

Supplemental Digital Content is available in the text
Abstract
X-ray repair complementing defective repair in Chinese hamster cells 2 (XRCC2) and poly(ADP-ribose) polymerase 1 (PARP1) both play important roles in homologous recombination DNA repair. According to the theory of synthetic lethality, XRCC2-deficient cells are more sensitive to PARP1 inhibitors compared to XRCC2-expressing cells. We investigated XRCC2 expression and function in colorectal cancer (CRC), and the characteristics of sensitivity to PARP1 inhibitor in CRC cells with different XRCC2 levels.
We enrolled 153 patients with CRC who had undergone surgery in this study. XRCC2 expression was assessed using immunohistochemistry. Stable CRC SW480 cell lines with low or high XRCC2 expression were constructed. Following treatment with the PARP1 inhibitor olaparib, the viability of cells with different XRCC2 levels was determined; cell cycle distribution and apoptosis were analyzed using flow cytometry. B-cell lymphoma-2 (Bcl-2) protein expression was measured by Western blotting.
The positive rates of XRCC2 in primary CRC tissue were significantly higher than that in the matched adjacent noncancerous tissue, and XRCC2 expression status in primary CRC was related to tumor site, Dukes’ stage, and tumor-nodes-metastasis (TNM) stage. XRCC2 overexpression inhibited CRC cell apoptosis and promoted proliferation by enriching cells in the G0/G1 phase. Moreover, olaparib suppressed proliferation, and olaparib sensitivity in CRC cells with high XRCC2 expression was greater.
High XRCC2 expression promotes CRC cell proliferation and enriches cells in the G0/G1 phase but inhibits apoptosis. High XRCC2 expression cells are more sensitive to olaparib, which inhibits their viability.
INTRODUCTION
Colorectal cancer (CRC) is one of the most common malignant tumors in clinical practice. It is the second most frequent cancer after lung cancer in the United States and Europe that leads to death.1 In China, the incidence of CRC has increased annually and will continue to rise in the next few years.2 Currently, about 1.25 million patients are diagnosed with CRC, and more than 600,000 patients will die from this disease every year worldwide.3 Progress has been made in the targeted therapies of CRC; however, better targeted drugs are required because the current drugs do not produce satisfactory effects.
X-ray repair complementing defective repair in Chinese hamster cells 2 (XRCC2) is located in 7q36.1 and encodes a member of the Rad51-related protein family that participates in homologous recombination (HR) to maintain chromosome stability and repair DNA damage. It is believed that XRCC2 repairs chromosomal fragmentation, translocations, and deletions; furthermore, this gene is involved in the HR repair (HRR) pathway of DNA double-stranded breaks (DSBs).4–6 Numerous studies have reported an association between XRCC2 single-nucleotide polymorphisms (SNPs) and cancer incidence risk; the most common mutation studied is rs3218536.7–9
The concept of synthetic lethality postulates that the functional inhibition of two proteins leads to cell death, but blockade of either protein alone does not.10 One of the greatest threats to genomic integrity is DSBs, which trigger repair proteins involved in the HRR and non-homologous end joining (NHEJ) pathways. Poly(ADP-ribose) polymerase (PARP) plays a key role in HRR and NHEJ. According to the synthetic lethality theory, HRR-defective tumors are highly sensitive to PARP1 inhibitors.11–13 PARP1 inhibitors mainly block the catalytic activity of PARP1 and increase the levels of persisting single-strand breaks that lead to DNA DSBs upon replication.12,14 In this study, we investigated the function of XRCC2 in CRC and the effect of PARP1 inhibitors in XRCC2-expressing or XRCC2-deficient CRC SW480 cells.
MATERIALS AND METHODS
Patients
The 153 malignant CRC tissues and 31 matching adjacent non-cancerous tissues used in this study were obtained from patients who underwent surgery (without preoperative chemotherapy and/or radiotherapy) at the First Affiliated Hospital of Sun Yat-Sen University from May 2012 to August 2013. The Sun Yat-Sen University and First Affiliated Hospital Institutional Ethical Board approved the use of clinical materials for research purposes in this study, and we obtained written informed consent from all patients.
Immunohistochemical Staining
Sections were obtained from the Pathology Department of the First Affiliated Hospital of Sun Yat-Sen University, and incubated with polyclonal primary antibody against XRCC2 (1:100; Abcam, Cambridge, UK) at 4°C overnight. After incubation with horseradish peroxidase-conjugated sheep anti-rabbit secondary antibody (Beyotime; Guangzhou, China) and diaminobenzidine, the slides were counterstained with Mayer's hematoxylin. A known positive tissue sample (primary CRC tissue slide) was used as the positive control; phosphate-buffered saline (PBS) buffer was used in place of the primary antibodies in negative control staining.
Cell Lines
Normal colonic mucosal epithelial cells were isolated and purified from the adjacent noncancerous tissues of patients who had undergone surgery at our hospital. The CRC cell lines SW620, SW480, LoVo, HT29, and LS174T were obtained from American Type Culture Collection (Manassas, VA, USA). The cells were cultured in RPMI 1640 medium (Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (HyClone, Logan, UT, USA) and 1% penicillin/streptomycin. All cells were maintained in a humidified atmosphere at 37°C containing 5% CO2.
Vectors and Retroviral Infection
We generated pBABE/XRCC2-overexpressing human XRCC2 by subcloning the PCR-amplified human XRCC2 coding sequence into a pBABE-puro vector. To silence endogenous XRCC2, two RNA interference (RNAi) oligonucleotides were cloned into pSuper-retro-puro vectors to generate the respective pSuper-retro-XRCC2-RNAi. Retroviral production and infection were performed as described previously.15 Stable cell lines expressing XRCC2 (SW480-XRCC2 or pBABE-puro-XRCC2; control, SW480-Vector or pBABE-puro, respectively) or XRCC2 RNAi (SW480-XRCC2/RNAi or pSuper-retro-puro-siXRCC2; control, SW480-Scramble or pSuper-retro-puro, respectively) were selected over 10 days using 0.5 μg/mL puromycin 48 hours after infection. SW480 cell lysates prepared from the pooled population of cells in sample buffer were fractionated using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) to detect XRCC2 protein levels.
Cell Proliferation Assay
SW480 cell proliferation was measured using the Cell Counting Kit-8 (CCK-8) cell proliferation kit (Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer's instructions. Cells were seeded in 96-well plates at 1 × 103 cells/well with 100 μL complete medium and cultured at 37°C, and then 10 μL CCK-8 solution was added to each well after 24, 72, and 96 hours. Plates were incubated at 37°C for 2 hours, and then the absorbance at 450 nm was measured with a microplate reader (Bio-Rad, La Jolla, CA, USA). The PARP1 inhibitor olaparib (AZD2281) was obtained from Selleckchem (Houston, TX, USA). All experiments were performed in triplicate, and three independent repeat experiments were performed.
Flow Cytometry Analysis of Cell Cycle and Apoptosis
The cell cycle was analyzed by flow cytometry. Briefly, cultured cells were trypsinized into single-cell suspensions and fixed with 70% ethanol for 30 min on ice. RNA was degraded by incubation with 20 mg/mL RNase (Sigma–Aldrich, St. Louis, MO, USA) for 1 hour at 37°C. DNA was labeled with 20 mg/mL propidium iodide (PI, Sigma–Aldrich); DNA content was assessed using a FACSCalibur unit (Becton Dickinson, Franklin Lakes, NJ, USA) equipped with ModFit LT v2.0 software. To analyze apoptosis, cultured cells were harvested by trypsinization and washed with PBS. Cells (1 × 106) from each sample were processed for annexin V–fluorescein isothiocyanate (FITC)/PI apoptosis detection according to the manufacturer's instructions (Becton Dickinson).
RNA Extraction, Reverse Transcription, and Real-Time Quantitative PCR
Total RNA was extracted from the cultured cells using TRIzol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Real-time quantitative reverse transcription–polymerase chain reaction (PCR) was performed using SYBR Green I (Invitrogen) with an ABI PRISM 7500 system (Applied Biosystems, Foster City, CA, USA). Expression data were normalized to the geometric mean of the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) to control expression level variability. The following primers were used: (GAPDH) forward: 5′-GACTCATGACCACAGTCCATGC-3′, reverse: 5′-AGAGGCAGGGATGATGTTCTG-3′; (XRCC2) forward: 5′-TCACCTGTGCATGGTGATATT-3′, reverse: 5′-TTCCAGGCCACCTTCTGATT-3′.
Western Blotting
Proteins from cell lysates were prepared, separated on SDS–PAGE, and transferred to polyvinyl difluoride (PVDF) membranes according to the manufacturers’ instructions. Anti-human XRCC2 mouse monoclonal antibody (1:1500; Abcam) and anti-β-actin mouse monoclonal antibody (1:1000; Sigma–Aldrich) were used as primary antibodies for detecting specific proteins. Goat anti-mouse antibody (1:2000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used as the secondary antibody. Signal amplification and detection were achieved by exposing the membrane to electrochemiluminescence reagent (GE Healthcare, Buckinghamshire, UK), followed by visualization using the Storm imaging system (Amersham Biosciences, Piscataway, NJ, USA).
Statistical Analysis
The statistical software package SPSS (version 17.0; IBM, New York, NY, USA) was employed for all analyses. The association between XRCC2 expression and clinicopathological parameters was analyzed with the Chi-square test. The Student's t test was used to evaluate significant differences between two groups of data in all relevant experiments. All results are presented as means ± standard deviation; the level of significance was set at P < 0.05.
RESULTS
XRCC2 Was Upregulated in CRC Clinical Tissues and Cell Lines
Figure 1 depicts the XRCC2 staining results in primary CRC tissue and the matched adjacent noncancerous tissue. Positive XRCC2 staining was detected in 107 of 153 (69.9%) primary CRC tissues. In marked contrast, the positive rate of XRCC2 staining in the matched adjacent noncancerous tissues was only 16.1% (5/31 samples, P = 0.000 compared to primary CRC tissue). Furthermore, Western blotting and real-time PCR showed that XRCC2 was markedly upregulated in all five CRC cell lines compared with the normal colonic mucosal epithelial cells (Figure 2A and B). These results suggest that XRCC2 is significantly upregulated in CRC (Table 1).
FIGURE 1.
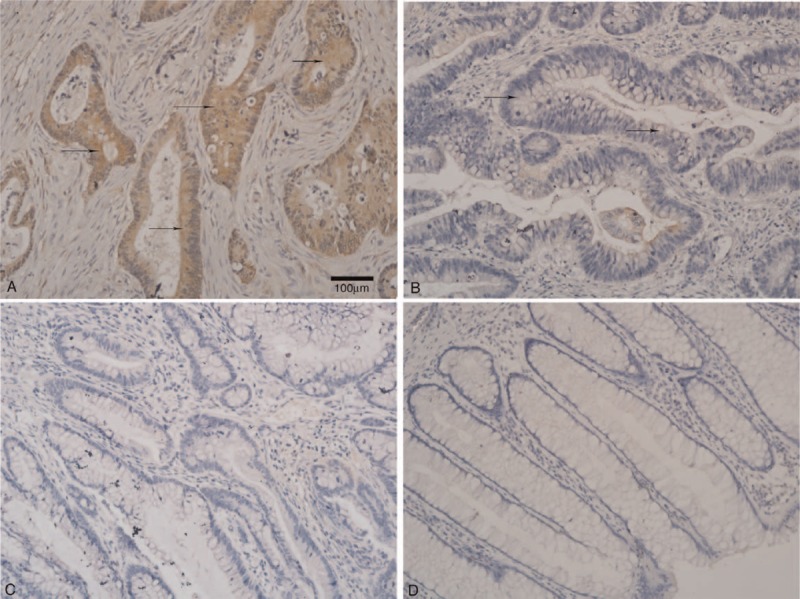
Immunohistochemical staining of XRCC2 expression in CRC and adjacent normal mucosal tissues (×200 magnification). There was (A) positive and (B) weakly positive XRCC2 expression in primary CRC. There was negative XRCC2 expression in (C) CRC and (D) normal colorectal mucosa tissue. Arrows indicate XRCC2-positive cells. The scale bar represents 100 μm.
FIGURE 2.
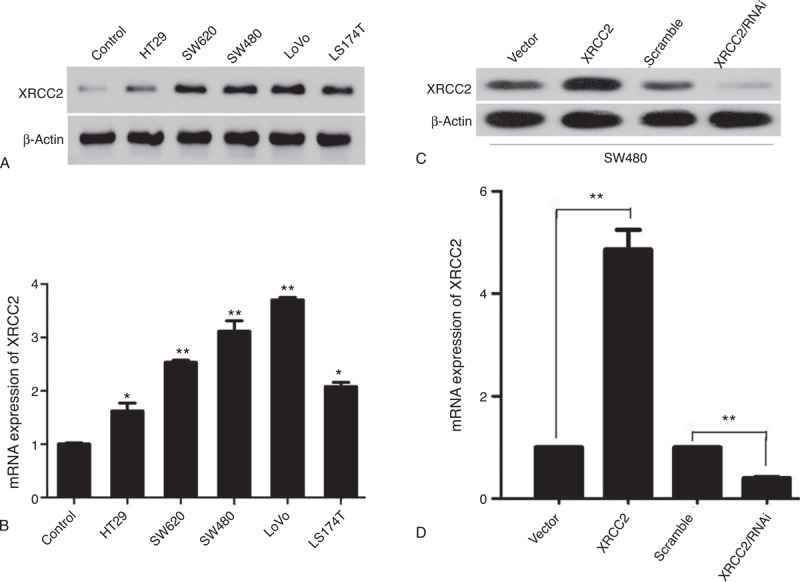
XRCC2 protein and gene expression in SW480 cells. (A) Western blot analysis of XRCC2 expression in normal colonic mucosal epithelial cells and SW620, SW480, LoVo, HT29, and LS174T cells. (B) QPCR analysis of XRCC2 mRNA expression in normal colonic mucosal epithelial cells and SW620, SW480, LoVo, HT29, and LS174T cells. (C) Western blot analysis of XRCC2 expression in SW480-Vector, SW480-XRCC2, SW480-Scramble, and SW480-XRCC2/RNAi cells. (D) QPCR analysis of XRCC2 mRNA expression in SW480 cells. ∗P < 0.05, ∗∗P < 0.01 (Student's t test).
TABLE 1.
XRCC2 Expression in Primary Colorectal Cancer Tissue and Adjacent Noncancerous Tissue

Correlation Between XRCC2 Expression and Clinicopathological Features of Patients With CRC
The correlation between XRCC2 expression and clinicopathological characteristics, which included gender, age, tumor site, tumor size, lymph node metastasis, distant metastasis, depth of invasion, differentiation, tumor-nodes-metastasis (TNM) stage, and Dukes’ stage, is described in Table 2. XRCC2 expression status in primary CRC was significantly related to tumor size, Dukes’ stage, and TNM stage (P = 0.012; P = 0.043; and P = 0.028; respectively).
TABLE 2.
Clinicopathological Characteristics and XRCC2 Expression Status of Patients With Colorectal Cancer
Successful Construction of Stable Cell Lines Expressing Low or High XRCC2
After constructing cell lines with high or low XRCC2 expression, we detected XRCC2 expression using Western blotting and quantitative PCR (QPCR). Western blotting showed that XRCC2 expression in SW480-XRCC2 cells was higher than that in SW480-Vector cells, and that XRCC2 expression in SW480-XRCC2/RNAi was lower than that in SW480-Scramble cells (Figure 2C). Furthermore, the QPCR results were consistent with those of Western blotting (Figure 2D; P = 0.000). These results demonstrate that we had successfully constructed stable cell lines with low or high XRCC2 expression.
XRCC2 Overexpression Promoted CRC Cell Proliferation by Enriching Cells in G0/G1
To investigate whether XRCC2 plays a role in the development of CRC, we examined cell proliferation using CCK-8 in SW480, SW480-XRCC2/RNAi, SW480-Scramble, SW480-XRCC2, and SW480-Vector cells (supplementary http://links.lww.com/MD/A108). However, we removed the SW480-Scramble and SW480-Vector cell lines from the experiment because their growth curves were almost identical to that of the SW480 cell line. After 96-hour incubation, XRCC2 downregulation significantly decreased the growth rate of SW480 cells, while XRCC2 upregulation markedly increased it compared with the controls (Figure 3A; P = 0.003; P = 0.000, respectively). Flow cytometry analysis showed that XRCC2 ectopic expression was associated with the G0/G1 population increasing by 2.9% compared to the control cells (XRCC2-overexpressing cells, 57% vs control, 54.1%). Low XRCC2 expression was associated with the G0/G1 population decreasing dramatically by 6.9% compared to the control cells (XRCC2-low expression cells, 47.2% vs control, 54.1%) (Figure 3B and C, P = 0.048; P = 0.007, respectively). Collectively, these results suggest that XRCC2 ectopic expression enhances CRC cell proliferation and enriches cells in the G0/G1 phase.
FIGURE 3.
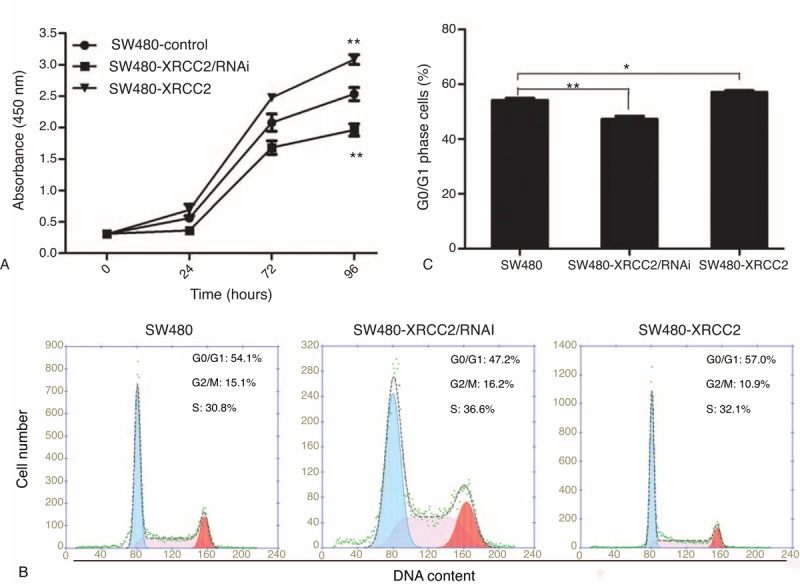
Cell proliferation and cell cycle analyses. (A) CCK-8 assay showing that viability was highest in the SW480-XRCC2 cell line and lowest in the SW480-XRCC2/RNAi cell line in the absence of olaparib. (B) Flow cytometry analysis of DNA content. The SW480-XRCC2 cell line had the most G0/G1 cells. (C) Histogram of the percentages of G0/G1 cells. ∗P < 0.05, ∗∗P < 0.01 (Student's t test).
XRCC2 Overexpression Inhibited CRC Cell Apoptosis
To quantify the anti-apoptotic effects of XRCC2 in CRC cells, we analyzed the apoptotic cell population using flow cytometry. XRCC2 downregulation increased the number of annexin V-positive cells compared with the controls; XRCC2 upregulation decreased the number of apoptotic cells (Figure 4A). A histogram of the annexin V-FITC-positive cells revealed that XRCC2 expression significantly altered the rates of apoptosis (Figure 4B; P = 0.006). Western blotting showed that XRCC2 downregulation increased Bcl-2 expression compared with the controls (Figure 4C). Therefore, XRCC2 overexpression inhibits CRC cell apoptosis.
FIGURE 4.
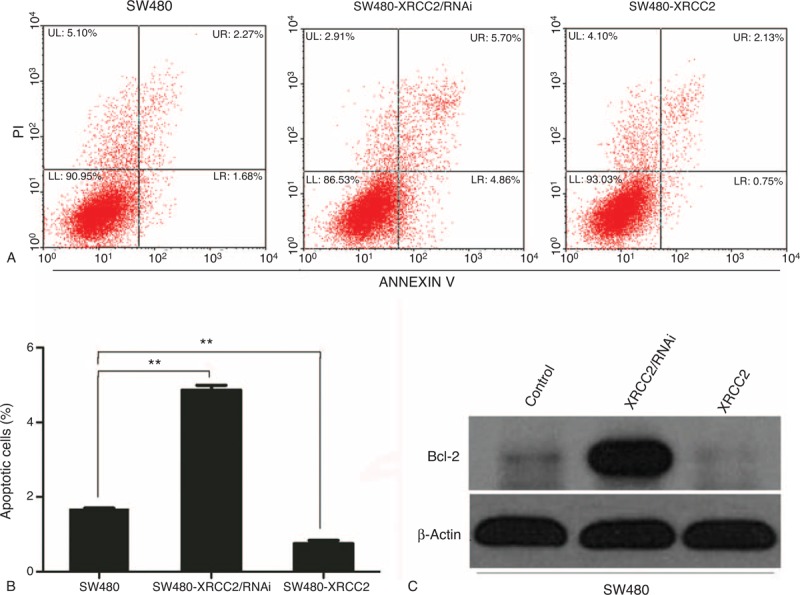
Analysis of apoptosis. (A) The number of apoptotic cells was highest in the SW480-XRCC2/RNAi cell line. (B) Histogram of apoptotic cells in the examined cell lines. (C) Western blotting results showing that XRCC2 downregulation increased bcl-2 expression compared with the controls. ∗∗P < 0.01 (Student's t test).
CRC Cells With High XRCC2 Expression Are More Sensitive to Olaparib
Figure 5D shows that cell viability decreased progressively as the olaparib concentration increased (P = 0.033). Following 96-hour treatment with 1 μM olaparib, SW480-XRCC2 cell viability was greater than that of SW480 cells, as was that of SW480 cells compared to SW480-XRCC2/RNAi cells (Figure 5A; P = 0.011; P = 0.001, respectively). The effect was consistent with the results depicted in Figure 3A (0 μM olaparib). Surprisingly, following treatment with 10 μM olaparib, the SW480-XRCC2 cell viability was lowest among the three cell lines (Figure 5B; P = 0.024). However, cell viability began to decline after 72 hours following treatment with 50 μM olaparib (Figure 5C). These results suggest that CRC cell sensitivity to olaparib increases progressively with the increased XRCC2 expression when the olaparib concentration is within a certain range. Moreover, although cell cycle analysis revealed that the number of G0/G1 cells was lowest among the SW480-XRCC2/RNAi cells in the absence of olaparib treatment (Figure 3B; P = 0.007), Figure 6A and B shows that the number of G0/G1 cells was lowest among the SW480-XRCC2 cells following olaparib treatment (10 μM olaparib; P = 0.042). Taken together, these results indicate that olaparib suppresses CRC cell proliferation, and CRC cells with high XRCC2 expression are more sensitive to it. Furthermore, treatment with 10 μM olaparib results in decreased G0/G1 cells with high XRCC2 expression (Figure 6C; P = 0.001).
FIGURE 5.
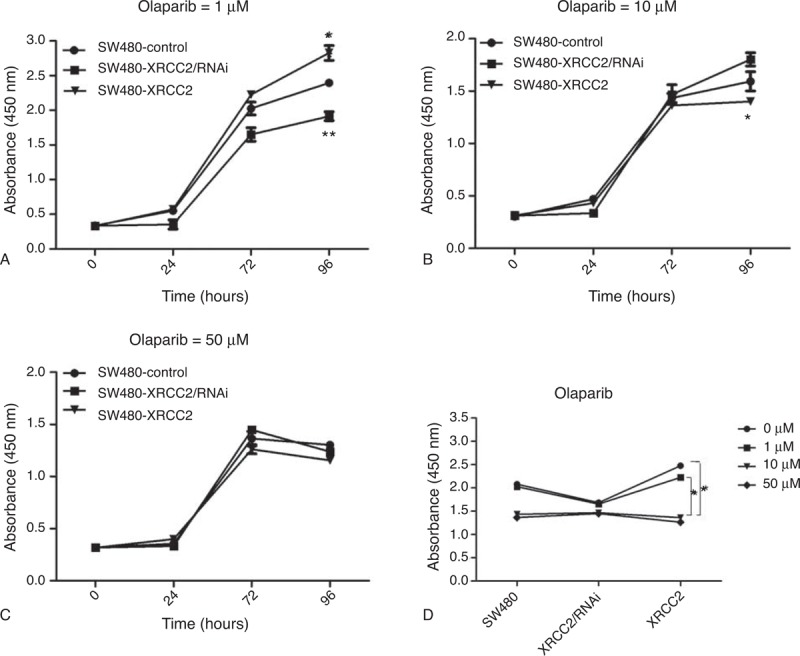
Olaparib inhibited cell proliferation, and CRC cells with high XRCC2 expression had higher olaparib sensitivity. The surviving fractions of SW480 cells treated with (A) 1 μM, (B) 10 μM, and (C) 50 μM olaparib were measured using CCK-8. (D) The relation between cell viability and olaparib concentration. ∗P < 0.05, ∗∗P < 0.01 (Student's t test).
FIGURE 6.
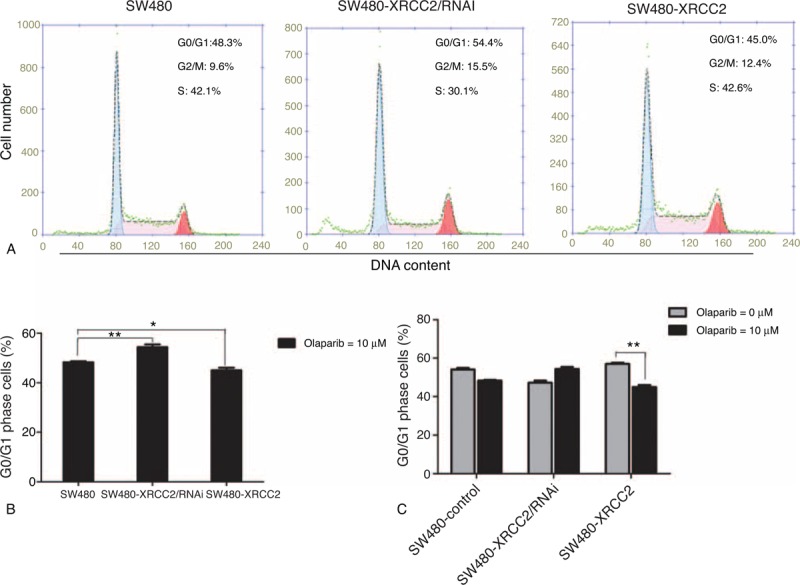
Flow cytometry cell cycle analysis following olaparib treatment. (A) The SW480-XRCC2 cell line had fewest G0/G1 cells following treatment with 10 μM olaparib. (B) Histogram of the percentage of cells in the G0/G1 phase following treatment with 10 μM olaparib. (C) Histogram of the percentage of cells in the G0/G1 phase in the absence of olaparib and following treatment with 10 μM olaparib. ∗P < 0.05, ∗∗P < 0.01 (Student's t test).
DISCUSSION
It is believed that XRCC2 is involved in the HRR pathway of double-stranded DNA and is responsible for the repair of chromosomal fragmentation, translocations, and deletions.4–6 Recent studies have shown that XRCC2 is associated with the development of CRC. For example, Curtin et al16 and Krupa et al17 revealed that XRCC2 SNPs increase the risk of CRC. Similarly, Haines et al18 proved that XRCC2 haploinsufficiency reduced spontaneous tumor incidence in multiple intestinal neoplasia mice but increased the tumorigenic response to radiation. Therefore, our findings demonstrate that the gain of XRCC2 expression is a common occurrence in the progression of CRC.
Our results revealed that positive XRCC2 expression and tumor size, Dukes’ stage, and TNM stage were significantly correlated, suggesting that increasing XRCC2 expression may promote the invasive behaviors as well as metastasis processes of CRC. While the mechanism by which XRCC2 affects CRC progression remains unclear, it is worth noting that previous studies support the premise that XRCC2 plays important roles in promoting tumor cell growth and proliferation. Wang et al19 reported that XRCC2 was overexpressed in colon cancer cell lines, and XRCC2 suppression rendered the cancer cells more sensitive to radiation therapy. Therefore, it is perhaps not surprising that XRCC2 is involved in CRC invasion and metastasis. However, further studies are warranted to elucidate the mechanism involved.
To further study the function of XRCC2 in CRC development and progression, we constructed stable cell lines with low (SW480-XRCC2/RNAi) or high XRCC2 expression (SW480-XRCC2). As uncontrolled growth is one of the important features of the cancer cell phenotype, we examined the effect of low or high XRCC2 expression on the growth of SW480 cells first. Our data indicated that suppressing XRCC2 decreased proliferation effectively, while increasing it increased proliferation significantly, suggesting that XRCC2 may be an important regulator of CRC cell proliferation.
Cell cycle arrest at any phase will consequently inhibit cell proliferation.20 However, how the cell cycle affects cell proliferation is yet to be fully understood. Generally speaking, cell lines enriched with G0/G1 cells have lower proliferation capacity than those lacking G0/G1 cells. In the present study, the fewer G0/G1 cells of the SW480-XRCC2/RNAi cell line appeared inconsistent in that the SW480-XRCC2/RNAi cell line grew more slowly than the SW480-XRCC2 cell line. It is generally believed that XRCC2 is an important role for HRR and the deficiency of it results in the loss of regulation of induction of DNA replication in the nuclei to prepare for further proliferation and differentiation. For that reason, we believe that ectopic XRCC2 enhancement of cell proliferation is partly attributable to cell enrichment in G0/G1, which provided the necessary elements for cell proliferation. Furthermore, we observed a significant increase in the percentage of apoptotic SW480-XRCC2/RNAi cells. Dramatically decreased cells in the G0/G1 phase and greatly increased apoptosis were observed in XRCC2-deficient cells, demonstrating that XRCC2 expression promotes CRC cell growth via the dual effect of enriching cells in the G0/G1 phase and decreasing apoptosis.
It has been proposed that simultaneous deficiencies in two genes introduce lethality in biological systems that would otherwise tolerate the loss of one of the two genes.21 According to this theory, inhibiting PARP is a potential therapeutic strategy for treating cancers with specific DNA repair gene defects, such as BRCA1/2, MRE11, XRCC2, and PARP1.22,23 In the present study, we investigated the effect of a PARP1 inhibitor on XRCC2-deficient CRC cells.
It has been reported that certain PARP1 inhibitors, including benzimidazole-4-carboxamides and tricyclic lactam indoles, inhibit cell growth by 50% at concentrations ranging 8–94 μM.24 In the present study, olaparib inhibited CRC cell proliferation in a dose- and time-dependent manner. Furthermore, Vilar et al reported a significant difference in cytotoxicity between biallelic mutants and wild-type cell lines following treatment with 10 μM veliparib, another PARP1 inhibitor.22 According to the theory of synthetic lethality and the findings of Vilar et al,22 SW480-XRCC2/RNAi cells should have been more sensitive to olaparib. However, we were surprised to find that, following 96-hour treatment with 10 μM olaparib, the olaparib sensitivity of SW480-XRCC2 cells was the highest among the three cell lines (P = 0.011). More interestingly, although the number of G0/G1 cells was lowest among SW480-XRCC2/RNAi cells in the absence of olaparib treatment, that among SW480-XRCC2 cells was the lowest following olaparib treatment (10 μM olaparib; P = 0.042). This indicates that decreasing cells in G0/G1 may contribute to the inhibition of proliferation following treatment with 10 μM olaparib. This effect is in agreement with the earlier results, where there were fewer cells in the G0/G1 phase among the low-viability cell lines.
Our results show that XRCC2 may be an oncogene in CRC, and that the PARP1 inhibitor has an obvious inhibitory effect on CRC cells. It is easy to believe that SW480-XRCC2/RNAi cells would have the lowest cell viability and highest sensitivity toward olaparib following olaparib treatment. Why, then, did the SW480-XRCC2 cells have greater olaparib sensitivity in the present study? First, XRCC2 and PARP1 are both involved in the HRR pathway of double-stranded DNA. It is possible that the effect of olaparib required XRCC2 to be present. However, there is no strong evidence proving the relationship between PARP1 inhibitors and XRCC2 so far. Second, Tentori et al25 found that the basal level of PARP1 may affect CRC cell sensitivity to PARP1 inhibitor monotherapy. The basal level of PARP1 may have differed among the three cell lines after the SW480-XRCC2/RNAi or SW480-XRCC2 cell lines were constructed. Third, as there are a number of PARP1 inhibitors, similar studies may yield different results.
In conclusion, the present results show that XRCC2 expression is high in CRC tissue, and there are significant correlations between positive XRCC2 expression and tumor size, Dukes’ stage, and TNM stage. Furthermore, XRCC2 expression promotes CRC cell proliferation and enriches cells in the G0/G1 phase, but inhibits apoptosis. Moreover, our results show that olaparib inhibits CRC cell proliferation in a dose- and time-dependent manner. Among the three cell lines examined, the viability of SW480-XRCC2 cells was the lowest following 96-hour treatment with 10 μM olaparib. However, further investigation is required to identify a clear relation between XRCC2 and PARP1 inhibitors.
ACKNOWLEDGMENTS
This study was supported by grants from the National Natural Science Foundation of China (NSFC-2011-81172339) and Guangdong Natural Science Foundation (2011B031800118).
Footnotes
Abbreviations: Bcl-2 = B-cell lymphoma-2, CCK-8 = Cell Counting Kit-8, CRC = colorectal cancer, DSBs = double-stranded breaks, GAPDH = glyceraldehyde-3-phosphate dehydrogenase, HRR = homologous recombination repair, NHEJ = non-homologous end joining, PARP1 = poly(ADP-ribose) polymerase 1, PBS = phosphate-buffered saline, PCR = polymerase chain reaction, PI = propidium iodide, PVDF = polyvinyl difluoride, RNAi = RNA interference, SDS–PAGE = sodium dodecyl sulfate–polyacrylamide gel electrophoresis, SNPs = single-nucleotide polymorphisms, SW480-Vector = SW480-pBABE-puro, SW480-Scramble = SW480-pSuper-retro-puro, SW480-XRCC2/RNAi = SW480-pSuper-retro-puro-siXRCC2, SW480-XRCC2 = SW480-pBABE-puro-XRCC2, TNM = tumor-nodes-metastasis, XRCC2 = X-ray repair complementing defective repair in Chinese hamster cells 2.
The authors declare that they have no conflict of interest.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Website (www.md-journal.com).
REFERENCES
- 1.Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev 2010; 19:1893–1907. [DOI] [PubMed] [Google Scholar]
- 2.Dai Z, Zheng RS, Zou XN, et al. Analysis and prediction of colorectal cancer incidence trend in China [article in Chinese]. Zhonghua Yu Fang Yi Xue Za Zhi 2012; 46:598–603. [PubMed] [Google Scholar]
- 3.Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010; 127:2893–2917. [DOI] [PubMed] [Google Scholar]
- 4.Jones NJ, Zhao Y, Siciliano MJ, Thompson LH. Assignment of the XRCC2 human DNA repair gene to chromosome 7q36 by complementation analysis. Genomics 1995; 26:619–622. [DOI] [PubMed] [Google Scholar]
- 5.Thacker J, Tambini CE, Simpson PJ, et al. Localization to chromosome 7q36.1 of the human XRCC2 gene, determining sensitivity to DNA-damaging agents. Hum Mol Genet 1995; 4:113–120. [DOI] [PubMed] [Google Scholar]
- 6.Liu N, Lamerdin JE, Tebbs RS, et al. XRCC2 and XRCC3, new human Rad51-family members, promote chromosome stability and protect against DNA cross-links and other damages. Mol Cell 1998; 1:783–793. [DOI] [PubMed] [Google Scholar]
- 7.Romanowicz-Makowska H, Smolarz B, Połać I, Sporny S. Single nucleotide polymorphisms of RAD51 G135C, XRCC2 Arg188His and XRCC3 Thr241Met homologous recombination repair genes and the risk of sporadic endometrial cancer in Polish women. J Obstet Gynaecol Res 2012; 38:918–924. [DOI] [PubMed] [Google Scholar]
- 8.Lin WY, Camp NJ, Cannon-Albright LA, et al. A role for XRCC2 gene polymorphisms in breast cancer risk and survival. J Med Genet 2011; 48:477–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yen CY, Liu SY, Chen CH, et al. Combinational polymorphisms of four DNA repair genes XRCC1, XRCC2, XRCC3, and XRCC4 and their association with oral cancer in Taiwan. J Oral Pathol Med 2008; 37:271–277. [DOI] [PubMed] [Google Scholar]
- 10.Whitehurst AW, Bodemann BO, Cardenas J, et al. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature 2007; 446:815–819. [DOI] [PubMed] [Google Scholar]
- 11.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-defective tumours with inhibitors of poly(ADPribose) polymerase. Nature 2005; 434:913–917. [DOI] [PubMed] [Google Scholar]
- 12.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434:917–921. [DOI] [PubMed] [Google Scholar]
- 13.Dedes KJ, Wilkerson PM, Wetterskog D, et al. Synthetic lethality of PARP inhibition in cancers lacking BRCA1 and BRCA2 mutations. Cell Cycle 2011; 10:1192–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med 2009; 361:1475–1485. [DOI] [PubMed] [Google Scholar]
- 15.Li J, Zhang N, Song LB, et al. Astrocyte elevated gene-1 is a novel prognostic marker for breast cancer progression and overall patient survival. Clin Cancer Res 2008; 14:3319–3326. [DOI] [PubMed] [Google Scholar]
- 16.Curtin K, Lin WY, George R, et al. Colorectal Cancer Study Group. Genetic variants in XRCC2: new insights into colorectalcancer tumorigenesis. Cancer Epidemiol Biomarkers Prev 2009; 18:2476–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krupa R, Sliwinski T, Wisniewska-Jarosinska M, et al. Polymorphisms in RAD51, XRCC2 and XRCC3 genes of the homologous recombination repair in colorectalcancer—a case control study. Mol Biol Rep 2011; 38:2849–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haines JW, Coster MR, Adam J, et al. Xrcc2 modulates spontaneous and radiation-induced tumorigenesis in Apcmin/+ mice. Mol Cancer Res 2010; 8:1227–1233. [DOI] [PubMed] [Google Scholar]
- 19.Wang Q, Wang Y, Du L, et al. shRNA-mediated XRCC2 gene knockdown efficiently sensitizes colon tumor cells to X-ray irradiation in vitro and in vivo. Int J Mol Sci 2014; 15:2157–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ujiki MB, Ding XZ, Salabat MR, et al. Apigenin inhibits pancreatic cancer cell proliferation through G2/M cell cycle arrest. Mol Cancer 2006; 5:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Connor MJ, Martin NM, Smith GC. Targeted cancer therapies based on the inhibition of DNA strand break repair. Oncogene 2007; 26:7816–7824. [DOI] [PubMed] [Google Scholar]
- 22.Vilar E, Bartnik CM, Stenzel SL, et al. MRE11 deficiency increases sensitivity to poly (ADP-ribose) polymerase inhibition in microsatellite unstable colorectal cancers. Cancer Res 2011; 71:2632–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly (ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 2009; 361:123–134. [DOI] [PubMed] [Google Scholar]
- 24.Calabrese CR, Batey MA, Thomas HD, et al. Identification of potent nontoxic poly(ADP-ribose) polymerase-1 inhibitors: chemopotentiation and pharmacological studies. Clin Cancer Res 2003; 9:2711–2718. [PubMed] [Google Scholar]
- 25.Tentori L, Muzi A, Dorio AS, et al. MSH3 expression does not influence the sensitivity of colon cancer HCT116 cell line to oxaliplatin and poly(ADP-ribose) polymerase (PARP) inhibitor as monotherapy or in combination. Cancer Chemother Pharmacol 2013; 72:117–125. [DOI] [PubMed] [Google Scholar]