

Abstract
Genetic and non-genetic predictors of 15-year survival in patients with chronic three-vessel disease (3VD) were investigated.
Coronary angiography was performed on 810 subjects with symptoms of stable ischemic heart disease in 1998. The patients with 3VD were genotyped for 23 candidate polymorphisms covering the PPAR-RXR pathway, matrix metalloproteinase-2, renin–angiotensin–aldosterone system, endothelin-1, cytokine genes, MTHFR and APO E variants. Fifteen-year survival data were obtained from the national insurance registry. All data were available in the case of 150 patients with 3VD. Statistical analysis used stepwise Cox regression with dominant, recessive, or additive mode of genetic expression. Involved variables included age, sex, BMI, blood pressure, diabetes, ejection fraction, left main stenosis, previously diagnosed coronary stenosis, myocardial infarction in personal history, and coronary bypass along with polymorphisms pre-selected by log-rank tests.
Out of the 23 polymorphisms, four were included in the model construction. SNP in the IL-6 gene rs1800795 (−174 G/C) has been found to be a significant predictor of survival. This SNP was in a linkage disequilibrium with rs1800797 (−597 G/A) in the same gene (D′ = 1.0), which was also found to constitute a significant predictor of survival when rs1800795 was not included in the model construction. Age, increased BMI, diabetes, low EF, and left main stenosis were also significant predictors in all models.
Age, increased BMI, diabetes, low ejection fraction, left main stenosis, and genetic variation in the IL-6 promoter were established as significant independent risk factors for the survival of patients with three-vessel disease.
INTRODUCTION
Coronary artery disease (CAD) and its complications – such as myocardial infarction or heart failure – is one of the leading causes of death in most world populations.1,2 Three-vessel disease (3VD) represents the most severe form of coronary atherosclerosis. Patients with 3VD and/or left main stenosis are considered a high-risk group according to therapeutic guidelines.3 In comparison with less severe forms of CAD, 3VD has been consistently associated with worse long-term prognosis.4–7 Genetic variation in several metabolic, inflammatory, and local signal pathways is worthy of consideration concerning possible effects on patients’ survival.
Overall heritability of CAD has been estimated at approximately 50% in population studies and over 30 genes have been associated with CAD onset in genome-wide association studies (GWAS).8 While the genome-wide association approach explains only a small fraction of total heritability,9 candidate gene-based studies often suffer from various types of bias which may lead to both false positive and false negative results.10–12 The role of various suspected genetic risk factors in the survival of patients already suffering from symptomatic CAD is not yet well understood.
As atherosclerosis is an inflammatory process, cytokines play an important role in its pathogenesis. Cytokines such as tumour necrosis factor (TNF) α and interleukin (IL) 6 have been extensively studied. While TNF-α seems to be clearly pro-atherogenic, the role of IL-6 is somewhat ambivalent in animal and human studies.13 The precursor of TNF-α is converted to its active form by its converting enzyme, TACE.14 TNF-β, also known as lymphotoxin α, is secreted by regulatory T-lymphocytes and exhibits anti-atherogenic effects.15
Regarding the variation of lipid metabolism pathways, one of the key molecules is apolipoprotein E (ApoE = protein, APO E = gene), a protein which ensures lipoprotein clearance, prevents lipid accumulation in the vessel wall,16 and has antioxidant,17 vasodilatory18 and anti-inflammatory19 effects. The peroxisome proliferator-activated receptor/retinoid X receptor (PPAR-RXR) pathway is involved in both the regulation of the lipid and glucose metabolism and in cytokine release.20 The lower expression of PPAR-γ and RXR-α has been associated with the faster progression of carotic atherosclerosis.21 Similarly, PPAR-α also has anti-atherogenic properties22
Contributing to local inflammation, matrix metalloproteinases (MMPs) are endopeptidases which degrade the extracellular matrix. Many MMPs are expressed in atherosclerotic vessels.23,24 Of these, MMP-2 has been found to participate in lesion formation in the animal model of atherosclerosis25 and its gene expression is higher in acute coronary syndrome patients compared to healthy subjects.26
The renin–angiotensin–aldosterone system (RAAS) plays also role in tissue remodelling and is an important regulator of blood pressure. The hyperactivity of RAAS is linked to cardiovascular diseases including hypertension and CAD. Angiotensin converting enzyme (ACE) is a key molecule activating angiotensin II, which is a strong vasoconstrictor.27 Endothelins are a group of other vasoconstriction peptides. Endothelin-1 (ET-1) is synthesized mostly in the vessel wall and is the most potent vasoconstrictor. Moreover, it exerts several other biological functions leading to elevated blood pressure.28
From other possible risk-modifying factors, methylene tetrahydrofolate reductase (MTHFR) is an enzyme important for homocysteine degradation. The overaccumulation of homocysteine is associated with higher risk of atherosclerosis, probably through various mechanisms.29
We conducted the study to establish the genetic and other factors contributing to all-cause death of patients with chronic symptomatic 3VD. The aim was to create a model predicting patients’ survival based on significant and independent risk factors. The non-genetic determinants that were considered to possibly play an important role in survival included the following clinical factors and characteristics of cardiac involvement: age at admission, sex, body mass index (BMI), systolic and diastolic blood pressure (SBP and DBP, respectively), diabetes mellitus (DM), hyperlipidemia, ejection fraction (EF), left main stenosis, extent of CAD, previously diagnosed stenosis of coronary artery, myocardial infarction in personal history, and mode of intervention – coronary artery bypass grafting (CABG), percutaneous coronary intervention (PCI), or pharmacological therapy. Potential genetic factors involved 23 candidate polymorphisms including the variants in genes coding RXR-a, RXR-b, PPAR-a, PPAR-g, endothelin-1, TNF-a, TACE, TNF-b, IL-6, MMP-2, angiotensinogen (AGT), ACE, MTHFR, and ApoE.
METHODS
Coronary Angiography and Patient Selection
Left and right coronary angiography and left ventriculography were performed on 810 consecutive subjects at the First Department of Internal Medicine – Cardioangiology at St. Anne's University Hospital in Brno in 1998. The subjects suffered from chest pain or other symptoms of stable ischemic heart disease. Coronary angiograms were assessed by four experienced invasive cardiologists. Of the total number of subjects, 196 suffered from 3VD, defined as ≥50% stenosis of the left anterior descendent branch (LAD), left circumflex branch (LCx), right coronary artery (RCA), and/or major branches of each artery. Extent of CAD was defined as the number of segments with ≥50% stenosis, according to the 16-segment scheme of American Heart Association.
Informed consent was obtained from all patients prior to their recruitment according to the requirements of Ethics committee of St. Anne's University Hospital, which approved the study. All the procedures were in accord with the Helsinki Declaration of 1975 as revised in 1983. Patients treated for concomitant significant valvular disease and those after heart transplantation were excluded. The remaining subjects were genotyped for 23 candidate polymorphisms and other clinical and laboratory data were collected. Data about 15-year survival were obtained from national insurance registry on May 23, 2013. All data were available in the case of 150 patients; only these subjects were included in subsequent analyses. The reasons of the exclusion of remaining 46 patients were incomplete genetic analysis of 23 polymorphisms (n = 40), incomplete clinical data (n = 5), and inability to obtain the data about patients survival from national insurance registry (n = 1). There was no difference in survival between patients included and excluded in the model (in case when survival data were available; Gehan Wilcoxon test P-value = .28).
Laboratory Methods
DNA was extracted from peripheral blood leukocytes using the phenol–chloroform method. Of the total number of 23 polymorphisms, 21 were identified using polymerase chain reaction (PCR), and restriction analysis. For the single nucleotide polymorphism (SNP) rs1536475 (intron 7, +70 A/G) in the RXR-α gene, PCR was carried out in a volume of 25 μl, containing 0.8 U of Taq polymerase and primers 5-AGACAGCTGAGTGACTGTGTG C-3 (forward) and 5-GAAATAATACTAGGCAGGATGTGC-3 (reverse). The method was described in30 and the resulting fragment was 269 base pairs (bp) in length. The process of restriction analysis was modified in our laboratory and included digestion by SatI enzyme in 37°C and electrophoresis in 2% agarose gel (Serva). The resulting fragments were 162 + 66 + 41 bp (A allele) or 123 + 66 + 41 + 39 bp (G allele) in length.
The parameters of methods used in the detection of other polymorphisms have been described in our previous publications. This includes polymorphisms rs148360070 (intron 5 39526 A/AA) and rs1805343 (intron 9 −25 G/A) in the RXR-α gene and all variants in genes coding RXR-β,31 PPAR-α, PPAR-γ,32 endothelin-1, TNF-α, TACE, TNF-β,33 IL-6,34,35 MMP-2,36 angiotensinogen,37 ACE,38 and MTHFR.39
Two polymorphisms in the Apo E were determined using real-time PCR monitored by SYBR Green.40 The method was optimized by our research group.41
Statistical Analysis
The Cox regression model was used in order to estimate the contribution of genetic polymorphisms and other risk factors to overall survival. The genetic variants had been pre-selected out of the total number of 23 polymorphisms in candidate genes using the Kaplan–Meier method and log-rank tests in dominant, recessive and co-dominant modes of allelic expression. A P-value of .1 was used as the cut-off for including the variable in the Cox regression analysis, the power of log-rank tests were 0.2 to 0.8 for each separate test, depending on allele and genotype frequency. Tests with lower power due to low number in one of the compared groups were not performed; the combination of three tests for each polymorphism increased power to at least 0.6 with the exception of APO E2. The Hardy–Weinberg equilibrium was calculated for each polymorphism using the χ2-test. To address potential selection bias, the log-rank tests were repeated for the variants included in the Cox regression model construction in all patients with 3VD, where the data about given variant and survival were available (150 < n < 196).
A stepwise construction of the Cox regression model with a P-value to include of .05 and P-value to remove of .051 was subsequently employed to determine the contribution of pre-selected genetic and other variables to overall survival. Non-genetic variables included in the stepwise construction were age at admission, sex, BMI, hyperlipidemia, systolic and diastolic blood pressure, DM, EF, left main stenosis, extent of CAD, previously diagnosed stenosis of coronary artery, myocardial infarction in personal history, and mode of treatment.
With the unknown allelic coefficient of dominance, three models of gene expressions were employed: dominant, where minor allele carriers were compared with major allele homozygotes, recessive, where major allele carriers were compared with minor allele homozygotes (calculated only when the number of minor allele homozygotes was more than five) and additive, a gene dose-based model where the values 0, 1 and 2 were attributed to major allele homozygotes, heterozygotes, and minor allele homozygotes.
Finally, an all-effects multivariate Cox model was used to determine the hazard ratio (HR) with 95% confidence interval (CI) of different genotypes of the SNPs determined as a significant factor in overall survival following adjustments for age, sex, body mass index (BMI), systolic and diastolic blood pressure (SBP and DBP, respectively), diabetes mellitus (DM), hyperlipidemia, left main stenosis, EF, extent of CAD, previously diagnosed stenosis of coronary artery, myocardial infarction in personal history, and mode of treatment. Bonferroni correction was used for multiple comparisons testing, the respective P-values are listed as pcorr. Comparisons of continuous variables between specified groups of patients were performed using Mann–Whitney U-test; for categorical variables Fisher exact test was used. Generally, α = 0.05 was used to determine statistically significant results in all analyses. STATISTICA software (StatSoft, version 12) was used for statistical analysis. MIDAS software (version 1.0)42 was used for linkage disequilibrium determination.
RESULTS
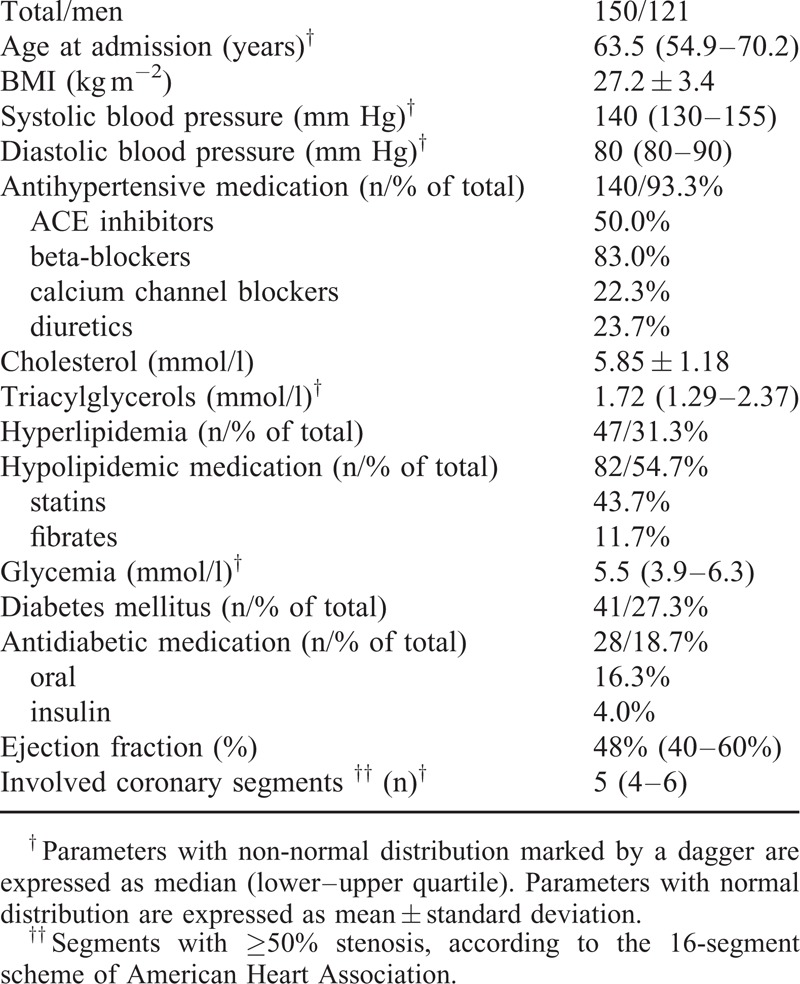
The basic characteristics of the group and its medication are shown in a table (Table 1). Approximately 50% of the patients with three-vessel disease died before the end of the study. The 5-year survival rate was 88%.
TABLE 1.
Basic Characteristics of the Subjects
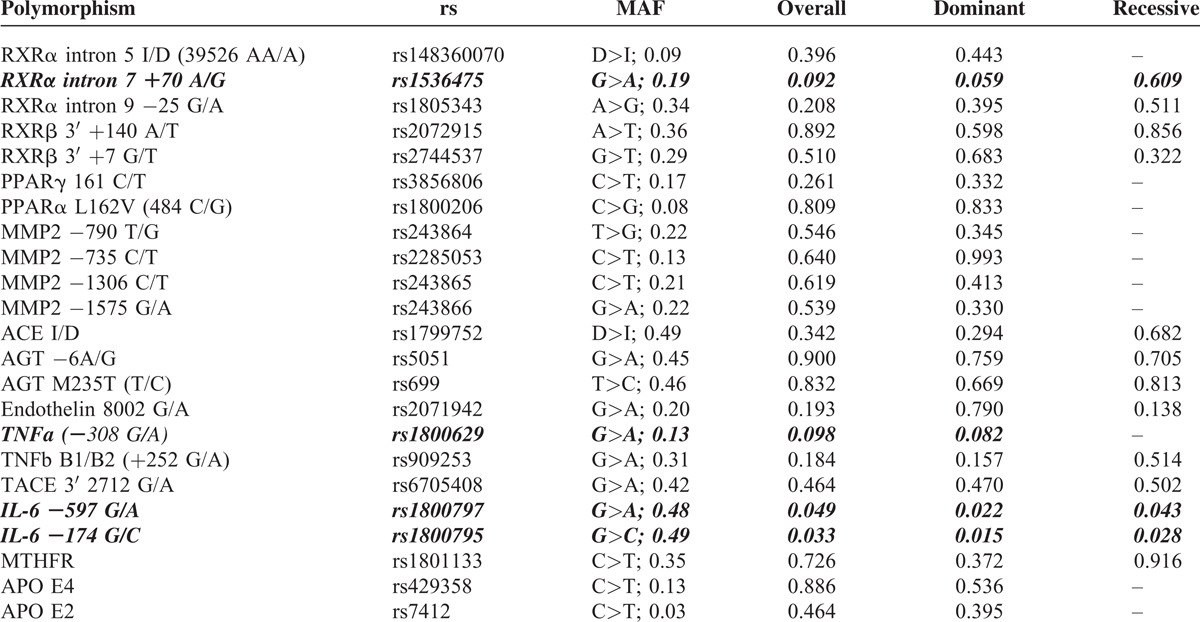
The pre-selection of polymorphisms included in final model construction was based on log-rank P-values. Only the genetic variants with P < .10 in at least one model were selected for further analysis (Table 2). Polymorphisms rs1536475 in RXR-α, 1800629 in TNF-α, and two SNPs in Il-6 (rs1800795 and 1800797) met the inclusion criteria. When the log-rank tests were repeated in all patients with 3VD where the data about given variant and survival were available, only the two latter polymorphisms showed the P-value <.10 (and <.05, actually).
TABLE 2.
Candidate Polymorphisms, Their Respective Minor Allele Frequencies (MAF) and Log-Rank P-Values. Polymorphisms Included in the Stepwise Cox Regression Model Construction are Indicated in BoldItalics
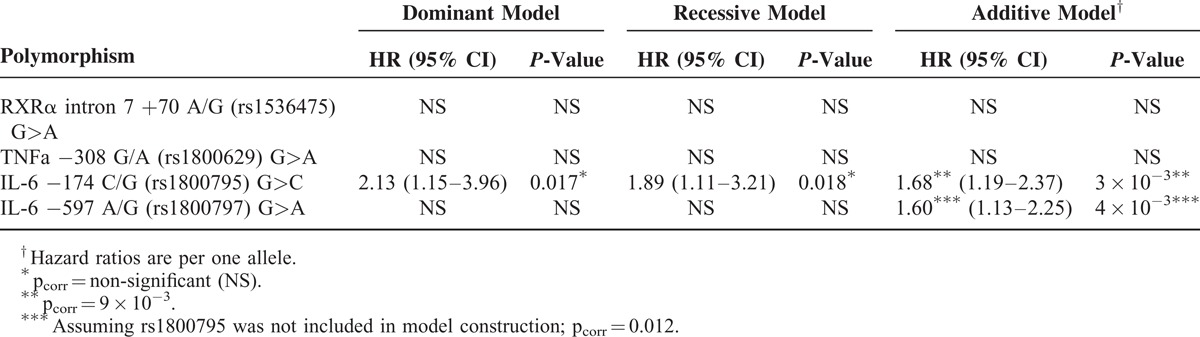
The stepwise Cox regression model construction identified the same significant predictors of survival consistently in all three models: age at admission, ejection fraction, left main stenosis, BMI, diabetes, and polymorphism rs1800795 (−174 G/C) in the IL-6 gene. In additive model, the effect of rs1800795 alleles remained significant after Bonferroni correction, the C allele was associated with worse prognosis. This SNP was in strong linkage disequilibrium with rs1800797 (−597 G/A) in the same gene (D′ = 1.0; r2 = .97; P < 10−20). The G–G haplotype was the most common (51%), followed by A–C (48%) and G–C (1%); A–G was missing in our group of patients. SNP rs1800797 was also a significant predictor of survival in the additive model when rs1800795 was not included in model construction. The G allele was protective and the A allele risky in this case. No other polymorphism was a significant independent predictor of survival (Table 3).
TABLE 3.
Effect of Selected Polymorphisms in Multivariate Analysis. P-Values and Hazard Ratios (HR) are Adjusted for Other Significant Independent Predictors of Survival. The Major Allele Was Taken as the Reference Variant
The final multivariate model obtained by stepwise Cox regression and including all significant predictors (with the additive effect of rs1800795 alleles) is shown in a table (Table 4).
TABLE 4.
Final Multivariate Cox Regression Model Including All Significant Independent Predictors of Survival

Following adjustments for all clinical covariates (all effects model) and compared to GG carriers of rs1800795, CG carriers had a HR of 2.19 (95% CI = 1.05–4.58) and CC homozygotes had a HR of 3.79 (95% CI = 1.78–8.10). The Kaplan–Meier survival curves of different genotypes of rs1800795 following adjustment for age, sex, diabetes, BMI, EF, extent of CAD, dyslipidemia, SBP, DBP, left main stenosis, previously diagnosed coronary stenosis, myocardial infarction in personal history, and mode of intervention are shown in a graph (Figure 1). Analogically, compared to GG homozygotes in rs1800797, AG heterozygotes had a HR of 2.09 (95% CI = 1.01–4.33) and AA homozygotes exhibited a HR of 3.40 (95% CI = 1.61–7.16); a P-value was <.05 in all the cases.
FIGURE 1.
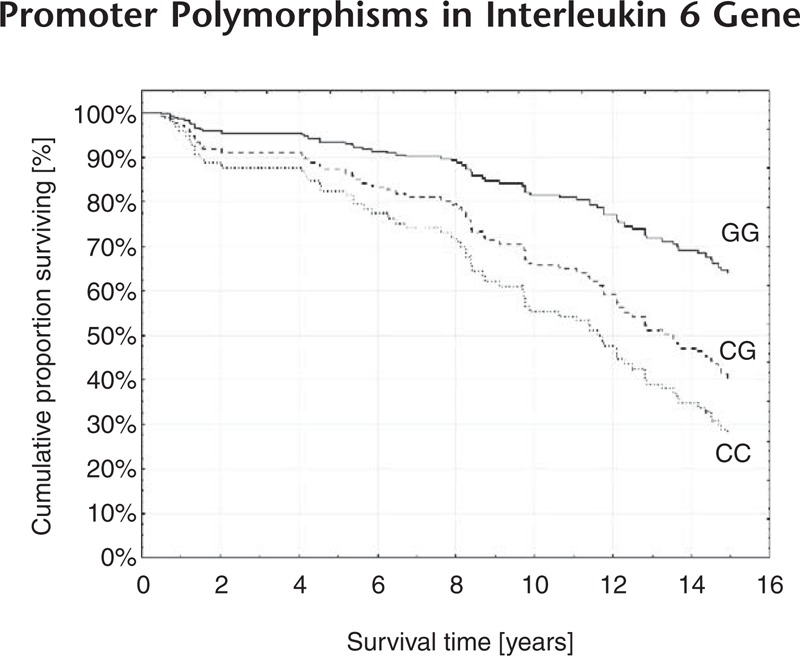
Effect of rs1800795 (−174 G/C) in the IL-6 gene on survival in multivariate analysis. Kaplan–Meier curves are adjusted for age, sex, diabetes, BMI, EF, SBP, DBP, left main stenosis, previously diagnosed coronary stenosis and myocardial infarction in personal history. Compared to GG homozygotes, both CG (HR = 2.19; 95% CI = 1.05–4.58; P = .04) and CC (HR = 3.79; 95% CI = 1.78–8.10; P = 6 × 10−4) carriers showed higher mortality.
Of the total number of 150 patients, a majority (n = 94) was treated by CABG, 28 patients underwent PCI, and the remaining subjects (n = 28) were treated pharmacologically. The CABG procedure was not a significant independent predictor of survival in the stepwise Cox regression model. In a separate analysis, CABG was superior to pharmacological therapy (log-rank test: P = .04); however, this effect became insignificant in a multivariate model (P = .18, HR = 0.65, 95% CI = 0.35–1.21) following adjustment for all clinical covariates. Patients treated by CABG had higher EF compared to patients treated by PCI or pharmacological therapy (P = 6 × 10−3), they did not differ in other investigated clinical parameters and characteristics of cardiac involvement.
DISCUSSION
Clinical and Angiographic Predictors of Mortality
This study revealed several significant independent predictors of mortality in patients with chronic 3VD. Characteristics of cardiac involvement, detected using coronary angiography, and ventriculography were important predictors of all-cause death. Specifically, left main stenosis increased the risk of death more than three-fold while each 5% of ejection fraction lowered the risk by approximately 20%. Other significant predictors of death – age, diabetes, and BMI – are considered to be established cardiovascular risk factors.43–45 Diabetes mellitus leads to hyperglycaemia which promotes the progression of atherosclerosis both directly, by non-enzymatic glycation of endothelial proteins,46 and indirectly, by lipoprotein modification and changes in their function.47–49
In this study, higher BMI was associated with a worse prognosis in multivariate analysis: an increase of 1 kg m−2 increased risk of death by over 12%. This result confirms the role of overweight as an independent risk factor in patients suffering from 3VD and is in contradiction with some studies and meta-analyses reporting on the so-called “obesity paradox,” that is, better prognosis in overweight cardiac patients.50,51 However, selection bias is most likely at least a component factor contributing to the obesity paradox.52 It must be noted that the study group assembled for the purposes of our study was relatively homogenous and that patients suffered from a severe form of coronary atherosclerosis. Prospective design, as used in our study, should reduce the possibility of selection bias which might influence the results of retrospective studies.
Promoter Polymorphisms in Interleukin 6 Gene
Out of the 23 potential genetic predictors (including genetic variants of the PPAR-RXR pathway, matrix metalloproteinase-2, renin–angiotensin–aldosterone system, endothelin-1, cytokine genes, MTHFR and APO E), only two closely linked polymorphisms in the IL-6 promoter proved to have both statistical and clinical significance for patient survival. The role of IL-6 and its genetic variants in atherosclerosis is not fully clarified and results of many in vitro, animal model and human studies are controversial. In vitro, IL-6 stimulates angiogenesis53 while animal models of atherosclerosis have shown IL-6 to be both anti-atherosclerotic54,55 and pro-atherosclerotic,56 depending on concentration. It has also been found to have pro-coagulation and pro-inflammatory effects57,58 and to contribute to heart remodelling after myocardial infarction in human patients.59 The long-term elevation of IL-6 levels has been associated with the risk of CAD60 and CAD severity.61 Elevated concentrations of IL-6 are also predictors of mortality in patients with CAD or heart failure,62,63 although the causality is uncertain and the concentration of IL-6 largely varies in the same individual.64 “Trans-signalling,” that is, cell activation by the soluble IL-6/IL-6 receptor complex instead of more specific IL-6 binding to its surface receptor, is probably responsible for many detrimental effects of IL-6.65,66
The promoter polymorphisms of IL-6 have been repeatedly associated with different gene expression. The promoter region from −225 to −113, that is, involving the common −174 G/C polymorphism, contains regulatory elements which ensure transcription induction by IL-1 or TNFα.67 Indeed, following stimulation by IL-1, transcription is enhanced in the IL-6 −174 G-allele containing transfected HeLa cells when compared to the C-allele; this SNP can therefore be considered functional.68 The carriers of different genotypes have different plasmatic concentration of IL-6, which decreases in a sequence of GG > GC > CC in healthy people68 or in patients with brain vessel malformations,69 while CC carriers were shown to have higher IL-6 levels in days following coronary artery bypass graft surgery, suggesting a different pattern during inflammatory responses.70 This effect may be partially explained by linkage disequilibrium with another functional promoter SNP in the IL-6 gene, that is, −6331 T/C, located in the binding site for enhancing transcription factor Oct-1.71 Other polymorphisms in the IL-6 gene have also been shown to influence its expression in interaction with −174 G/C. They include −597 G/A, SNP in strong linkage disequilibrium with −174 G/C, which was confirmed in our study.72,73 Following the administration of lipopolysaccharides, bacterial surface molecules capable of inducing foam cell formation,74 a lower secretion of IL-6 was observed in subjects with −597 G to −174 G haplotypes compared to −597 A to −174 C haplotypes.73
The relationship between IL-6 polymorphisms and CAD has been investigated for more than 20 years and may provide insight into causality in the association between high IL-6 levels and CAD. In a recent extensive meta-analysis of 50 studies involving over 30,000 patients, no significant association between IL-6 −174 G/C and coronary artery disease onset was established in patients of Caucasian origin. However, there was substantial heterogeneity among the studies. In subgroup analysis the G allele was protective in studies where population-based control subjects were used and in non-Caucasian populations.75 Data regarding survival of patients with confirmed CAD are scarcer. In patients presenting with acute coronary syndrome, the G allele was found to have detrimental effects on 1-year survival.76 On the other hand, in another study including 218 patients with chronic CAD and renal failure, subjects carrying the G allele had a significantly better prognosis which was found to be consistent with our results.77 It is possible that mildly elevated IL-6 levels in −597 G to −174 G carriers could be beneficial due to their support of angiogenesis in chronic 3VD, while the increased risk of plaque destabilization is more important after acute coronary syndrome.
Other Investigated Polymorphisms
In the present study, none of the remaining 21 investigated polymorphisms constituted significant independent risk factors of death in patients suffering from 3VD, although the association with CAD has been previously reported in many cases. For example, promoter polymorphisms of the MMP-2 gene −1306 C/T and −790 T/G have been associated with the presence of CAD in a meta-analysis of 9 studies78 including a study conducted by our research group which compared patients with 3VD to controls.36 The genetic variation of RAAS has also been extensively studied in cardiovascular research. Polymorphisms of ACE and angiotensinogen have been associated with different angiotensin II levels, as well as with cardiovascular risk.79 A common polymorphism in MTHFR, 677 C/T, has been associated with the risk of atherosclerosis in Asian populations. In Europeans, who have a relatively higher folate intake, no significant contribution of MTHFR 677 C/T to the onset of CAD has been established80; however, there might be greater risk for the T allele in otherwise risky subgroups.81 The intron polymorphism of the ET-1 gene 8002 G/A has been associated with myocardial infarction by our group.33 In our previous research we failed to prove the effect of polymorphisms in PPAR-α PPAR-γ and RXR-α on prognosis after PCI32; however, variation in the RXR-α intron was associated with all-cause heart failure.82 Furthermore, 161 C/T in PPAR-γ has been linked to CAD among the Chinese (but not Caucasians) in a recent meta-analysis.83
The APO E locus belongs to 35 loci which have been associated with CAD in GWAS.8 While ε4 carriers are at a slightly higher risk of atherosclerosis compared to most frequent ε3/ε3 homozygotes, the ε2 allele is protective in heterozygotes and ε2/ε2 homozygotes have a variable risk of atherosclerosis.84 With respect to tumour necrosis factors, association studies of TNF-α promoter variants have produced various results85–87 while the role of polymorphism B1/B2 (+252 G/A) in TNF-β, previously suggested as contributing to CAD onset, was found to be insignificant in a meta-analysis of 22 studies.88
Since data from other studies regarding the effect of the mentioned variants on CAD patient survival are largely missing, this study contributes to understanding their role in the progression of atherosclerosis. The long-term prospective approach should be a useful tool with respect to the evaluation of the contribution of these variants to the prognosis of patients suffering from severe CAD. In our study, only the genetic variation of the IL-6 promoter added new, independent information besides the characteristics of cardiac involvement and the traditional cardiovascular risk factors.
Limitations
The study has several limitations. Firstly, all patients were enrolled in single institution, which limits the generalizability of our results. Other limitation is relatively low number of participants with all available data which reduces the power of the study; however the power is gained by relative homogeneity of the study group and long follow-up time. The power could have not been high enough to detect all potential genetic and non-genetic effects and larger multicentre studies are needed in this respect. The disappearance of the CABG effect in a multivariate model suggests the role of patient selection for the surgery; in subsequent analysis, patients with higher EF were more frequently selected for CABG, and EF was the most significant predictor of survival in stepwise model. However, the fact that CABG was not a significant independent predictor does not contradict the established therapeutic benefit of the procedure; it is rather a consequence of the small size of the PCI and pharmacological treatment groups, and therefore low power of the test.
CONCLUSION
Age, increased BMI, diabetes, low ejection fraction, left main stenosis, and genetic variation in the IL-6 promoter were established as significant independent risk factors for the survival of patients with three-vessel disease. The G alleles of promoter polymorphisms rs1800795 (−174 G/C) and rs1800797 (−597 G/A) were associated with lower mortality.
Acknowledgments
The authors would like to express acknowledgement to David Konečný, M.A., for valuable help with language revision.
Footnotes
Abbreviations: ACE = angiotensin converting enzyme, AGT = angiotensinogen, ApoE = apolipoprotein E, APO E = apolipoprotein E gene, bp = base pair, BMI = body mass index, CI = confidence interval, CABG = coronary artery bypass graft, CAD = coronary artery disease, DM = diabetes mellitus, DBP = diastolic blood pressure, EF = ejection fraction, ET-1 = endothelin-1, GWAS = genome-wide association studies, HR = hazard ratio, IL = interleukin, LAD = left anterior descendent branch, LCx = left circumflex branch, MMP = matrix metalloproteinase, MTHFR = methylene tetrahydrofolate reductase, MAF = minor allele frequency, PCI = percutaneous coronary intervention, PPAR = peroxisome proliferator-activated receptor, PCR = polymerase chain reaction, RAAS = renin–angiotensin–aldosterone system, RXR = retinoid X receptor, RCA = right coronary artery, SNP = single nucleotide polymorphism, SBP = systolic blood pressure, 3VD = three-vessel disease, TACE = TNF-α converting enzyme, TNF = tumour necrosis factor.
The study was funded by Project VS96-097 “Promotion of Research in Universities” from Ministry of Education, Youth and Physical Training of the Czech Republic, by European Regional Development Fund – Project FNUSA ICRC (No. CZ.1.05/1.1.00/02.0123) and by Specific Research grant of Masaryk University MUNI/A/0948/2011 and by Research Grant IGA MZ ČR NT11412-5/2010
There is no potential conflict of interests.
REFERENCES
- 1.Gersh BJ, Sliwa K, Mayosi BM, Yusuf S. Novel therapeutic concepts: the epidemic of cardiovascular disease in the developing world: global implications. Eur Heart J 2010; 31:642–648. [DOI] [PubMed] [Google Scholar]
- 2.WHO. World Health Organization – Global atlas on CVD prevention and control. 2011; Available from: http://whqlibdoc.who.int/publications/2011/9789241564373_eng.pdf (accessed 16/03/2012). [Google Scholar]
- 3.Mohr FW, Morice MC, Kappetein AP, et al. Coronary artery bypass graft surgery versus percutaneous coronary intervention in patients with three-vessel disease and left main coronary disease: 5-year follow-up of the randomised, clinical SYNTAX trial. Lancet 2013; 381:629–638. [DOI] [PubMed] [Google Scholar]
- 4.Lopes NH, Paulitsch FdaS, Gois AF, et al. Impact of number of vessels disease on outcome of patients with stable coronary artery disease: 5-year follow-up of the Medical, Angioplasty, and bypass Surgery study (MASS). Eur J Cardiothorac Surg 2008; 33:349–354. [DOI] [PubMed] [Google Scholar]
- 5.Lawrie GM, Morris GC, Jr, Calhoon JH, et al. Clinical results of coronary bypass in 500 patients at least 10 years after operation. Circulation 1982; 66 (Pt 2):I1–5. [PubMed] [Google Scholar]
- 6.van Domburg RT, Kappetein AP, Bogers AJ. The clinical outcome after coronary bypass surgery: a 30-year follow-up study. Eur Heart J 2009; 30:453–458. [DOI] [PubMed] [Google Scholar]
- 7.Chauhan MS, Kuntz RE, Ho KL, et al. Coronary artery stenting in the aged. J Am Coll Cardiol 2001; 37:856–862. [DOI] [PubMed] [Google Scholar]
- 8.McPherson R. From genome-wide association studies to functional genomics: new insights into cardiovascular disease. Can J Cardiol 2013; 29:23–29. [DOI] [PubMed] [Google Scholar]
- 9.So HC, Gui AH, Cherny SS, Sham PC. Evaluating the heritability explained by known susceptibility variants: a survey of ten complex diseases. Genet Epidemiol 2011; 35:310–317. [DOI] [PubMed] [Google Scholar]
- 10.Casas JP, Cooper J, Miller GJ, et al. Investigating the genetic determinants of cardiovascular disease using candidate genes and meta-analysis of association studies. Ann Hum Genet 2006; 70 (Pt 2):145–169. [DOI] [PubMed] [Google Scholar]
- 11.Ioannidis JP, Tarone R, McLaughlin JK. The false-positive to false-negative ratio in epidemiologic studies. Epidemiology 2011; 22:450–456. [DOI] [PubMed] [Google Scholar]
- 12.Colhoun HM, McKeigue PM, Davey Smith G. Problems of reporting genetic associations with complex outcomes. Lancet 2003; 361:865–872. [DOI] [PubMed] [Google Scholar]
- 13.Ait-Oufella H, Taleb S, Mallat Z, Tedgui A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler Thromb Vasc Biol 2011; 31:969–979. [DOI] [PubMed] [Google Scholar]
- 14.Mohan MJ, Seaton T, Mitchell J, et al. The tumor necrosis factor-alpha converting enzyme (TACE): a unique metalloproteinase with highly defined substrate selectivity. Biochemistry 2002; 41:9462–9469. [DOI] [PubMed] [Google Scholar]
- 15.Chistiakov DA, Sobenin IA, Orekhov AN. Regulatory T cells in atherosclerosis and strategies to induce the endogenous atheroprotective immune response. Immunol Lett 2013; 151:10–22. [DOI] [PubMed] [Google Scholar]
- 16.Kolovou GD, Anagnostopoulou KK. Apolipoprotein E polymorphism, age and coronary heart disease. Ageing Res Rev 2007; 6:94–108. [DOI] [PubMed] [Google Scholar]
- 17.Miyata M, Smith JD. Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta-amyloid peptides. Nat Genet 1996; 14:55–61. [DOI] [PubMed] [Google Scholar]
- 18.Sacre SM, Stannard AK, Owen JS. Apolipoprotein E (apoE) isoforms differentially induce nitric oxide production in endothelial cells. FEBS Lett 2003; 540:181–187. [DOI] [PubMed] [Google Scholar]
- 19.Baitsch D, Bock HH, Engel T, et al. Apolipoprotein e induces antiinflammatory phenotype in macrophages. Arterioscler Thromb Vasc Biol 2011; 31:1160–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nohara A, Kobayashi J, Mabuchi H. Retinoid X receptor heterodimer variants and cardiovascular risk factors. J Atheroscler Thromb 2009; 16:303–318. [DOI] [PubMed] [Google Scholar]
- 21.Giaginis C, Klonaris C, Katsargyris A, et al. Correlation of Peroxisome Proliferator-Activated Receptor-gamma (PPAR-gamma) and Retinoid X Receptor-alpha (RXR-alpha) expression with clinical risk factors in patients with advanced carotid atherosclerosis. Med Sci Monit 2011; 17:Cr381–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fruchart JC. Peroxisome proliferator-activated receptor-alpha (PPARalpha): at the crossroads of obesity, diabetes and cardiovascular disease. Atherosclerosis 2009; 205:1–8. [DOI] [PubMed] [Google Scholar]
- 23.Back M, Ketelhuth DF, Agewall S. Matrix metalloproteinases in atherothrombosis. Prog Cardiovasc Dis 2010; 52:410–428. [DOI] [PubMed] [Google Scholar]
- 24.Ketelhuth DF, Back M. The role of matrix metalloproteinases in atherothrombosis. Curr Atheroscler Rep 2011; 13:162–169. [DOI] [PubMed] [Google Scholar]
- 25.Kuzuya M, Nakamura K, Sasaki T, et al. Effect of MMP-2 deficiency on atherosclerotic lesion formation in apoE-deficient mice. Arterioscler Thromb Vasc Biol 2006; 26:1120–1125. [DOI] [PubMed] [Google Scholar]
- 26.Dabek J, Glogowska-Ligus J, Szadorska B. Transcription activity of MMP-2 and MMP-9 metalloproteinase genes and their tissue inhibitor (TIMP-2) in acute coronary syndrome patients. J Postgrad Med 2013; 59:115–120. [DOI] [PubMed] [Google Scholar]
- 27.Ferrario CM, Strawn WB. Role of the renin–angiotensin–aldosterone system and proinflammatory mediators in cardiovascular disease. Am J Cardiol 2006; 98:121–128. [DOI] [PubMed] [Google Scholar]
- 28.Giannessi D, Del Ry S, Vitale RL. The role of endothelins and their receptors in heart failure. Pharmacol Res 2001; 43:111–126. [DOI] [PubMed] [Google Scholar]
- 29.Eldibany MM, Caprini JA. Hyperhomocysteinemia and thrombosis: an overview. Arch Pathol Lab Med 2007; 131 6:872–884. [DOI] [PubMed] [Google Scholar]
- 30.Hegele RA, Cao H. Single nucleotide polymorphisms of RXRA encoding retinoid X receptor alpha. J Hum Genet 2001; 46:423–425. [DOI] [PubMed] [Google Scholar]
- 31.Vasku V, Bienertova Vasku J, Pavkova Goldbergova M, Vasku A. Three retinoid X receptor gene polymorphisms in plaque psoriasis and psoriasis guttata. Dermatology 2007; 214:118–124. [DOI] [PubMed] [Google Scholar]
- 32.Neugebauer P, Goldbergova-Pavkova M, Kala P, et al. Nuclear receptors gene polymorphisms and risk of restenosis and clinical events following coronary stenting. Vnitr Lek 2009; 55:1135–1140. [PubMed] [Google Scholar]
- 33.Spinarova L, Spinar J, Vasku A, et al. Genetics of humoral and cytokine activation in heart failure and its importance for risk stratification of patients. Exp Mol Pathol 2008; 84:251–255. [DOI] [PubMed] [Google Scholar]
- 34.Vasků A, Soucek M, Goldbergová M, Vácha J. Office blood pressure, heart rate and A(−596)G interleukin-6 gene polymorphism in apparently healthy Czech middle-aged population. Physiol Res 2003; 52:291–297. [PubMed] [Google Scholar]
- 35.Vasku JA, Vasku A, Goldbergova M, Vasku V. Heterozygote AG variant of −596 A/G IL-6 gene polymorphism is a marker for cutaneous T-cell lymphoma (CTCL). Clin Immunol 2004; 113:256–260. [DOI] [PubMed] [Google Scholar]
- 36.Vasku A, Goldbergova M, Izakovicova Holla L, et al. A haplotype constituted of four MMP-2 promoter polymorphisms (−1575G/A, −1306C/T, −790T/G and −735C/T) is associated with coronary triple-vessel disease. Matrix Biol 2004; 22:585–591. [DOI] [PubMed] [Google Scholar]
- 37.Goldbergova M, Spinarova L, Spinar J, et al. Association of two angiotensinogen gene polymorphisms, M235T and G(−6)A, with chronic heart failure. Int J Cardiol 2003; 89:267–272. [DOI] [PubMed] [Google Scholar]
- 38.Panovsky R, Vasku A, Meluzin J, et al. Association of polymorphisms of zinc metalloproteinases with clinical response to stem cell therapy. Herz 2010; 35:309–316. [DOI] [PubMed] [Google Scholar]
- 39.Benes P, Kankova K, Muzik J, et al. Methylenetetrahydrofolate reductase polymorphism, type II diabetes mellitus, coronary artery disease, and essential hypertension in the Czech population. Mol Genet Metab 2001; 73:188–195. [DOI] [PubMed] [Google Scholar]
- 40.Calero O, Hortigüela R, Bullido MJ, Calero M. Apolipoprotein E genotyping method by real time PCR, a fast and cost-effective alternative to the TaqMan and FRET assays. J Neurosci Methods 2009; 183:238–240. [DOI] [PubMed] [Google Scholar]
- 41.Machal J, Vasku A, Hlinomaz O, et al. Apolipoprotein E polymorphism is associated with both number of diseased vessels and extent of coronary artery disease in Czech patients with CAD. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2012; 156:151–158. [DOI] [PubMed] [Google Scholar]
- 42.Gaunt TR, Rodriguez S, Zapata C, Day IN. MIDAS: software for analysis and visualisation of interallelic disequilibrium between multiallelic markers. BMC Bioinformatics 2006; 7:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yusuf S, Hawken S, Ounpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet 2004; 364:937–952. [DOI] [PubMed] [Google Scholar]
- 44.Najjar SS, Scuteri A, Lakatta EG. Arterial aging: is it an immutable cardiovascular risk factor? Hypertension 2005; 46:454–462. [DOI] [PubMed] [Google Scholar]
- 45.Rheaume C, Leblanc ME, Poirier P. Adiposity assessment: explaining the association between obesity, hypertension and stroke. Expert Rev Cardiovasc Ther 2011; 9:1557–1564. [DOI] [PubMed] [Google Scholar]
- 46.Basta G, Schmidt AM, De Caterina R. Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc Res 2004; 63:582–592. [DOI] [PubMed] [Google Scholar]
- 47.Younis N, Sharma R, Soran H, et al. Glycation as an atherogenic modification of LDL. Curr Opin Lipidol 2008; 19:378–384. [DOI] [PubMed] [Google Scholar]
- 48.Shuvaev VV, Fujii J, Kawasaki Y, et al. Glycation of apolipoprotein E impairs its binding to heparin: identification of the major glycation site. Biochim Biophys Acta 1999; 1454:296–308. [DOI] [PubMed] [Google Scholar]
- 49.Nobecourt E, Davies MJ, Brown BE, et al. The impact of glycation on apolipoprotein A-I structure and its ability to activate lecithin:cholesterol acyltransferase. Diabetologia 2007; 50:643–653. [DOI] [PubMed] [Google Scholar]
- 50.Hamer M, Stamatakis E. Overweight and obese cardiac patients have better prognosis despite reporting worse perceived health and more conventional risk factors. Prev Med 2013; 57:12–16. [DOI] [PubMed] [Google Scholar]
- 51.Romero-Corral A, Montori VM, Somers VK, et al. Association of bodyweight with total mortality and with cardiovascular events in coronary artery disease: a systematic review of cohort studies. Lancet 2006; 368:666–678. [DOI] [PubMed] [Google Scholar]
- 52.Schooling CM, Cowling BJ, Jones HE. Selection bias in cohorts of cases. Prev Med 2013; 57:247–248. [DOI] [PubMed] [Google Scholar]
- 53.Fan Y, Ye J, Shen F, et al. Interleukin-6 stimulates circulating blood-derived endothelial progenitor cell angiogenesis in vitro. J Cereb Blood Flow Metab 2008; 28:90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schieffer B, Selle T, Hilfiker A, et al. Impact of interleukin-6 on plaque development and morphology in experimental atherosclerosis. Circulation 2004; 110:3493–3500. [DOI] [PubMed] [Google Scholar]
- 55.Xing Z, Gauldie J, Cox G, et al. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest 1998; 101:311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huber SA, Sakkinen P, Conze D, et al. Interleukin-6 exacerbates early atherosclerosis in mice. Arterioscler Thromb Vasc Biol 1999; 19:2364–2367. [DOI] [PubMed] [Google Scholar]
- 57.Yudkin JS, Kumari M, Humphries SE, Mohamed-Ali V. Inflammation, obesity, stress and coronary heart disease: is interleukin-6 the link? Atherosclerosis 2000; 148:209–214. [DOI] [PubMed] [Google Scholar]
- 58.Hurst SM, Wilkinson TS, McLoughlin RM, et al. IL-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity 2001; 14:705–714. [DOI] [PubMed] [Google Scholar]
- 59.Ohtsuka T, Hamada M, Inoue K, et al. Relation of circulating interleukin-6 to left ventricular remodeling in patients with reperfused anterior myocardial infarction. Clin Cardiol 2004; 27:417–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Danesh J, Kaptoge S, Mann AG, et al. Long-term interleukin-6 levels and subsequent risk of coronary heart disease: two new prospective studies and a systematic review. PLoS Med 2008; 5:e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gotsman I, Stabholz A, Planer D, et al. Serum cytokine tumor necrosis factor-alpha and interleukin-6 associated with the severity of coronary artery disease: indicators of an active inflammatory burden? Isr Med Assoc J 2008; 10:494–498. [PubMed] [Google Scholar]
- 62.Su D, Li Z, Li X, et al. Association between serum interleukin-6 concentration and mortality in patients with coronary artery disease. Mediators Inflamm 2013; 2013:726178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haugen E, Gan LM, Isic A, et al. Increased interleukin-6 but not tumour necrosis factor-alpha predicts mortality in the population of elderly heart failure patients. Exp Clin Cardiol 2008; 13:19–24. [PMC free article] [PubMed] [Google Scholar]
- 64.Fisman EZ, Tenenbaum A. The ubiquitous interleukin-6: a time for reappraisal. Cardiovasc Diabetol 2010; 9:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schuett H, Luchtefeld M, Grothusen C, et al. How much is too much? Interleukin-6 and its signalling in atherosclerosis. Thromb Haemost 2009; 102:215–222. [DOI] [PubMed] [Google Scholar]
- 66.Schuett H, Oestreich R, Waetzig GH, et al. Transsignaling of interleukin-6 crucially contributes to atherosclerosis in mice. Arterioscler Thromb Vasc Biol 2012; 32:281–290. [DOI] [PubMed] [Google Scholar]
- 67.Ray A, Tatter SB, May LT, Sehgal PB. Activation of the human “beta 2-interferon/hepatocyte-stimulating factor/interleukin 6” promoter by cytokines, viruses, and second messenger agonists. Proc Natl Acad Sci U S A 1988; 85:6701–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fishman D, Faulds G, Jeffery R, et al. The effect of novel polymorphisms in the interleukin-6 (IL-6) gene on IL-6 transcription and plasma IL-6 levels, and an association with systemic-onset juvenile chronic arthritis. J Clin Invest 1998; 102:1369–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen Y, Pawlikowska L, Yao JS, et al. Interleukin-6 involvement in brain arteriovenous malformations. Ann Neurol 2006; 59:72–80. [DOI] [PubMed] [Google Scholar]
- 70.Brull DJ, Montgomery HE, Sanders J, et al. Interleukin-6 gene −174 G>C and −572 G>C promoter polymorphisms are strong predictors of plasma interleukin-6 levels after coronary artery bypass surgery. Arterioscler Thromb Vasc Biol 2001; 21:1458–1463. [DOI] [PubMed] [Google Scholar]
- 71.Smith AJ, D’Aiuto F, Palmen J, et al. Association of serum interleukin-6 concentration with a functional IL6-6331T>C polymorphism. Clin Chem 2008; 54:841–850. [DOI] [PubMed] [Google Scholar]
- 72.Terry CF, Loukaci V, Green FR. Cooperative influence of genetic polymorphisms on interleukin 6 transcriptional regulation. J Biol Chem 2000; 275:18138–18144. [DOI] [PubMed] [Google Scholar]
- 73.Muller-Steinhardt M, Ebel B, Hartel C. The impact of interleukin-6 promoter −597/−572/−174 genotype on interleukin-6 production after lipopolysaccharide stimulation. Clin Exp Immunol 2007; 147:339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Peluso I, Morabito G, Urban L, et al. Oxidative stress in atherosclerosis development: the central role of LDL and oxidative burst. Endocr Metab Immune Disord Drug Targets 2012; 12:351–360. [DOI] [PubMed] [Google Scholar]
- 75.Yin YW, Li JC, Zhang M, et al. Influence of interleukin-6 gene −174G>C polymorphism on development of atherosclerosis: a meta-analysis of 50 studies involving 33,514 subjects. Gene 2013; 529:94–103. [DOI] [PubMed] [Google Scholar]
- 76.Antonicelli R, Olivieri F, Bonafe M, et al. The interleukin-6 −174 G>C promoter polymorphism is associated with a higher risk of death after an acute coronary syndrome in male elderly patients. Int J Cardiol 2005; 103:266–271. [DOI] [PubMed] [Google Scholar]
- 77.Aker S, Bantis C, Reis P, et al. Influence of interleukin-6 G-174C gene polymorphism on coronary artery disease, cardiovascular complications and mortality in dialysis patients. Nephrol Dial Transplant 2009; 24:2847–2851. [DOI] [PubMed] [Google Scholar]
- 78.Niu W, Qi Y. Matrix metalloproteinase family gene polymorphisms and risk for coronary artery disease: systematic review and meta-analysis. Heart 2012; 98:1483–1491. [DOI] [PubMed] [Google Scholar]
- 79.Gluba A, Banach M, Mikhailidis DP, Rysz J. Genetic determinants of cardiovascular disease: the renin–angiotensin–aldosterone system, paraoxonases, endothelin-1, nitric oxide synthase and adrenergic receptors. In Vivo 2009; 23:797–812. [PubMed] [Google Scholar]
- 80.Lewis SJ, Ebrahim S, Davey Smith G. Meta-analysis of MTHFR 677C->T polymorphism and coronary heart disease: does totality of evidence support causal role for homocysteine and preventive potential of folate? BMJ 2005; 331:1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sarecka-Hujar B, Zak I, Krauze J. The TT genotype of the MTHFR 677C>T polymorphism increases susceptibility to premature coronary artery disease in interaction with some of the traditional risk factors. Acta Medica (Hradec Kralove) 2012; 55:172–179. [DOI] [PubMed] [Google Scholar]
- 82.Goldbergova MP, Spinarova L, Spinar J, et al. RXRA introne polymorphism and ABO blood groups in chronic heart failure. Central Eur J Biol 2010; 5:749–756. [Google Scholar]
- 83.Wu Z, Lou Y, Jin W, et al. The C161T polymorphism in the peroxisome proliferator-activated receptor gamma gene (PPARgamma) is associated with risk of coronary artery disease: a meta-analysis. Mol Biol Rep 2013; 40:3101–3112. [DOI] [PubMed] [Google Scholar]
- 84.Bennet AM, Di Angelantonio E, Ye Z, et al. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA 2007; 298:1300–1311. [DOI] [PubMed] [Google Scholar]
- 85.Keso T, Perola M, Laippala P, et al. Polymorphisms within the tumor necrosis factor locus and prevalence of coronary artery disease in middle-aged men. Atherosclerosis 2001; 154:691–697. [DOI] [PubMed] [Google Scholar]
- 86.Rodriguez-Rodriguez L, Gonzalez-Juanatey C, Palomino-Morales R, et al. TNFA −308 (rs1800629) polymorphism is associated with a higher risk of cardiovascular disease in patients with rheumatoid arthritis. Atherosclerosis 2011; 216:125–130. [DOI] [PubMed] [Google Scholar]
- 87.Szabo GV, Acsady G. Tumornecrosis-factor-alpha 308 GA polymorphism in atherosclerotic patients. Pathol Oncol Res 2011; 17:853–857. [DOI] [PubMed] [Google Scholar]
- 88.Li W, Xu J, Wang X, et al. Lack of association between lymphotoxin-alpha, galectin-2 polymorphisms and coronary artery disease: a meta-analysis. Atherosclerosis 2010; 208:433–436. [DOI] [PubMed] [Google Scholar]