

Abstract
Despite documented laboratory and clinical benefits of hydroxyurea for children with sickle cell anemia (SCA), the drug's long-term safety and efficacy remains poorly defined. The HUSOFT trial and extension study examined feasibility, toxicity, and hematological efficacy of hydroxyurea in infants with SCA.
This report describes HUSOFT participants who have continued hydroxyurea therapy for 15 years. With IRB approval, medical records were reviewed for clinical, laboratory, and growth parameters.
Twenty-eight infants enrolled in the original 2-year HUSOFT study received open-label liquid hydroxyurea at 20 mg/kg/day; 17 completed the extension study with dose escalation to 30 mg/kg/day. Eight of these 17 (6 girls and 2 boys, all HbSS) have continued on daily hydroxyurea for at least 15 years (median age at last follow-up 17.6 years) without interruption. All hematologic indices (Hb concentration, mean corpuscular volume (MCV), fetal hemoglobin) showed sustained effect after 15 years. The median maximum tolerated dose of hydroxyurea has decreased from 30 to 26 mg/kg/day (range 19.5–31.2); neutropenia [absolute neutrophil count (ANC) < 1.0 × 109/L] prompting temporary drug discontinuation occurred a total of 10 times in 4 subjects and there was no severe neutropenia (ANC < 0.5 × 109/L). Growth rates over 15 years continued at the 50th percentile for both height and weight, and puberty occurred without delay (age range 10–14 years). There were 5.1 vaso-occlusive events (pain and acute chest syndrome)/100 patient years, 7.3 packed red blood cell transfusions/100 patient years. No malignancies, strokes, or deaths occurred. At last follow up, all subjects were at appropriate grade level (10–12 grade) with no history of repeated grades.
A cohort of young teenagers with SCA who initiated treatment in infancy have had sustained and continued hematological benefits for a decade and a half. Growth and sexual development are normal and comparable to the general pediatric population. Continuous hydroxyurea therapy since infancy appears safe and efficacious in SCA.
INTRODUCTION
Hydroxyurea has been utilized for more than 20 years to prevent complications of sickle cell anemia (SCA). Due to its ability to increase fetal hemoglobin (HbF) production and reduce white blood cell count (WBC) and platelet counts, among other effects, hydroxyurea can ameliorate the disease phenotype.1–3 Among adults, the long-term use of hydroxyurea and its impact on mortality were examined in participants of the multicenter study of hydroxyurea in SCA (MSH) as well as in a Greek cohort who participated in the Laikon study of hydroxyurea in sickle cell syndromes (LaSHS).4,5 Both studies had prolonged follow-up (approximately 17 years) and both showed elevated HbF, reduced acute events, and decreased mortality with hydroxyurea utilization. However, the drug's long-term efficacy and toxicity have not been described in a pediatric cohort treated with hydroxyurea from a very young age.
The hydroxyurea safety and organ toxicity (HUSOFT) study was a phase I/II clinical trial in a cohort of 28 infants with SCA, not selected for severity, who started treatment at 6 to 24 months of age. HUSOFT demonstrated that administration of liquid hydroxyurea at 20 mg/kg/day for 2 years was feasible, well tolerated, and associated with expected hematologic effects and possibly improved splenic function.6 Twenty-one of these children then entered a follow-up study, receiving hydroxyurea dose escalation to maximum tolerated dose (MTD) or 30 mg/kg/day (whichever is highest). Eleven of these 21 children completed an additional 4 years of follow-up, and had sustained hematologic effects, fewer acute vaso-occlusive events and less splenic dysfunction compared with untreated historical controls.7 Although children treated with hydroxyurea for up to 7 years have been described,8,9 the present study provides data on 8 children from the original HUSOFT trial, who were treated with hydroxyurea therapy for a minimum of 15 years at MTD starting at a very young age. We have examined the risks and benefits of long-term continuous hydroxyurea treatment in this cohort.
METHODS
Patient Selection and Evaluations
In the HUSOFT extension study cohort, 11 subjects who started hydroxyurea in infancy completed 6 years of therapy.7 Since that report, three subjects have discontinued hydroxyurea: 1 due to parental request, 1 due to the patient's decision to interrupt therapy, and 1 due to loss of follow-up (Figure 1). The data for the 8 patients who have continuously been treated with hydroxyurea for ≥15 years without interruption are presented. The clinical and laboratory data of those original HUSOFT subjects who received therapy for longer than 6 years were collected under the longitudinal observational hydroxyurea study of long-term effects (HUSTLE, NCT00305175), which prospectively monitors the function of the spleen, brain, and kidneys at 3-year intervals, in addition to growth and development, during hydroxyurea therapy. These subjects were followed clinically with standard-of-care visits every 1 or 2 months at St. Jude Children's Research Hospital (St. Jude) or Duke University, but all research evaluations including organ function were conducted at St. Jude.
FIGURE 1.
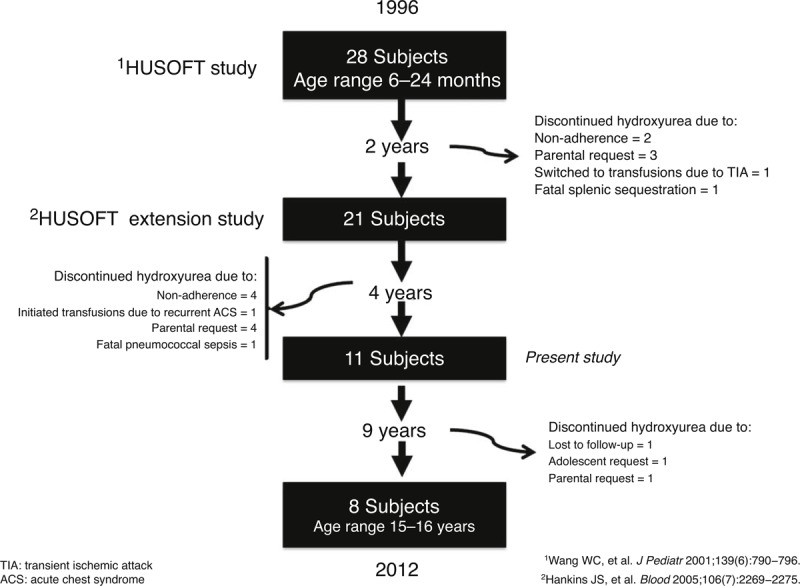
Reasons for premature discontinuation of hydroxyurea therapy.
Complete blood counts were obtained at every clinic visit and chemistry panel and HbF measurements (using high performance liquid chromatography) were performed every 2 months. Post-pubertal females were offered contraception and/or received monthly pregnancy tests. Adherence was not formally measured, but was estimated by calculation of the medication possession ratio (MPR). This was determined by dividing the amount of drug dispensed (in days of treatment covered) by the interval period between refills (in days), providing a measure of the proportion of days of medication availability. An MPR >80% has been considered a surrogate for good adherence with hydroxyurea therapy.10 Pill counts were not consistently available for these subjects.
Spleen function was assessed by 99mTc-liver/spleen scan, and uptake was graded as normal, decreased, or absent. The presence or absence of silent cerebral infarcts (SCI) and cerebral vasculopathy were assessed by brain magnetic resonance imaging (MRI) and angiography (MRA). Transcranial Doppler ultrasonography (TCD) was performed to monitor velocity in the major cerebral arteries. Renal function was evaluated through quantification of urine protein and measurement of glomerular filtration rate (GFR) by 99mTc-DTPA plasma clearance. Data regarding sickle cell-related clinical events, including pain, acute chest syndrome (ACS), stroke, and blood transfusions, and hematologic toxicity were captured at routine visits by review of emergency department, hospital, and clinic records.
The present study was approved by the St. Jude Institutional Review Board IRB in accordance with the current version of the Helsinki Declaration. All HUSTLE participants had informed consent signed by their parents or legal guardians. Assent was obtained from all participants older than 7 years of age.
Statistical Considerations
The mean and standard deviation of laboratory parameters, including Hb concentration, mean corpuscular volume (MCV), Hb F, WBC, absolute neutrophil count (ANC), reticulocyte count, platelet count, and lactate dehydrogenase (LDH), were calculated for each of the 15 years of hydroxyurea therapy. Weight and height were plotted using standard pediatric growth curves.
RESULTS
Patient Characteristics
As of December 31, 2012, 8 of the original HUSOFT subjects (all HbSS, 6 females and 2 males, median age 17.1 years, range 16.4–17.8 years) had received hydroxyurea continuously for a median of 15.9 years (range, 15.5–16.0 years). After the initial 2 years of hydroxyurea therapy, their dose was escalated to MTD and dose adjustments for weight gain were performed. Their median dose after 15 years was 26.6 mg/kg/day (range 18.0–33.1 mg/kg/day), a slight reduction from the initial dose escalation to 30 mg/kg/day.6
Hematologic Efficacy and Hydroxyurea Adherence
After 15 years of hydroxyurea therapy, hematologic indices, including mean annual values for Hb, MCV, HbF, reticulocyte count, WBC, and platelet counts showed sustained long-term hydroxyurea effect (Table 1). HbF levels remained above the expected values for untreated individuals with SCA of the same age (Figure 2). Monthly MPR was available for the 5 subjects treated at St. Jude. Their median MPR between the 6th and 15th year of treatment was 90% (range 88.2–97.4%).
TABLE 1.
Annual Means for Laboratory Indices and Hydroxyurea Dose by Year of Treatment Among 8 Children With SCA Treated ≥15 Years
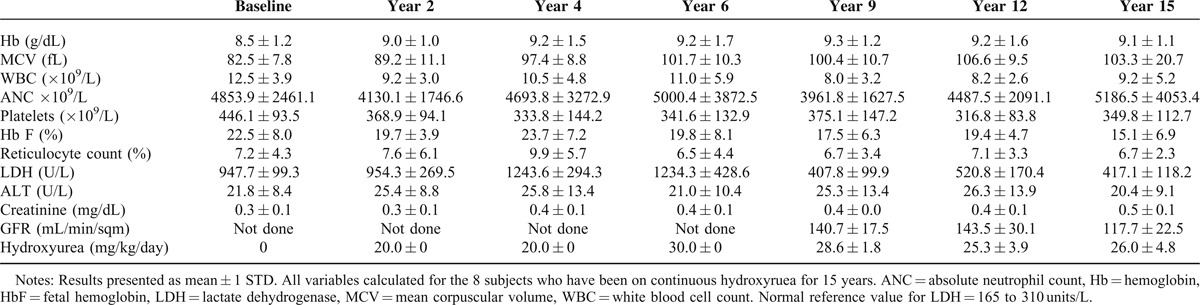
FIGURE 2.
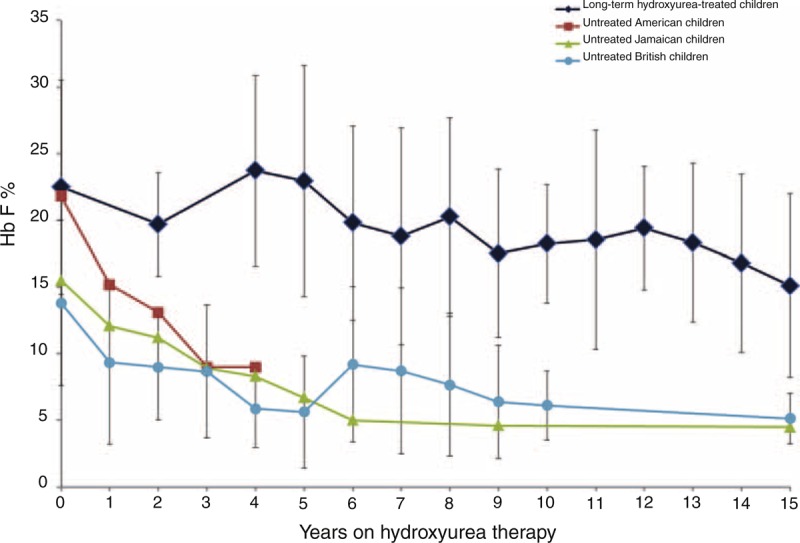
Fetal hemoglobin (HbF) levels during extended hydroxyurea therapy. Children with SCA treated with hydroxyurea for a minimum of 15 years have sustained HbF levels in comparison with untreated children. Legend: diamonds represent 8 children with SCA treated with hydroxyurea for a minimum of 15 years, squares represent untreated American children,18 triangles untreated Jamaican children,19 and circles untreated British children.20 HbF values depicted are mean (±1 STD) for the populations.
Hematologic Toxicity
Toxicity from hydroxyurea was rare. Neutropenia ANC) < 1.0 × 109/L prompting temporary drug discontinuation occurred 10 times in 4 subjects over a median of 15.9 years. No severe neutropenia (ANC < 0.5 × 109/L) was recorded. Two episodes of thrombocytopenia (in 2 subjects) with platelet counts below 80 × 109/L, and one instance of parvovirus B19-induded reticulocytopenia with worsened anemia (absolute reticulocyte count < 80 × 109/L with Hb concentration < 7 g/dL) occurred. In all occurrences of presumed hematologic toxicity, hydroxyurea treatment was held for 1 week, and blood counts completely recovered. Treatment was restarted at the same dose or at a dose reduced by 2.5 mg/kg if toxicity had been recurrent. There was no evidence of renal or hepatic toxicity (Table 1).
Growth and Development
In the 9 years since the last report, HUSOFT subjects have, on average, remained at the 50th percentile for both height and weight. They started pubertal development between 10 and 14 years of age (median of 12 years), and in which is the expected age in the general pediatric population. All subjects were attending school at an appropriate grade level (grades 10, 11, or 12) with no history of repeated grades.
Organ Function
Two subjects underwent splenectomy during the first 6 years of hydroxyurea therapy. Of the remaining 6, 1 (17%) had normal splenic uptake on Tc99 LSscan, and the other 5 had absent splenic function. None of the subjects had proteinuria (>300 mcg albumin/g creatinine), but microalbuminuria was present in all; the median protein/creatinine ratio was 120 mcg albumin/g creatinine (range, 53–145). The mean GFR was normal at 116.0 ± 20.6 mL/min/m2, albeit decreased from Years 9 and 12 (Table 1). Hb oxygen saturation was normal (median 100%, range 99–100%).
MRI of the brain revealed 2 children with SCI. The first was a male with bilateral frontal lesions diagnosed at 17 months of age (at HUSOFT study entry) that have remained unchanged for the past 15 years. The second was a female who developed a small focus of increased MRI T2-weighted signal in the right periatrial white matter at 7 years of age7 (5 years after starting hydroxyurea); this lesion did not change in the subsequent 9 years. No subjects had cerebral artery stenosis on brain MRA. TCD velocities were in the conditional range during hydroxyurea therapy in 2 (25%) subjects in the HUSOFT group, but later normalized; none had abnormal TCD velocities.
Acute Clinical Events
Life-time ACS and vaso-occlusive pain events were 5.1 events/100 patient-years and blood transfusion utilization was 7.3 packed red blood cell transfusions/100 patient/years. There were no malignancies, strokes, or deaths among these subjects.
DISCUSSION
Hydroxyurea therapy is the only FDA-approved “anti-sickling” medication for adults with SCA, and the only one, that is, commercially available (with off-label indication) for children with SCA. In the most recent National Heart Lung and Blood Institute (NHLBI) evidence-based guidelines, hydroxyurea is recommended for all children ≥9 months of age with SCA, regardless of disease severity, to reduce disease-related complications.11
Hydroxyurea has been shown to reduce vaso-occlusive events (pain and ACS) in phase III randomized clinical trials in both adults and children,2,3 but its long-term benefits in children are incompletely characterized. Our data indicate that hydroxyurea therapy, when started at a very young age (<2 years) and given for an extended period of time, appears safe and promotes the expected hematologic and clinical effects for more than 15 years. These subjects, who are now adolescents/young adults, have had normal growth and development, are in school at appropriate grade level, and continue to experience very few vaso-occlusive episodes and minimal blood utilization. This is the first cohort of children to be reported who have had hydroxyurea therapy continuously prescribed for this length of time.
Hematologic effects of hydroxyurea were sustained over the prolonged period of drug exposure, which was more than twice as long as previously reported.8 The median hydroxyurea doses lightly decreased from 30 to 26.5 mg/kg/day, perhaps reflecting the physiologic decrease in marrow cellularity that occurs with increasing age, and/or changes in body habitus, renal function, or muscle mass. There were no apparent cases of tolerance or refractoriness to hydroxyurea therapy, as hematologic effects continued even after 15 years of drug exposure among those with adequate drug adherence. This is consistent with the lack of evidence for pharmacologic tolerance or resistance discussed in a recent review.12
Organ dysfunction is an expected long-term complication of SCA. Although no conclusions can be drawn from the small cohort of HUSOFT patients, their current brain, kidney, and spleen evaluations suggest some degree of protection from dysfunction related to prolonged hydroxyurea exposure. Finally, growth and development were normal for those treated with long-term hydroxyurea. Pubertal onset occurred at the expected age, contrasting with the published literature that reports an average delay in pubertal development in SCA children of approximately 2 years.13
An increased cancer risk in individuals with SCA who utilize hydroxyurea has not been demonstrated in children or adults. Cross-sectional and prospective studies (involving chromosomal karyotype, illegitimate VDJ recombination events, cytotoxicity development, and genotoxicity measured by micronucleated reticulocyte formation) have failed to demonstrate increases in mutagenesis when compared with measurements made prior to initiation of therapy or with individuals who were receiving placebo.14–17 Although no relationships have been demonstrated to date, prospective monitoring of cancer risk, and hydroxyurea genotoxicity should continue.
Our study has many limitations, including a small sample size, no formal prospective treatment adherence assessment, and lack of data on those who interrupted treatment prior to 15 years. Despite our efforts, we were not able to capture the clinical and laboratory data of the HUSOFT participants who discontinued hydroxyurea prior to 15 years. Therefore, we cannot make comparisons of the incidence of events or laboratory findings between the original HUSOFT participants who did not interrupt treatment and those who did, somewhat limiting the interpretation of long-term efficacy and safety results. In addition, because these adolescents were followed at only 2 institutions, our results may not be generalizable. This study, however, offers the strengths of prospective monitoring of laboratory and organ function at fixed intervals in a unique cohort of children who have been treated with hydroxyurea from a very early age with consistent prolonged treatment.
In conclusion, our study showed that in a cohort of children not selected for disease severity and treated with hydroxyurea continuously since infancy for a minimum of 15 years, hematologic effects of the drug persist without major toxicity. Growth is normal, and pubertal development timely. Future research should focus on continued and long-term exposure to hydroxyurea, as well as collection of data on those who have intermittent use of this drug. As current generations of individuals treated with hydroxyurea enter puberty and mature into adulthood on this medication, its effects and toxicities, including effects on fertility (especially spermatogenesis), must be monitored. The BABYHUG follow-up observational studies (clinicaltrials.gov #NCT00890396 and #NCT01783990) will be of great value in describing the long-term efficacy and toxicity of hydroxyurea in a larger cohort of children who also have been treated since infancy, generally without interruption. Adult studies, such as the MSH and LaSHS, should continue to provide us with important information regarding the risks and benefits of hydroxyurea in older individuals with SCA.
Acknowledgments
The authors are indebted to Terri Davis for preparation of graphs and formatting of the manuscript, Patricia Bass, PharmD for MPR adherence data collection, Gail Fortner, RN, Lynn Wynn, PNP, Amy Kimble, FNP, and Nicole Mortier, PA, MHS for their support with clinical care of the HUSOFT and HUSTLE patients. Dr. Courtney Thornburg is currently at the University of California, San Diego.
Footnotes
Abbreviations: ACS = acute chest syndrome, ANC = neutrophil count, FDA = Federal Drug Administration, GFR = glomerular filtration rate, Hb = hemoglobin, HbF = fetal hemoglobin, HUSOFT = hydroxyurea safety and organ toxicity study, HUSTLE = hydroxyurea study of long-term effects study, LaSHS = Laikon study of hydroxyurea in sickle cell syndromes, LDH = lactate dehydrogenase, MCV = corpuscular volume, MRA = magnetic resonance angiography, MRI = magnetic resonance imaging, MPR = medication possession ratio, MSH = multicenter study of hydroxyurea in SCA, MTD = maximum tolerated dose, SCA = sickle cell anemia, TCD = transcranial doppler ultrasonography, WBC = white blood cell count.
The authors have no conflicts of interest to disclose.
The study was supported in part by ALSAC.
Author contributions: Jane S. Hankins, MD, MS: study concept, data collection and interpretation, and writing of the manuscript. Banu Aygun, MD: study concept, data interpretation, and editing of the manuscript. Kerri Nottage, MD, MPH: data interpretation, data collection, and editing of the manuscript. Courtney Thornburg, MD, MS: data collection, data interpretation, and editing of the manuscript. Matthew Smeltzer, PhD, MS: data analysis and editing of the manuscript. Russell E. Ware, MD, PhD: study concept and editing of the manuscript. Winfred C. Wang, MD: study concept, data interpretation, and editing of the manuscript.
REFERENCES
- 1.Platt OS, Orkin SH, Dover G, et al. Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia. J Clin Invest 1984; 74:652–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the multicenter study of hydroxyurea in sickle cell anemia. N Engl J Med 1995; 332:1317–1322. [DOI] [PubMed] [Google Scholar]
- 3.Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet 2011; 377:1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: a 17.5 year follow-up. Am J Hematol 2010; 85:403–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Voskaridou E, Christoulas D, Bilalis A, et al. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: results of a 17-year, single-center trial (LaSHS). Blood 2010; 115:2354–2363. [DOI] [PubMed] [Google Scholar]
- 6.Wang WC, Wynn LW, Rogers ZR, et al. A two-year pilot trial of hydroxyurea in very young children with sickle-cell anemia. J Pediatr 2001; 139:790–796. [DOI] [PubMed] [Google Scholar]
- 7.Hankins JS, Ware RE, Rogers ZR, et al. Long-term hydroxyurea therapy for infants with sickle cell anemia: the HUSOFT extension study. Blood 2005; 106:2269–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zimmerman SA, Schultz WH, Davis JS, et al. Sustained long-term hematologic efficacy of hydroxyurea at maximum tolerated dose in children with sickle cell disease. Blood 2004; 103:2039–2045. [DOI] [PubMed] [Google Scholar]
- 9.DeMontalembert M, Belloy M, Bernaudin F, et al. Three-year follow-up of hydroxyurea treatment in severely ill children with sickle cell disease. The French Study Group on Sickle Cell Disease. J Pediatr Hematol Oncol 1997; 19:313–318. [DOI] [PubMed] [Google Scholar]
- 10.Candrilli SD, O’Brien SH, Ware RE, et al. Hydroxyurea adherence and associated outcomes among Medicaid enrollees with sickle cell disease. Am J Hematol 2011; 86:273–277. [DOI] [PubMed] [Google Scholar]
- 11.Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014; 312:1033–1048. [DOI] [PubMed] [Google Scholar]
- 12.Ware RE. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood 2010; 115:5300–5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Platt OS, Rosenstock W, Espeland MA. Influence of sickle hemoglobinopathies on growth and development. N Engl J Med 1984; 311:7–12. [DOI] [PubMed] [Google Scholar]
- 14.McGann PT, Flanagan JM, Howard TA, et al. Genotoxicity associated with hydroxyurea exposure in infants with sickle cell anemia: results from the BABY-HUG phase III clinical trial. Pediatr Blood Cancer 2012; 59:254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGann PT, Howard TA, Flanagan JM, et al. Chromosome damage and repair in children with sickle cell anaemia and long-term hydroxycarbamide exposure. Br J Haematol 2011; 154:134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khayat AS, Antunes LM, Guimaraes AC, et al. Cytotoxic and genotoxic monitoring of sickle cell anaemia patients treated with hydroxyurea. Clin Exp Med 2006; 6:33–37. [DOI] [PubMed] [Google Scholar]
- 17.Maluf S, Pra D, Friedrisch JR, et al. Length of treatment and dose as determinants of mutagenicity in sickle cell disease patients treated with hydroxyurea. Environ Toxicol Pharmacol 2009; 27:26–29. [DOI] [PubMed] [Google Scholar]
- 18.Brown AK, Sleeper LA, Miller ST, et al. Reference values and hematologic changes from birth to 5 years in patients with sickle cell disease. Cooperative Study of Sickle Cell Disease. Arch Pediatr Adolesc Med 1994; 148:796–804. [DOI] [PubMed] [Google Scholar]
- 19.Hayes RJ, Beckford M, Grandison Y, et al. The haematology of steady state homozygous sickle cell disease: frequency distributions, variation with age and sex, longitudinal observations. Br J Haematol 1985; 59:369–382. [DOI] [PubMed] [Google Scholar]
- 20.Davis LR. Changing blood picture in sickle-cell anaemia from shortly after birth to adolescence. J Clin Pathol 1976; 29:898–901. [DOI] [PMC free article] [PubMed] [Google Scholar]