity in central serous chorioretinopathy: a case-control study. Invest Ophthamol Vis Sci. 2006; 47:3474–3478. [DOI] [PubMed] [Google Scholar]
- 100.Yamada R, Yamada S, Ishii A, Tane S. Evaluation of tissue plasminogen activator and plasminogen activator inhibitor-1 in blood obtained from patients of idiopathic central serous chorioretinopathy. Nippon Ganka Gakkai Zasshi. 1993; 97:955–960. [PubMed] [Google Scholar]
- 101.Shin MC, Lim JW. Concentration of cytokines in the aqueous humor of patients with central serous chorioretinopathy. Retina. 2011; 31:1937–1943. [DOI] [PubMed] [Google Scholar]
- 102.Bouzas EA, Karadimas P, Pournaras CJ. Central serous chorioretinopathy and glucocorticoids. Surv Ophthalmol. 2002; 47:431–448. [DOI] [PubMed] [Google Scholar]
- 103.Jones JP., Jr Intravascular coagulation and osteonecrosis. Clin Orthop. 1992; 277:41–53. [PubMed] [Google Scholar]
- 104.Walker BR, Williams BC. Corticosteroids and vascular tone: mapping the messenger maze. Clin Sci. 1992; 82:597–605.PMID: [1320537]. [DOI] [PubMed] [Google Scholar]
- 105.Caccavale A, Romanazzi F, Imparato M, et al. Central serous chorioretinopathy: a pathogenetic model. Clin Ophthalmol. 2011; 5:239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nadeau S, Nguyen F, Guigou S. Serous central chorioretinopathy and tadalafil: a case report. J Fr Ophtalmol. 2012; 35:121. [DOI] [PubMed] [Google Scholar]
- 107.Moshirfar M, Hsu M, Schulman J, et al. The incidence of central serous chorioretinopathy after photorefractive keratectomy and laser in situ keratomileusis. J Ophthalmol. 2012; 2012:904215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kloos P, Laube I, Thoelen A. Obstructive sleep apnea in patients with central serous chorioretinopathy. Graefes Arch Clin Exp Ophthalmol. 2008; 246:1225–1228. [DOI] [PubMed] [Google Scholar]
- 109.Haimovici R, Koh S, Gagnon DR, et al. Risk factors for central serous chorioretinopathy: a case-control study. Ophthalmology. 2004; 111:244–249. [DOI] [PubMed] [Google Scholar]
- 110.Allibhai ZA, Gale JS, Sheidow TS. Central serous chorioretinopathy in a patient taking sildenafil citrate. Ophthalmic Surg Lasers Imaging. 2004; 35:165–167. [PubMed] [Google Scholar]
- 111.Koyama M, Mizota A, Igarashi Y. Seventeen cases of central serous chorioretinopathy associated with systemic corticosteroid therapy. Ophthalmologica. 2004; 218:107–110. [DOI] [PubMed] [Google Scholar]
- 112.Carvalho-Recchia CA, Yannuzzi LA, Negrão S, et al. Corticosteroids and central serous chorioretinopathy. Ophthalmology. 2002; 109:1834–1837. [DOI] [PubMed] [Google Scholar]
- 113.Costen MT, Parkin BT, Davison CR, Crick MP. Central serous chorioretinopathy and antiphospholipid antibodies-results of a pilot study. Eye (Lond). 2004; 18:938. [DOI] [PubMed] [Google Scholar]
- 114.Dorenboim Y, Rehany U, Rumelt S. Central serous chorioretinopathy associated with retinitis pigmentosa. Graefes Arch Clin Exp Ophthalmol. 2004; 242:346–349. [DOI] [PubMed] [Google Scholar]
- 115.Nucci C, Corsi A, Mancino R, Macri G. Central serous chorioretinopathy in patients with psoriasis. Acta Ophthalmol Scand. 2004; 82:105–107. [DOI] [PubMed] [Google Scholar]
- 116.Michael JC, Pak J, Pulido J, de Venecia G. Central serous chorioretinopathy associated with administration of sympathomimetic agents. Am J Ophthalmol. 2003; 136:182–185. [DOI] [PubMed] [Google Scholar]
- 117.Haimovici R, Rumelt S, Melby J. Endocrine abnormalities in patients with central serous chorioretinopathy. Ophthalmology. 2003; 110:698–703. [DOI] [PubMed] [Google Scholar]
- 118.Spahn C, Wiek J, Burger T, Hansen L. Psychosomatic aspects in patients with central serous chorioretinopathy. Br J Ophthalmol. 2003; 87:704–708. 10.1136/bjo.87.6.704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yannuzzi LA. Type A behavior and central serous chorioretinopathy. Trans Am Ophthalmol Soc. 1986; 84:799–845. [PMC free article] [PubMed] [Google Scholar]
- 120.Katsimpris JM, Vandoros M, Petropoulos IK, Andrikopoulos P. Central serous chorioretinopathy associated with adrenal myelolipoma. Klin Monbl Augenheilkd. 2003; 220:199–203. [DOI] [PubMed] [Google Scholar]
- 121.Karashima K, Fujioka S, Harino S. Two cases of central serous chorioretinopathy treated with photocoagulation after bone marrow transplantation. Retina. 2002; 22:651–653. [DOI] [PubMed] [Google Scholar]
- 122.Giusti C. Central serous chorioretinopathy: a new extragastric manifestation of Helicobacter pylori? Analysis of a clinical case. Clin Ter. 2001; 152:393–397. [PubMed] [Google Scholar]
- 123.Weenink AC, Borsje RA, Oosterhuis JA. Familial chronic central serous chorioretinopathy. Ophthalmologica. 2001; 215:183–187. [DOI] [PubMed] [Google Scholar]
- 124.Park DW, Schatz H, Gaffney MM, et al. Central serous chorioretinopathy in two families. Eur J Ophthalmol. 1998; 8:42–47. [DOI] [PubMed] [Google Scholar]
- 125.Zamir E, Chowers I. Central serous chorioretinopathy in a patient with cryoglobulinaemia. Eye (Lond). 1999; 13:265–266. [DOI] [PubMed] [Google Scholar]
- 126.Cunningham ET, Jr, Alfred PR, Irvine AR. Central serous chorioretinopathy in patients with systemic lupus erythematosus. Ophthalmology. 1996; 103:2081–2090. [DOI] [PubMed] [Google Scholar]
- 127.Liew G, Quin G, Gillies M, Fraser-Bell S. Central serous chorioretinopathy: a review of epidemiology and pathophysiology. Clin Experiment Ophthalmol. 2013; 41:201–214. [DOI] [PubMed] [Google Scholar]
- 128.Giusti C. Association of Helicobacter pylori with central serous chorioretinopathy: hypotheses regarding pathogenesis. Medical Hypotheses. 2004; 63:524–527. [DOI] [PubMed] [Google Scholar]
- 129.Cotticelli L, Borrelli M, D’Alessio AC, et al. Central serous chorioretinopathy and Helicobacter pylori. Eur J Ophthalmol. 2006; 16:274–278. [DOI] [PubMed] [Google Scholar]
- 130.Casella AM, Berbel RF, Bressanim GL, et al. Helicobacter pylori as a potential target for the treatment of central serous chorioretinopathy. Clinics (Sao Paulo). 2012; 67:1047–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Oshima T, Ozono R, Yano Y, et al. Association of Helicobacter pylori infection with systemic inflammation and endothelial dysfunction in healthy male subjects. J Am Coll Cardiol. 2005; 45:1219–1222. [DOI] [PubMed] [Google Scholar]
- 132.Szklo M, Ding J, Tsai MY, et al. Individual pathogens, pathogen burden and markers of subclinical atherosclerosis: the Multi-Ethnic Study of Atherosclerosis. J Cardiovasc Med (Hagerstown). 2009; 10:747–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Coskun S, Kasirga E, Yilmaz O, et al. Is Helicobacter pylori related to endothelial dysfunction during childhood? Pediatr Int. 2008; 50:150–153. [DOI] [PubMed] [Google Scholar]
- 134.Rahbani-Nobar MB, Javadzadeh A, Ghojazadeh L, et al. The effect of Helicobacter pylori treatment on remission of idiopathic central serous chorioretinopathy. Mol Vis. 2011; 17:99–103. [PMC free article] [PubMed] [Google Scholar]
- 135.Dang Y, Mu Y, Zhao M, et al. Ther Clin Risk Manag. 2013; 9:355–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lehmann Ch, Bac VH, Pavlovic D, et al. Metronidazole improves intestinal microcirculation in septic rats independently of bacterial burden. Clin Hemorheol Microcirc. 2006; 34:427–438. [PubMed] [Google Scholar]
- 137.Stone AF, Mendall MA, Kaski JC, et al. Effect of treatment for Chlamydia pneumoniae and Helicobacter pylori on markers of inflammation and cardiac events in patients with acute coronary syndromes: South Thames Trial of Antibiotics in Myocardial Infarction and Unstable Angina (STAMINA). Circulation. 2002; 106:1219–1223. [DOI] [PubMed] [Google Scholar]
- 138.Ferry JA, Fung CY, Zukerberg L, et al. Lymphoma of the ocular adnexa: a study of 353 cases. Am J Surg Pathol. 2007; 31:170–184. [DOI] [PubMed] [Google Scholar]
- 139.Cahill M, Barnes C, Moriarty P, et al. Ocular adnexal lymphoma-comparison of MALT lymphoma with other histological types. Br J Ophthalmol. 1999; 83:742–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ferreri AJ, Dolcetti R, Du MQ, et al. Ocular adnexal MALT lymphoma: an intriguing model for antigen-driven lymphomagenesis and microbial-targeted therapy. Ann Oncol 2008; 19:835–846. [DOI] [PubMed] [Google Scholar]
- 141.Sarraf D, Jain A, Dubovy S, et al. Mucosa-associated lymphoid tissue lymphoma with intraocular involvement. Retina 2005; 25:94–98. [DOI] [PubMed] [Google Scholar]
- 142.Wotherspoon AC, Ortiz-Hidalgo C, Falzon MR, Isaacson PG. Helicobacter pylori-associated gastritis and primary B-cell gastric lymphoma. Lancet. 1991; 338:1175–1176. [DOI] [PubMed] [Google Scholar]
- 143.Mobley HL, Hu LT, Foxal PA. Helicobacter pylori urease: properties and role in pathogenesis. Scand J Gastroenterol 1991; 187:39–46. [PubMed] [Google Scholar]
- 144.Wroblewski LE, Peek RM, Jr, Wilson KT. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev 2010; 23:713–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Benktander J, Ångström J, Breimer ME, Teneberg S. Redefinition of the carbohydrate binding specificity of Helicobacter pylori BabA adhesin. J Biol Chem. 2012; 287:31712–31724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Peek RM, Jr, Crabtree JE. Helicobacter infection and gastric neoplasia. J Pathol. 2006; 208:233–248. [DOI] [PubMed] [Google Scholar]
- 147.Blaser MJ, Parsonnet J. Parasitism by the “slow” bacterium Helicobacter pylori leads to altered gastric homeostasis and neoplasia. J Clin Invest 1994; 94:4–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bodger K, Crabtree JE. Helicobacter pylori and gastric inflammation. Br Med Bull 1998; 54:139–150. [DOI] [PubMed] [Google Scholar]
- 149.Jung HC, Kim JM, Song IS, Kim CY. Helicobacter pylori induces an array of pro-inflammatory cytokines in human gastric epithelial cells: quantification of mRNA for interleukin-8-1α/β, granulocyte-macro-phage colony-stimulating factor, monocyte chemoattractant protein-1and tumour necrosis factor-α. J Gastroenterol Hepatol 1997; 12:473–480. [DOI] [PubMed] [Google Scholar]
- 150.Kranzer K, Eckhardt A, Aigner M, et al. Induction of maturation and cytokine release of human dendritic cells by Helicobacter pylori. Infect Immun 2004; 72:4416–4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.O’Keeffe J, Moran AP. Conventional, regulatory, and unconventional T cells in the immunologic response to Helicobacter pylori. Helicobacter 2008; 13:1–19. [DOI] [PubMed] [Google Scholar]
- 152.Yeh PY, Kuo SH, Yeh KH, et al. A pathway for tumor necrosis factor-α-induced Bcl10 nuclear translocation: Bcl10 is up-regulated by NF-κB and phosphorylated by Akt1 and then complexes with Bcl3 to enter the nucleus. J Biol Chem. 2006; 281:167–175. [DOI] [PubMed] [Google Scholar]
- 153.Suzuki T, Kato K, Ohara S, et al. Localization of antigen-presenting cells in Helicobacter pylori-infected gastric mucosa. Pathol Int 2002; 52:265–271. [DOI] [PubMed] [Google Scholar]
- 154.Necchi V, Candusso ME, Tava F, et al. Intracellular, intercellular, and stromal invasion of gastric mucosa, preneoplastic lesions, and cancer by Helicobacter pylori. Gastroenterology 2007; 132:1009–1023. [DOI] [PubMed] [Google Scholar]
- 155.Borlace GN, Butler RN, Brooks DA. Monocyte and macrophage killing of Helicobacter pylori: relationship to bacterial virulence factors. Helicobacter 2008; 13:380–387. [DOI] [PubMed] [Google Scholar]
- 156.Wong BC, Lam SK, Wong WM, et al. Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: a randomized controlled trial. JAMA 2004; 291:187–194. [DOI] [PubMed] [Google Scholar]
- 157.Hussell T, Isaacson PG, Crabtree JE, Spencer J. The response of cells from low-grade B-cell gastric lymphomas of mucosa-associated lymphoid tissue to Helicobacter pylori. Lancet 1993; 34:571–574. [DOI] [PubMed] [Google Scholar]
- 158.Bergman MP, D’Elios MM Cytotoxic T cells in Hp-related gastric autoimmunity and gastric lymphoma. J Biomed Biotechnol. 2010; 2010:104918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hussell T, Isaacson PG, Crabtree JE, Spencer JO. Helicobacter pylori-specific tumour-infiltrating T cells provide contact dependent help for the growth of malignant B cells in low-grade gastric lymphoma of mucosa-associated lymphoid tissue. J Pathol. 1996; 178:122–127. [DOI] [PubMed] [Google Scholar]
- 160.Munari F, Lonardi S, Cassatella MA, et al. Tumor-associated macrophages as major source of APRIL in gastric MALT lymphoma. Blood. 2011; 117:6612–6616. [DOI] [PubMed] [Google Scholar]
- 161.Witkowska M, Smolewski P. Helicobacter pylori infection, chronic inflammation, and genomic transformations in gastric MALT lymphoma. Mediators Inflamm. 2013; 2013:523170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.D’Elios MM, Amedei A, Manghetti M, et al. Impaired T-cell regulation of B-cell growth in Helicobacter pylori-related gastric low-grade MALT lymphoma. Gastroenterology. 1999; 117:1105–1112. [DOI] [PubMed] [Google Scholar]
- 163.Isaacson PG, Du MQ. MALT lymphoma: from morphology to molecules. Nat Rev Cancer 2004; 4:644–653. [DOI] [PubMed] [Google Scholar]
- 164.Suarez F, Lortholary O, Hermine O, Lecuit M. Infection-associated lymphomas derived from marginal zone B cells: a model of antigen-driven lymphoproliferation. Blood. 2006; 107:3034–3044. [DOI] [PubMed] [Google Scholar]
- 165.Zinzani PL. The many faces of marginal zone lymphoma. Hematology Am Soc Hematol Educ Program. 2012; 2012:426–432. [DOI] [PubMed] [Google Scholar]
- 166.Royer B, Cazals-Hatem D, Sibilia J, et al. Lymphomas in patients with Sjogren's syndrome are marginal zone B-cell neoplasms, arise in diverse extranodal and nodal sites, and are not associated with viruses. Blood. 1997; 90:766–775. [PubMed] [Google Scholar]
- 167.Bloch-Michel E, Nussenblatt RB. International Uveitis Study Group recommendations for the evaluation of intraocular inflammatory disease. Am J Ophthalmol. 1987; 103:234–235. [DOI] [PubMed] [Google Scholar]
- 168.Wakefield D, Chang JH. Epidemiology of uveitis. Int Ophthalmol Clin. 2005; 45:1–13. [DOI] [PubMed] [Google Scholar]
- 169.Barisani-Asenbauer T, Maca SM, Mejdoubi L, et al. Uveitis—a rare disease often associated with systemic diseases and infections- a systematic review of 2619 patients. Orphanet J Rare Dis. 2012; 7:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Otasevic L, Zlatanovic G, Stanojevic-Paovic A, et al. Helicobacter pylori: an underestimated factor in acute anterior uveitis and spondyloarthropathies? Ophthalmologica. 2007; 221:6–13. [DOI] [PubMed] [Google Scholar]
- 171.Kim JM, Park KH, Choi MJ, et al. Eye (Lond). 2012; 26:1503–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Martin TM, Smith JR, Rosenbaum JT. Anterior uveitis: current concepts of pathogenesis and interactions with the spondyloarthropathies. Curr Opin Rheumatol. 2002; 14:337–341. 10.1097/01.BOR.0000017926.78715.FD [DOI] [PubMed] [Google Scholar]
- 173.Gueudry J, Wechsler B, Terrada C, et al. Long-term efficacy and safety of low-dose interferon alpha-2a therapy in severe uveitis associated with Behçet disease. Am J Ophthalmol. 2008; 146:837–844. [DOI] [PubMed] [Google Scholar]
- 174.Orchard TR, Chua CN, Ahmad T, et al. Uveitis and erythema nodosum in inflammatory bowel disease: clinical features and the role of HLA genes. Gastroenterology. 2002; 123:714–718. [DOI] [PubMed] [Google Scholar]
- 175.Rosenbaum JT, McDevitt HO, Guss RB, Egbert PR. Endotoxin-induced uveitis in rats as a model for human disease. Nature. 1980; 286:611–613. 10.1038/286611a0 [DOI] [PubMed] [Google Scholar]
- 176.Yadav UC, Kalariya NM, Ramana KV. Emerging role of antioxidants in the protection of uveitis complications. Curr Med Chem. 2011; 18:931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Nagata M. Inflammatory cells and oxygen radicals. Curr Drug Targets Inflamm Allergy. 2005; 4:503–504. [DOI] [PubMed] [Google Scholar]
- 178.Srivastava SK, Ramana KV. Focus on molecules: nuclear factor-kappaB. Exp Eye Res. 2009; 88:2–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Han D, Ybanez MD, Ahmadi S, et al. Redox regulation of tumor necrosis factor signaling. Antioxid Redox Signal. 2009; 11:2245–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Gloire G, Piette J. Redox regulation of nuclear post-translational modifications during NF-kappaB activation. Antioxid Redox Signal. 2009; 11:2209–2222. [DOI] [PubMed] [Google Scholar]
- 181.Oliveira-Marques V, Marinho HS, Cyrne L, Antunes F. Role of hydrogen peroxide in NF-kappaB activation: from inducer to modulator. Antioxid Redox Signal. 2009; 11:2223–2243. [DOI] [PubMed] [Google Scholar]
- 182.Trouba KJ, Hamadeh HK, Amin RP, Germolec DR. Oxidative stress and its role in skin disease. Antioxid Redox Signal 2002; 4:665–673. [DOI] [PubMed] [Google Scholar]
- 183.Harris PR, Mobley HLT, Perez-Perez GI, et al. Helicobacter pylori urease is a potent stimulus of mononuclear phagocyte activation and imflammatory cytokine production. Gastroenterology 1996; 11:419–425. [DOI] [PubMed] [Google Scholar]
- 184.Verma MJ, Lloyd A, Rager H, et al. Chemokines in acute anterior uveitis. Curr Eye Res 1997; 16:1202–1208.PMID: [9426952]. [DOI] [PubMed] [Google Scholar]
- 185.Appelmelk BJ, Simoons-Smit I, Negrini R, et al. Potential role of molecular mimicry between Helicobacter pylori lipopolysaccharide and host Lewis blood group antigens in autoimmunity. Infect Immun 1996; 64:2031–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Otasevic L, Walduck A, Meyer TF, et al. Helicobacter pylori infection in anterior uveitis. Infection. 2005; 33:82–85.PMID: [15827876]. [DOI] [PubMed] [Google Scholar]
- 187.DeLano FA, Balete R, Schmid-Schonbein GW. Control of oxidative stress in microcirculation of spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2005; 288:H805–H812. [DOI] [PubMed] [Google Scholar]
- 188.Halliday GM. Inflammation, gene mutation and photoimmunosuppression in response to UVR-induced oxidative damage contributes to photocarcinogenesis. Mutat Res. 2005; 571:107–120. [DOI] [PubMed] [Google Scholar]
- 189.Sun YQ, Girgensone I, Leanderson P, et al. Effects of antioxidant vitamin supplements on Helicobacter pylori-induced gastritis in Mongolian gerbils. Helicobacter. 2005; 10:33–42. [DOI] [PubMed] [Google Scholar]
- 190.Calvino Fernández M, Parra Cid TH. pylori and mitochondrial changes in epithelial cells The role of oxidative stress. Rev Esp Enferm Dig. 2010; 102:41–50. [DOI] [PubMed] [Google Scholar]
- 191.May JM. Is ascorbic acid an antioxidant for the plasma membrane? FASEB J. 1995; 13,:1999e1006. [DOI] [PubMed] [Google Scholar]
- 192.Xu P, Lin Y, Porter K, Liton PB. Ascorbic acid modulation of iron homeostasis and lysosomal function in trabecular meshwork cells. J Ocul Pharmacol Ther. 2014; 30:246–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Aditi A, Graham DY. Vitamin C, gastritis, and gastric disease: a historical review and update. Dig Dis Sci. 2012; 57:2504–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]