

Abstract
SSc is a multiorgan disease with significant morbidity that is associated with poor health-related quality of life. Treatment of this condition is often organ based and non-curative. However, there are newer, potentially disease-modifying therapies available to treat certain aspects of the disease. This review focuses on old and new therapies in the management of SSc in clinical practice.
Keywords: systemic sclerosis, interstitial lung disease, digital ulcers, Raynaud’s phenomenon, pulmonary arterial hypertension, renal crisis, skin fibrosis, treatment, immunosuppressive therapy
Introduction
SSc is a pleomorphic autoimmune disorder that can affect multiple organs, most commonly the skin, lungs, kidneys, gastrointestinal tract and vasculature. Inflammatory, vascular and fibrotic processes often occur simultaneously and can cause severe dysfunction of these organs [1]. The aetiopathogenesis of SSc is poorly understood, hence treatment of this condition is difficult and often organ based. This review discusses the old and new therapies available for management of SSc and how to manage patients in clinical care (Fig. 1). Early screening and diagnosis were discussed recently in another review [2].
Fig. 1.
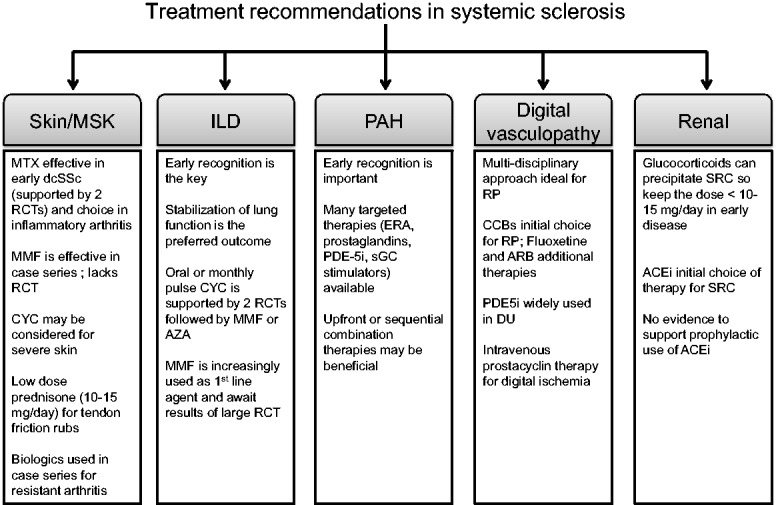
Treatment recommendations in SSc
Expert consensus and a growing evidence base underpin the management of SSc, as shown schematically with focus on individual organ systems and current approaches used in practice. MSK: musculoskeletal; ILD: interstitial lung disease; PAH: pulmonary arterial hypertension; RCT: randomized controlled trial; ERA: endothelin receptor antagonist; PDE-5i: phosphodiesterase-5 inhibitor; sGC: soluble guanylate cyclase; CCBs: calcium channel blockers; ARB: angiotensin receptor blocker; DU: digital ulcer; SRC: scleroderma renal crisis; ACEi: angiotensin converting enzyme inhibitor.
Fibrotic complications
Skin
Patients with SSc are subclassified into dcSSc or lcSSc based on the extent of skin involvement. The majority of trials have focused on dcSSc. The modified Rodnan skin score (mRSS), a measure of the extent of skin involvement, has been used as the primary outcome measure in clinical trials of dcSSc [3]. Measurement of skin thickness is used as a surrogate measure of disease severity and mortality in patients with dcSSc; an increase in skin thickening is associated with involvement of internal organs and increased mortality [4]. It is generally accepted that mRSS tends to worsen in early disease and improve in late disease. In our experience, practising clinicians do not routinely perform mRSS unless they have a special interest in scleroderma.
Treatment options have targeted different pathogenic processes, including inflammation, immune dysregulation and fibrosis. Currently there is no regulatory approved treatment for skin fibrosis.
MTX
The European League Against Rheumatism (EULAR) and EULAR Scleroderma Trials and Research (EUSTAR) group recommendations endorse the use of MTX in early dcSSc. Two randomized controlled trials (RCTs) have shown that MTX improves the mRSS in early dcSSc [5, 6]. In a multicentre, double-blind RCT, 71 patients with dcSSc of <3 years’ duration (mean duration 6.3 months in the MTX group, 7.3 months in the placebo group) were randomized to receive either MTX (n = 35) or placebo (n = 36) for a 12 month period [6]. Oral weekly MTX up to 15 mg/week [the average dose of MTX was 14.9 (s.d. 1.8) mg/week] was used. At the end of the study the primary outcomes favoured MTX over placebo, with an improvement in mean mRSS (6.3 U for MTX, 1.1 U for placebo, P < 0.17). Bayesian analysis of this trial showed that the probability that treatment with MTX results in better mean outcomes than placebo was 94% for mRSS and 88% for the physician global assessment [5].
CYC
There are no RCTs comparing CYC vs placebo for the management of skin disease as a primary indication. In the post hoc analysis of the Scleroderma Lung Study-I (SLS-I) [7], 85 patients with dcSSc who received oral CYC for 12 months showed significant difference mRSS between the two groups favouring CYC (improvement in mean mRSS 3.06 U, P = 0.008), but it was non-significant at 24 months (P = 0.23) [8]. An open-label case series supports low-dose pulse CYC for aggressive skin disease [9].
MMF
There have been mixed results from open-label and retrospective studies with regard to the effect of MMF on skin disease [10, 11]. There is no RCT assessing its efficacy. In a retrospective study at a single academic centre, the change in mRSS in 98 dcSSc patients who were treated with MMF was compared with the mRSS from historical controls [d-penicillamine (d-pen) and oral bovine type I collagen] obtained from pooled analysis of RCTs [12]. In a subgroup of 20 patients, a decrease or discontinuation of MMF resulted in increased skin activity that subsequently improved after re-initiation.
d-Pen
A double-blind RCT was conducted to compare high-dose (750–1000 mg/day) and low dose d-pen (125 mg every other day) in 134 patients with early dcSSc [13]. There were modest improvements in the mRSS, although not significantly different between the two groups, thus the RCT does not support the use of d-pen in dcSSc.
Other agents
IFN-α [14], recombinant human relaxin [15], recombinant human anti-TGF-β1 antibody [16], infliximab [17] and imatinib [18] have not been shown to be beneficial in RCTs or open-label trials.
Treatment of skin involvement in SSc
There is some evidence that MTX is beneficial in the treatment of early dcSSc. The authors utilize up to 30 mg/week using either an oral or s.c. route. Another group includes patients with SSc who also have concomitant clinical inflammatory arthritis (16%) [19]. CYC may be effective in early dcSSc but is usually reserved for patients with concomitant interstitial lung disease (ILD). MMF appears to have beneficial effects in dcSSc but lacks RCTs. d-pen should not be used for the treatment of skin involvement in SSc. There is no evidence to support immunosuppressive treatment of skin involvement in lcSSc.
ILD
Pulmonary disease in SSc (ILD and pulmonary hypertension) is the leading cause of death in patients with SSc [20]. High-resolution CT (HRCT) of the lungs reveals features of ILD in up to 90% of patients with SSc [21, 22]. Forced vital capacity (FVC) on pulmonary function tests (PFTs) has been used as a surrogate for restrictive lung disease, although patients can have ILD on HRCT with normal spirometry. Restrictive lung disease (FVC <75%) develops in ∼40% of SSc patients. Severe restrictive lung disease (defined as FVC <50%) is found in ∼10–15% of SSc patients. Therefore not every patient requires immunosuppressive therapy.
When to initiate immunosuppressive therapy
The decision on the timing of immunosuppression can be quite challenging. It is important to identify these SSc-ILD patients and predict their disease course, as the treatment needs to be individualized. In SSc patients, progressive ILD is a predictor of mortality [23]. Severe and progressive ILD, as demonstrated by rapid decline in the FVC (>10% of the FVC%) usually occurs in the first 3–4 years of disease onset. The rate of decline in FVC is greater in patients with early onset disease, FVC <70% at presentation and greater extent of lung fibrosis on HRCT (>25% involvement of lung zones or >20% total lung involvement) [24, 25]. Thus immunosuppressive therapy is largely indicated for SSc patients with early disease who have (i) >10% decrease in FVC in the previous 3–12 months, (ii) moderate to severe lung involvement on HRCT and/or (iii) FVC <70% at presentation.
CYC
CYC has been evaluated in two placebo-controlled RCTs involving SSc-ILD patients [7, 26]. In the multicentric SLS [7], 158 patients with disease duration of <7 years and with symptomatic ILD received either oral CYC (≤2 mg/kg of body weight/day) or placebo for 1 year and were followed up for an additional year. At 12 months (primary outcome) there was a modest but statistically significant improvement in both the FVC (adjusted mean absolute difference in FVC 2.53% favouring CYC, P < 0.03). Small improvements in lung function were associated with significant improvements in dyspnoea and health-related quality of life [27]. At 24 months the mean values of FVC in the two groups were almost identical. In contrast, the positive effect on dyspnoea persisted through 24 months [8].
In another double-blind RCT, the Fibrosing Alveolitis in Scleroderma Trial (FAST), 22 of 45 patients with SSc (lcSSc or dcSSc) were given six doses of i.v. CYC (600 mg/m2) at 4-week intervals followed by oral AZA (2.5 mg/kg/day) [26]. Prednisolone (20 mg on alternate days) was co-administered in the active treatment group. At the end of 1 year there was an improvement in FVC (adjusted mean difference in FVC 4.19%, P = 0.08).
Based on the results of these trials, EULAR/EUSTAR recommends that CYC should be considered for the treatment of SSc-ILD [28].
MMF
There are no published RCTs of MMF in SSc-ILD. In several small, open-label studies, MMF at a dose of 1–2 g/day (alone or with steroids) either improved or stabilized lung function up to 12 months [29, 30]. In a retrospective analysis of 172 SSc patients (mainly dcSSc) MMF was compared with other immunosuppressive agents [11]. A significantly lower incidence of clinically significant ILD was noted in patients treated with MMF and there was no significant between-group difference in FVC change up to 5 years. The SLS II, a randomized, double-blind study evaluating MMF vs CYC in patients with SSc-ILD is currently under way.
Glucocorticoids
There are sparse and unclear data regarding the role of glucocorticoids in the management of SSc-ILD. A single-centre open-label study comparing high-dose vs low-dose prednisone therapy with monthly i.v. CYC showed improvement in the FVC and the percentage of ground glass parenchymal lung involvement only in the high-dose prednisone group [31]. In the FAST study, prednisolone was prescribed at 20 mg on alternate days [26]. There is a lack of evidence to support high-dose prednisone in SSc-ILD. In addition, there are retrospective data to support an association of renal crisis with moderate to high doses of glucocorticoids [32]. Based on these studies, low-dose prednisone (10 mg/day or 20 mg every other day) may be reasonable for SSc-ILD.
Other agents
There is limited evidence with regard to other immunosuppressive agents such as MTX, AZA, CYC and tacrolimus. In small, open-label studies, rituximab has been shown to improve or stabilize lung function [33]. Bosentan was studied in a large RCT for SSc-ILD with negative results [34]. Imatinib was studied in a phase 1–2a open-label pilot study involving 20 patients with SSc-ILD. Oral imatinib therapy (up to 600 mg/day) was given for a period of 1 year [35]. There was a trend towards improvement in lung function (FVC increased by 1.74%), however, 7 of 20 patients discontinued drug use due to adverse effects. At present the role of tyrosine kinase inhibitors in SSc-ILD is unclear. Pirfenidone is being evaluated in an ongoing open-label, randomized, phase 2 study of its safety and tolerability in SSc-ILD (clinical trial registration number NCT01933334).
Treatment of ILD in SSc
Treatment of ILD should be reserved for patients with progressive ILD. Various analyses suggest that early disease (the first 3–4 years of disease onset), moderate to severe lung involvement [24, 25] and worsening pulmonary physiology should lead to initiation of immunosuppressive therapy [25, 36]. Bronchoalveolar lavage is not needed to make a clinical decision on treatment [37]. The authors prefer monthly pulse CYC at doses of 500–750 mg/m2 for 6–12 months. The response to therapy is measured by PFTs every 3–4 months and patient-reported improvement in dyspnoea. The therapeutic response tends to be slow. Stabilization of the FVC as opposed to improvement is the norm and is considered a favourable response. Therefore early identification and treatment are needed. The optimal duration of therapy is not known. At the end of 6 months of CYC therapy, if there is improvement or stabilization of lung function, the practice of the authors is to transition to either MMF or AZA for 1–3 more years, although some patients may require a longer duration. The decision to continue CYC beyond the 6 month period is based on the rate of decline in FVC: if there is a slower decline, CYC may be continued for another 3–6 months; if there is continuation of a rapid decline, then CYC therapy has probably not been beneficial.
There is increasing use of MMF in patients with mild ILD (ILD on HRCT and mild to moderate restrictive pulmonary physiology on PFT). Although MMF is being used in clinical care, SLS II will provide data on the efficacy of MMF vs CYC in an RCT. The authors recommend care coordination with a pulmonologist.
Early fibrotic disease in scleroderma
Human autologous stem cell transplantation
To date there have been three controlled trials on human autologous stem cell transplantation (HSCT) in the treatment of SSc patients. An open-label, phase 2 RCT, the American Scleroderma Stem Cell vs Immune Suppression Trial (ASSIST), was conducted to assess the efficacy and safety of HSCT vs monthly pulse i.v. CYC in patients with SSc [38]. The HSCT patients improved, with a significant decrease in mean mRSS (from 28 to 15, P = 0.0004) and a significant increase in mean FVC (from 62% to 74%, P = 0.004). In the first phase 3 HSCT trial, the Autologous Stem cell Transplantation International Scleroderma trial (ASTIS), 156 SSc patients were recruited from 2001 to 2009 and randomized to receive either HSCT (n = 79) or 12 monthly pulses of i.v. CYC 750 mg/m2 (n = 77) [39]. There was higher treatment-related mortality in the HSCT arm and the event-free survival favoured HSCT. The Scleroderma: CYC or Transplant trial is currently under way in the USA.
Currently, HSCT is not ready to be incorporated in clinical practice. However, recent data are supportive for a subset of dcSSc patients, with or without progressive internal organ involvement, who have not responded to conventional immunosuppressive agents. In addition, one may also consider HSCT in lcSSc patients with progressive ILD, but there is HSCT-associated mortality.
Vascular complications
Pulmonary arterial hypertension
Pulmonary arterial hypertension (PAH) is a fatal complication seen in ∼8–12% of SSc patients and has a poor prognosis, with a 3 year survival rate of 50–60% [40]. Three pathogenic pathways have been the targets of therapy: endothelin [endothelin receptor antagonists (ERAs)], nitric oxide [phosphodiesterase-5 inhibitors (PDE-5is)] and prostacyclin (prostanoids). There are a limited number of RCTs specifically targeting the treatment of CTD-PAH or SSc-PAH [41, 42]. There are multiple regulatory agency-approved medications that target the endothelin pathway [bosentan, macitentan and ambrisentan (all oral therapies)], nitric oxide/soluble guanylate cyclase pathway [sildenafil, tadalafil and riociguat (all oral therapies)] and prostacyclin pathway [iloprost (inhaled and i.v.), epoprostenol (i.v.) and treprostinil (inhaled, i.v., s.c.)]. Recommendations are for early screening and diagnosis of SSc-PAH [43] and treatment guidelines based on the severity of disease have been published [44, 45].
Digital vasculopathy (RP and digital ulcers)
RP is the most common clinical manifestation of SSc, affecting ∼95% of patients. Up to 30% of SSc patients with RP can progress to have digital ulceration by 1 year [46]. Digital ischaemia in SSc is multifactorial in aetiology, including imbalances in neuroendocrine control mechanisms, structural changes in the vessels and haematological factors such as hypercoagulability [47].
Calcium channel blockers
Calcium channel blockers (CCBs) are the cornerstone of treatment of RP and DU. Among the CCBs, nifedipine has the most supportive evidence at this time. Nifedipine is a dihydropyridine-type CCB that has a direct effect on vascular smooth muscles and inhibits platelet activation. In a meta-analysis including eight RCTs (seven with nifedipine and one with nicardipine), the weighted mean reduction in RP attacks over a 2 week period with CCBs (compared with placebo) was −8.3 U (range −15.7 to −0.9 U) [48]. CCBs reduced the severity of these attacks by 35%. In a small RCT, nifedipine was shown to reduce the number of digital ulcers (Dus) [mean 4.3 (s.e. 0.8) to 1.4 (0.5)] over a 16 week period [49]. Other therapies include topical nitroglycerine patches to reduce the frequency and severity of RP attacks in both primary and secondary RP [50]. Based on the available evidence, oral CCBs like nifedipine or amlodipine should be considered as first-line therapy for RP.
Prostaglandins
Continuous i.v. iloprost infusion (6 h/day for 5 consecutive days) resulted in healing of DUs over a 10 week period in SSc-RP patients, in addition to reducing the frequency and severity of RP attacks [51]. Centres in the USA use i.v. epoprostenol daily for 5 days due to the non-availability of iloprost (personal communication: Johns Hopkins, UCLA and University of Michigan Scleroderma Centres). Continuous infusion of i.v. epoprostenol for patients with severe SSc-PAH revealed a tendency towards a reduction in the number of new DUs (secondary outcome measure) by 50% [41].
ERAs
Endothelin levels are elevated in the serum of patients with SSc, especially in those patients with DUs [52]. In two clinical trials that evaluated the efficacy of bosentan in SSc-DU, the prevention of new DUs was noted, especially in patients with multiple ulcers [53, 54]. ERAs are not approved in the USA for the management of DUs.
PDE-5is
PDE-5is such as sildenafil have been studied for use in patients with RP and have shown a significant benefit in terms of the frequency, duration and severity of attacks [55, 56]. In the first few months of treatment, maximally tolerated doses of sildenafil seem to help with ulcer healing [57, 58]. In a double-blind RCT of 25 patients, alternate-day tadalafil (20 mg) as add-on therapy for 6 weeks improved RP symptoms (reduction in frequency, duration and severity of RP) and healed DUs [59].
Other agents
The selective serotonin reuptake inhibitor fluoxetine (20 mg/day) has been shown to reduce the frequency and severity of RP attacks [60].
Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers
There is little evidence to support the use of angiotensin-converting enzyme (ACE) inhibitors in treating RP. A randomized trial in 210 patients with secondary RP did not support the use of the ACE inhibitor quinapril, as there was no reduction in the incidence of DUs or in the frequency or severity of RP attacks [61, 62]. Interestingly, in an RCT comparing the efficacy of losartan (an angiotensin receptor blocker) with nifedipine (40 mg/day), losartan (50 mg/day) produced a greater reduction in the severity and frequency of attacks in a 12 week period in both primary and secondary RP [63].
Other therapies
Surgical digital sympathectomy can be used in patients with DUs and critical ischaemia when oral and/or topical vasodilatory therapy does not quickly result in improvement in digital blood flow [64–67]. Initial reports on the use of botulinum toxin A in the treatment of RP have been promising [68]. Larger RCTs with better designs are needed to recommend this in clinical care.
Treatment of RP and DUs in SSc
The authors include a multidisciplinary approach in the management of RP and DU (Fig. 2). Non-pharmacological management including core and peripheral warming is critical and infection must be excluded. Evidence suggests the use of CCB as initial treatment for RP and DUs. The authors use amlodipine up to 20 mg/day, although lower extremity oedema and dizziness are an issue with doses >10 mg/day. Fluoxetine or losartan as additional therapy is employed for RP. For DUs, PDE-5i is incorporated [57], despite lack of a well-powered RCT. If the patient has digital ischaemia, he/she is admitted and started on prostacyclin therapy for 5 days with concomitant ASA or heparin and PDE-5i, if tolerated. Botulinum toxin and sympathectomy are used in resistant cases. Analgesia is critical, as these ulcers are usually very painful.
Fig. 2.

Current management of digital vasculopathy in SSc
Treatment approaches may target individual pathological processes in SSc and include agents that are undergoing clinical evaluation or may have potential. Anti-fibrotic strategies are currently less developed than vascular or immunological candidate therapies. *If indicated. MRA: magnetic resonance angiography; CCB: calcium channel blocker; ARB: angiotensin receptor blocker. Modified from the Royal Free Hospital protocol.
Scleroderma renal crisis
SSc can cause many abnormalities in the renal tract, with scleroderma renal crisis (SRC) being the most important and serious manifestation. More than 70% of the severe kidney involvement occurs within the first 3 years of disease onset in SSc patients [23], thus early SSc patients (especially dcSSc) and patients with anti-RNA polymerase III antibody are at highest risk. SRC was once the most common cause of death in SSc; however, with the advent of ACE inhibitors, it is much more treatable. Although there are no RCTs to support the use of ACE inhibitors in SRC, there are numerous case reports and uncontrolled studies that have reported very favourable results. In a prospective analysis of 108 patients with SRC in a single centre, patients on ACE inhibitors [captopril (n = 47) and enalapril (n = 8)] had a significantly better survival rate at 1 year (76%) and 5 years (66%) compared with patients not on ACE inhibitors (survival rate at 1 year 15% and at 5 years 10%) [69]. In another prospective trial, 145 patients with SRC treated with ACE inhibitors demonstrated survival rates of 90% and 85% at 5 and 8 years, respectively, after the onset of SRC [70]. Further, treatment with ACE inhibitors decreased the need for permanent dialysis.
Given the perceived decrease in the incidence of SRC with the use of ACE inhibitors, their prophylactic use to prevent SRC has been considered [71]. However, one of the main concerns is that prophylactic use of ACE inhibitors may mask hypertension, leading to worse outcomes after SRC onset. In a recent prospective cohort study of incident SRC patients, exposure to ACE inhibitors prior to the onset of SRC was associated with increased risk of death (hazard ratio 2.42, P < 0.05) [72]. In another study utilizing an international web-based cohort design, ACE inhibitor exposure prior to SRC onset was associated with a >2-fold increase in the risk of death after the onset of SRC [73].
Iloprost by continuous i.v. infusion has been used in the management of SRC and may have specific benefits in renal perfusion [74]. At present there is insufficient evidence to recommend the use of iloprost in the treatment of SRC.
Prevention and treatment of SRC in SSc
Patients with early disease (especially dcSSc and anti-RNA polymerase III antibodies) are associated with incident cases of renal crisis. We recommend blood pressure measurement at home three times a week and providing the patient with parameters that should alert them to contact their physician [1]. Prophylactic ACE inhibitors have not been shown to improve outcomes. For treatment, our current approach to SRC is to admit all SSc patients with new-onset accelerated-phase hypertension with evidence of renal injury, microangiopathic haemolysis or significant end-organ damage and treat them with ACE inhibitors. We suggest starting with a short-acting ACE inhibitor (such as oral captopril) and aggressively up-titrate the dose every few hours with the goal of normalizing blood pressure (reduction of 10–20 mmHg systolic pressure per 24 h, even if there is continued deterioration in renal function). If the blood pressure remains elevated, we suggest adding other agents like CCBs, angiotensin receptor blockers, hydralazine or furosemide. Intermittent dialysis should be considered to salvage renal function, and we suggest at least 18 months before considering a renal transplant. Glucocorticoids have often been implicated in precipitating SRC [75–77] and patients on steroids should be carefully monitored for blood pressure and renal function.
New potential disease-modifying therapies for SSc
This review also highlights important progress that has been made in conducting high-quality controlled trials for the treatment of skin, lung, PAH and DU disease in SSc that have provided an evidence base to support current and some emerging therapies. There are potential future treatments that link to novel therapeutics and arise from a better understanding of key pathogenic pathways (Fig. 3). One logical pathway to target is the TGF-β axis, which includes a large number of ligands, receptors and accessory molecules. This pathway is essential for normal growth and development and tissue repair and has been implicated in many fibrotic disorders, including SSc. Attempts to attenuate TGF-β signalling using CAT-192, a neutralizing recombinant monoclonal antibody directed against one isoform, TGF-β1, were somewhat disappointing, although, importantly for such a potentially pleiotropic target, there were no safety concerns [16]. Other strategies, including topical administration of β-glycan-derived peptide fragments that may block ligand–receptor interaction in the skin [78] or more potent pan-specific antibodies such as GC1008, are being tested and may have much more potential [79]. One of the attractions of targeting TGF-β pathways is that they also regulate or modulate many other potentially important molecular mediators that may be important in SSc, including chemokines, vascular endothelial growth factor and endothelin. These can be identified through a better understanding of key pathways and mediators that may drive fibrosis in SSc and other diseases. Thus, in addition to later-stage clinical trials that are under way and were discussed in this article, there are small studies under way that target pathways as diverse as lipid mediators implicated in fibroblast recruitment and myofibroblast differentiation [80] (especially LPA1), fibrocyte differentiation, integrin signalling or cannabinoid receptors [81]. Blockade of the αv integrin has the potential to modulate TGF-β activation in target organs, and potential biologic therapies are in clinical development [82]. Additionally, there are ongoing studies evaluating the potential role for more established therapeutic agents such as endothelin receptor antagonists in renal disease in SSc, including a small pilot safety study of bosentan [74], and evaluation of the novel endothelin A receptor selective agent zibotentan [83] is under way. Other novel agents include hyperimmune goat serum (AIMSPRO; Daval International, Eastbourne, UK), which was recently subjected to a safety study in established late-stage dcSSc and demonstrated potential benefit for skin sclerosis [84]. Platelet-derived serotonin has been implicated as a potential therapeutic target in experimental models of scleroderma [85]. Other trials include assessment of tocilizumab (IL-6 receptor antagonist; NCT01532869) and abatacept in dcSSc. New and emerging therapeutic approaches are summarized in Fig. 2, which groups them according to their major actions on pathogenic processes in SSc.
Fig. 3.

Potential novel therapies in SSc
Digital ischaemia underlies important complications of systemic sclerosis and is amenable to multifaceted treatment including an increasing number of potentially disease-modifying agents that may work through a combination of vasodilator, anti-infective or structural vascular mechanisms. ETA: endothelin A; ETB: endothelin B; IP: prostacyclin; CD20: cluster of differentiation 20; CTLA4: cytotoxic T lymphocyte antigen 4; c-ABL: Abelson murine leukaemia viral oncogene; c-KIT: stem cell factor; CTGF: connective tissue growth factor; FGF: fibroblast growth factor; LPA: lysophosphatidic acid receptor; CB2: cannabinoid receptor 2; HT: 5-hydroxytryptamine receptor.
Rheumatology key messages.
Detailed evaluation of SSc patients is necessary to identify internal involvement.
Treatment of SSc is often organ based.
Novel agents are currently being investigated to treat different disease manifestations in SSc.
Acknowledgements
D.K. is supported by a National Institute of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases K24 grant (AR063120). V.N. has salary support from the US Department of Veterans Affairs.
Disclosure statement: D.K. is a consultant to Actelion, Bayer, Biogen-Idec, BMS, DIGNA, Genentech/Roche, Genzyme, InterMune, Merck/EMD Serono and Sanofi-Aventis. C.P.D. has been a consultant to Actelion, GSK, Sanofi-Aventis, Merck-Serono, Genentech-Roche, Pfizer, CSL Behring, Biogen-Idec and Bayer. The other author has declared no conflicts of interest.
Funding: No specific funding was received from any funding bodies in the public, commercial or not-for-profit sectors to carry out the work described in this manuscript.
References
- 1.Khanna D, Denton CP. Evidence-based management of rapidly progressing systemic sclerosis. Best Pract Res Clin Rheumatol. 2010;24:387–400. doi: 10.1016/j.berh.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avouac J, Fransen J, Walker UA, et al. Preliminary criteria for the very early diagnosis of systemic sclerosis: results of a Delphi Consensus Study from EULAR Scleroderma Trials and Research Group. Ann Rheum Dis. 2011;70:476–81. doi: 10.1136/ard.2010.136929. [DOI] [PubMed] [Google Scholar]
- 3.Khanna D, Merkel PA. Outcome measures in systemic sclerosis: an update on instruments and current research. Curr Rheumatol Rep. 2007;9:151–7. doi: 10.1007/s11926-007-0010-5. [DOI] [PubMed] [Google Scholar]
- 4.Clements PJ, Hurwitz EL, Wong WK, et al. Skin thickness score as a predictor and correlate of outcome in systemic sclerosis: high-dose versus low-dose penicillamine trial. Arthritis Rheum. 2000;43:2445–54. doi: 10.1002/1529-0131(200011)43:11<2445::AID-ANR11>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 5.Johnson SR, Feldman BM, Pope JE, Tomlinson GA. Shifting our thinking about uncommon disease trials: the case of methotrexate in scleroderma. J Rheumatol. 2009;36:323–9. doi: 10.3899/jrheum.071169. [DOI] [PubMed] [Google Scholar]
- 6.Pope JE, Bellamy N, Seibold JR, et al. A randomized, controlled trial of methotrexate versus placebo in early diffuse scleroderma. Arthritis Rheum. 2001;44:1351–8. doi: 10.1002/1529-0131(200106)44:6<1351::AID-ART227>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 7.Tashkin DP, Elashoff R, Clements PJ, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006;354:2655–66. doi: 10.1056/NEJMoa055120. [DOI] [PubMed] [Google Scholar]
- 8.Tashkin DP, Elashoff R, Clements PJ, et al. Effects of 1-year treatment with cyclophosphamide on outcomes at 2 years in scleroderma lung disease. Am J Respir Crit Care Med. 2007;176:1026–34. doi: 10.1164/rccm.200702-326OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D’Angelo S, Cuomo G, Paone C, et al. Low-dose intravenous cyclophosphamide in systemic sclerosis: a preliminary safety study. Clin Rheumatol. 2003;22:393–6. doi: 10.1007/s10067-003-0756-8. [DOI] [PubMed] [Google Scholar]
- 10.Herrick AL, Lunt M, Whidby N, et al. Observational study of treatment outcome in early diffuse cutaneous systemic sclerosis. J Rheumatol. 2010;37:116–24. doi: 10.3899/jrheum.090668. [DOI] [PubMed] [Google Scholar]
- 11.Nihtyanova SI, Brough GM, Black CM, Denton CP. Mycophenolate mofetil in diffuse cutaneous systemic sclerosis—a retrospective analysis. Rheumatology. 2007;46:442–5. doi: 10.1093/rheumatology/kel244. [DOI] [PubMed] [Google Scholar]
- 12.Le EN, Wigley FM, Shah AA, Boin F, Hummers LK. Long-term experience of mycophenolate mofetil for treatment of diffuse cutaneous systemic sclerosis. Ann Rheum Dis. 2011;70:1104–7. doi: 10.1136/ard.2010.142000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clements PJ, Furst DE, Wong WK, et al. High-dose versus low-dose D-penicillamine in early diffuse systemic sclerosis: analysis of a two-year, double-blind, randomized, controlled clinical trial. Arthritis Rheum. 1999;42:1194–203. doi: 10.1002/1529-0131(199906)42:6<1194::AID-ANR16>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 14.Black CM, Silman AJ, Herrick AI, et al. Interferon-alpha does not improve outcome at one year in patients with diffuse cutaneous scleroderma: results of a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 1999;42:299–305. doi: 10.1002/1529-0131(199902)42:2<299::AID-ANR12>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 15.Khanna D, Clements PJ, Furst DE, et al. Recombinant human relaxin in the treatment of systemic sclerosis with diffuse cutaneous involvement: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2009;60:1102–11. doi: 10.1002/art.24380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Denton CP, Merkel PA, Furst DE, et al. Recombinant human anti-transforming growth factor beta1 antibody therapy in systemic sclerosis: a multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum. 2007;56:323–33. doi: 10.1002/art.22289. [DOI] [PubMed] [Google Scholar]
- 17.Denton CP, Engelhart M, Tvede N, et al. An open-label pilot study of infliximab therapy in diffuse cutaneous systemic sclerosis. Ann Rheum Dis. 2009;68:1433–9. doi: 10.1136/ard.2008.096123. [DOI] [PubMed] [Google Scholar]
- 18.Pope J, McBain D, Petrlich L, et al. Imatinib in active diffuse cutaneous systemic sclerosis: results of a six-month, randomized, double-blind, placebo-controlled, proof-of-concept pilot study at a single center. Arthritis Rheum. 2011;63:3547–51. doi: 10.1002/art.30549. [DOI] [PubMed] [Google Scholar]
- 19.Avouac J, Clements PJ, Khanna D, Furst DE, Allanore Y. Articular involvement in systemic sclerosis. Rheumatology. 2012;51:1347–56. doi: 10.1093/rheumatology/kes041. [DOI] [PubMed] [Google Scholar]
- 20.Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis. 2007;66:940–4. doi: 10.1136/ard.2006.066068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schurawitzki H, Stiglbauer R, Graninger W, et al. Interstitial lung disease in progressive systemic sclerosis: high-resolution CT versus radiography. Radiology. 1990;176:755–9. doi: 10.1148/radiology.176.3.2389033. [DOI] [PubMed] [Google Scholar]
- 22.Warrick JH, Bhalla M, Schabel SI, Silver RM. High resolution computed tomography in early scleroderma lung disease. J Rheumatol. 1991;18:1520–8. [PubMed] [Google Scholar]
- 23.Steen VD, Medsger TA., Jr Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum. 2000;43:2437–44. doi: 10.1002/1529-0131(200011)43:11<2437::AID-ANR10>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 24.Khanna D, Tseng CH, Farmani N, et al. Clinical course of lung physiology in patients with scleroderma and interstitial lung disease: analysis of the Scleroderma Lung Study Placebo Group. Arthritis Rheum. 2011;63:3078–85. doi: 10.1002/art.30467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goh NS, Desai SR, Veeraraghavan S, et al. Interstitial lung disease in systemic sclerosis: a simple staging system. Am J Respir Crit Care Med. 2008;177:1248–54. doi: 10.1164/rccm.200706-877OC. [DOI] [PubMed] [Google Scholar]
- 26.Hoyles RK, Ellis RW, Wellsbury J, et al. A multicenter, prospective, randomized, double-blind, placebo-controlled trial of corticosteroids and intravenous cyclophosphamide followed by oral azathioprine for the treatment of pulmonary fibrosis in scleroderma. Arthritis Rheum. 2006;54:3962–70. doi: 10.1002/art.22204. [DOI] [PubMed] [Google Scholar]
- 27.Khanna D, Yan X, Tashkin DP, et al. Impact of oral cyclophosphamide on health-related quality of life in patients with active scleroderma lung disease: results from the scleroderma lung study. Arthritis Rheum. 2007;56:1676–84. doi: 10.1002/art.22580. [DOI] [PubMed] [Google Scholar]
- 28.Kowal-Bielecka O, Landewe R, Avouac J, et al. EULAR recommendations for the treatment of systemic sclerosis: a report from the EULAR Scleroderma Trials and Research group (EUSTAR) Ann Rheum Dis. 2009;68:620–8. doi: 10.1136/ard.2008.096677. [DOI] [PubMed] [Google Scholar]
- 29.Derk CT, Grace E, Shenin M, et al. A prospective open-label study of mycophenolate mofetil for the treatment of diffuse systemic sclerosis. Rheumatology. 2009;48:1595–9. doi: 10.1093/rheumatology/kep295. [DOI] [PubMed] [Google Scholar]
- 30.Gerbino AJ, Goss CH, Molitor JA. Effect of mycophenolate mofetil on pulmonary function in scleroderma-associated interstitial lung disease. Chest. 2008;133:455–60. doi: 10.1378/chest.06-2861. [DOI] [PubMed] [Google Scholar]
- 31.Pakas I, Ioannidis JP, Malagari K, et al. Cyclophosphamide with low or high dose prednisolone for systemic sclerosis lung disease. J Rheumatol. 2002;29:298–304. [PubMed] [Google Scholar]
- 32.Steen VD, Medsger TA., Jr Case-control study of corticosteroids and other drugs that either precipitate or protect from the development of scleroderma renal crisis. Arthritis Rheum. 1998;41:1613–9. doi: 10.1002/1529-0131(199809)41:9<1613::AID-ART11>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 33.Aggarwal R, Nagaraja V, Khanna D. B-cell targeted therapies in systemic sclerosis and inflammatory myopathies. In: Bosch X, Ramos-Casals M, Khamashta MA, editors. Drugs Targeting B-Cells in Autoimmune Diseases. Basel, Switzerland: Springer; 2014. pp. 153–80. [Google Scholar]
- 34.Seibold JR, Denton CP, Furst DE, et al. Randomized, prospective, placebo-controlled trial of bosentan in interstitial lung disease secondary to systemic sclerosis. Arthritis Rheum. 2010;62:2101–8. doi: 10.1002/art.27466. [DOI] [PubMed] [Google Scholar]
- 35.Khanna D, Saggar R, Mayes MD, et al. A one-year, phase I/IIa, open-label pilot trial of imatinib mesylate in the treatment of systemic sclerosis-associated active interstitial lung disease. Arthritis Rheum. 2011;63:3540–6. doi: 10.1002/art.30548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khanna DT, Tashkin DP. Treatment of interstitial lung disease. In: Varga J, Denton CP, Wigley FM, editors. Scleroderma: From Pathogenesis to comprehensive Management. New York: Springer; 2012. [Google Scholar]
- 37.Goh NS, Veeraraghavan S, Desai SR, et al. Bronchoalveolar lavage cellular profiles in patients with systemic sclerosis-associated interstitial lung disease are not predictive of disease progression. Arthritis Rheum. 2007;56:2005–12. doi: 10.1002/art.22696. [DOI] [PubMed] [Google Scholar]
- 38.Burt RK, Shah SJ, Dill K, et al. Autologous non-myeloablative haemopoietic stem-cell transplantation compared with pulse cyclophosphamide once per month for systemic sclerosis (ASSIST): an open-label, randomised phase 2 trial. Lancet. 2011;378:498–506. doi: 10.1016/S0140-6736(11)60982-3. [DOI] [PubMed] [Google Scholar]
- 39.Van Laar JM, Farge D, Sont JK, et al. Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial. JAMA. 2014;311:2490–8. doi: 10.1001/jama.2014.6368. [DOI] [PubMed] [Google Scholar]
- 40.Hachulla E, Carpentier P, Gressin V, et al. Risk factors for death and the 3-year survival of patients with systemic sclerosis: the French ItinerAIR-Sclerodermie study. Rheumatology. 2009;48:304–8. doi: 10.1093/rheumatology/ken488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Badesch DB, Tapson VF, McGoon MD, et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann Intern Med. 2000;132:425–34. doi: 10.7326/0003-4819-132-6-200003210-00002. [DOI] [PubMed] [Google Scholar]
- 42.Oudiz RJ, Schilz RJ, Barst RJ, et al. Treprostinil, a prostacyclin analogue, in pulmonary arterial hypertension associated with connective tissue disease. Chest. 2004;126:420–7. doi: 10.1378/chest.126.2.420. [DOI] [PubMed] [Google Scholar]
- 43.Khanna D, Gladue H, Channick R, et al. Recommendations for screening and detection of connective tissue disease-associated pulmonary arterial hypertension. Arthritis Rheum. 2013;65:3194–201. doi: 10.1002/art.38172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Galie N, Corris PA, Frost A, et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62(Suppl):D60–72. doi: 10.1016/j.jacc.2013.10.031. [DOI] [PubMed] [Google Scholar]
- 45.McLaughlin VV, Gaine SP, Howard LS, et al. Treatment goals of pulmonary hypertension. J Am Coll Cardiol. 2013;62(Suppl):D73–81. doi: 10.1016/j.jacc.2013.10.034. [DOI] [PubMed] [Google Scholar]
- 46.Ingraham K, Steen VD. Morbidity of digital tip ulcerations in scleroderma. Arthritis Rheum. 2006;54(Suppl 9):P578. [Google Scholar]
- 47.Herrick AL. Pathogenesis of Raynaud’s phenomenon. Rheumatology. 2005;44:587–96. doi: 10.1093/rheumatology/keh552. [DOI] [PubMed] [Google Scholar]
- 48.Thompson AE, Shea B, Welch V, Fenlon D, Pope JE. Calcium-channel blockers for Raynaud’s phenomenon in systemic sclerosis. Arthritis Rheum. 2001;44:1841–7. doi: 10.1002/1529-0131(200108)44:8<1841::AID-ART322>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 49.Rademaker M, Cooke ED, Almond NE, et al. Comparison of intravenous infusions of iloprost and oral nifedipine in treatment of Raynaud’s phenomenon in patients with systemic sclerosis: a double blind randomised study. BMJ. 1989;298:561–4. doi: 10.1136/bmj.298.6673.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Teh LS, Manning J, Moore T, et al. Sustained-release transdermal glyceryl trinitrate patches as a treatment for primary and secondary Raynaud’s phenomenon. Br J Rheumatol. 1995;34:636–41. doi: 10.1093/rheumatology/34.7.636. [DOI] [PubMed] [Google Scholar]
- 51.Wigley FM, Seibold JR, Wise RA, McCloskey DA, Dole WP. Intravenous iloprost treatment of Raynaud’s phenomenon and ischemic ulcers secondary to systemic sclerosis. J Rheumatol. 1992;19:1407–14. [PubMed] [Google Scholar]
- 52.Biondi ML, Marasini B, Bassani C, Agastoni A. Increased plasma endothelin levels in patients with Raynaud’s phenomenon. N Engl J Med. 1991;324:1139–40. [PubMed] [Google Scholar]
- 53.Korn JH, Mayes M, Matucci Cerinic M, et al. Digital ulcers in systemic sclerosis: prevention by treatment with bosentan, an oral endothelin receptor antagonist. Arthritis Rheum. 2004;50:3985–93. doi: 10.1002/art.20676. [DOI] [PubMed] [Google Scholar]
- 54.Matucci-Cerinic M, Denton CP, Furst DE, et al. Bosentan treatment of digital ulcers related to systemic sclerosis: results from the RAPIDS-2 randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 2011;70:32–8. doi: 10.1136/ard.2010.130658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fries R, Shariat K, von Wilmowsky H, Bohm M. Sildenafil in the treatment of Raynaud’s phenomenon resistant to vasodilatory therapy. Circulation. 2005;112:2980–5. doi: 10.1161/CIRCULATIONAHA.104.523324. [DOI] [PubMed] [Google Scholar]
- 56.Herrick AL, van den Hoogen F, Gabrielli A, et al. Modified-release sildenafil reduces Raynaud’s phenomenon attack frequency in limited cutaneous systemic sclerosis. Arthritis Rheum. 2011;63:775–82. doi: 10.1002/art.30195. [DOI] [PubMed] [Google Scholar]
- 57.Brueckner CS, Becker MO, Kroencke T, et al. Effect of sildenafil on digital ulcers in systemic sclerosis: analysis from a single centre pilot study. Ann Rheum Dis. 2010;69:1475–8. doi: 10.1136/ard.2009.116475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gore J, Silver R. Oral sildenafil for the treatment of Raynaud’s phenomenon and digital ulcers secondary to systemic sclerosis. Ann Rheum Dis. 2005;64:1387. doi: 10.1136/ard.2004.034488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shenoy PD, Kumar S, Jha LK, et al. Efficacy of tadalafil in secondary Raynaud’s phenomenon resistant to vasodilator therapy: a double-blind randomized cross-over trial. Rheumatology. 2010;49:2420–8. doi: 10.1093/rheumatology/keq291. [DOI] [PubMed] [Google Scholar]
- 60.Coleiro B, Marshall SE, Denton CP, et al. Treatment of Raynaud’s phenomenon with the selective serotonin reuptake inhibitor fluoxetine. Rheumatology. 2001;40:1038–43. doi: 10.1093/rheumatology/40.9.1038. [DOI] [PubMed] [Google Scholar]
- 61.Gliddon AE, Dore CJ, Black CM, et al. Prevention of vascular damage in scleroderma and autoimmune Raynaud’s phenomenon: a multicenter, randomized, double-blind, placebo-controlled trial of the angiotensin-converting enzyme inhibitor quinapril. Arthritis Rheum. 2007;56:3837–46. doi: 10.1002/art.22965. [DOI] [PubMed] [Google Scholar]
- 62.Tosi S, Marchesoni A, Messina K, et al. Treatment of Raynaud’s phenomenon with captopril. Drugs Exp Clin Res. 1987;13:37–42. [PubMed] [Google Scholar]
- 63.Dziadzio M, Denton CP, Smith R, et al. Losartan therapy for Raynaud’s phenomenon and scleroderma: clinical and biochemical findings in a fifteen-week, randomized, parallel-group, controlled trial. Arthritis Rheum. 1999;42:2646–55. doi: 10.1002/1529-0131(199912)42:12<2646::AID-ANR21>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 64.Drake DB, Kesler RW, Morgan RF. Digital sympathectomy for refractory Raynaud’s phenomenon in an adolescent. J Rheumatol. 1992;19:1286–8. [PubMed] [Google Scholar]
- 65.Tomaino MM, Goitz RJ, Medsger TA. Surgery for ischemic pain and Raynaud’s phenomenon in scleroderma: a description of treatment protocol and evaluation of results. Microsurgery. 2001;21:75–9. doi: 10.1002/micr.1013. [DOI] [PubMed] [Google Scholar]
- 66.Yee AM, Hotchkiss RN, Paget SA. Adventitial stripping: a digit saving procedure in refractory Raynaud’s phenomenon. J Rheumatol. 1998;25:269–76. [PubMed] [Google Scholar]
- 67.Kotsis SV, Chung KC. A systematic review of the outcomes of digital sympathectomy for treatment of chronic digital ischemia. J Rheumatol. 2003;30: 1788–92. [PubMed] [Google Scholar]
- 68.Iorio ML, Masden DL, Higgins JP. Botulinum toxin A treatment of Raynaud’s phenomenon: a review. Semin Arthritis Rheum. 2012;41:599–603. doi: 10.1016/j.semarthrit.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 69.Steen VD, Costantino JP, Shapiro AP, Medsger TA., Jr Outcome of renal crisis in systemic sclerosis: relation to availability of angiotensin converting enzyme (ACE) inhibitors. Ann Intern Med. 1990;113:352–7. doi: 10.7326/0003-4819-113-5-352. [DOI] [PubMed] [Google Scholar]
- 70.Steen VD, Medsger TA., Jr Long-term outcomes of scleroderma renal crisis. Ann Intern Med. 2000;133:600–3. doi: 10.7326/0003-4819-133-8-200010170-00010. [DOI] [PubMed] [Google Scholar]
- 71.Denton CP, Black CM. Scleroderma—clinical and pathological advances. Best Pract Res Clin Rheumatol. 2004;18:271–90. doi: 10.1016/j.berh.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 72.Hudson M, Baron M, Tatibouet S, et al. Exposure to ACE inhibitors prior to the onset of scleroderma renal crisis—results from the International Scleroderma Renal Crisis Survey. Semin Arthritis Rheum. 2014;43:666–72. doi: 10.1016/j.semarthrit.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 73.Hudson M, Baron M, Lo E, et al. An international, web-based, prospective cohort study to determine whether the use of ACE inhibitors prior to the onset of scleroderma renal crisis is associated with worse outcomes-methodology and preliminary results. Int J Rheumatol. 2010;2010:347402. doi: 10.1155/2010/347402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Penn H, Quillinan N, Khan K, et al. Targeting the endothelin axis in scleroderma renal crisis: rationale and feasibility. QJM. 2013;106:839–48. doi: 10.1093/qjmed/hct111. [DOI] [PubMed] [Google Scholar]
- 75.DeMarco PJ, Weisman MH, Seibold JR, et al. Predictors and outcomes of scleroderma renal crisis: the high-dose versus low-dose D-penicillamine in early diffuse systemic sclerosis trial. Arthritis Rheum. 2002;46:2983–9. doi: 10.1002/art.10589. [DOI] [PubMed] [Google Scholar]
- 76.Helfrich DJ, Banner B, Steen VD, Medsger TA., Jr Normotensive renal failure in systemic sclerosis. Arthritis Rheum. 1989;32:1128–34. doi: 10.1002/anr.1780320911. [DOI] [PubMed] [Google Scholar]
- 77.Teixeira L, Servettaz A, Mehrenberger M, et al. [Scleroderma renal crisis] Presse Med. 2006;35(12 Pt 2):1966–74. doi: 10.1016/s0755-4982(06)74931-4. [DOI] [PubMed] [Google Scholar]
- 78.Santiago B, Gutierrez-Canas I, Dotor J, et al. Topical application of a peptide inhibitor of transforming growth factor-beta1 ameliorates bleomycin-induced skin fibrosis. J Invest Dermatol. 2005;125:450–5. doi: 10.1111/j.0022-202X.2005.23859.x. [DOI] [PubMed] [Google Scholar]
- 79.Stevenson JP, Kindler HL, Papasavvas E, et al. Immunological effects of the TGFbeta-blocking antibody GC1008 in malignant pleural mesothelioma patients. Oncoimmunology. 2013;2:e26218. doi: 10.4161/onci.26218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tager AM, LaCamera P, Shea BS, et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat Med. 2008;14:45–54. doi: 10.1038/nm1685. [DOI] [PubMed] [Google Scholar]
- 81.Gonzalez EG, Selvi E, Balistreri E, et al. Synthetic cannabinoid ajulemic acid exerts potent antifibrotic effects in experimental models of systemic sclerosis. Ann Rheum Dis. 2012;71:1545–51. doi: 10.1136/annrheumdis-2011-200314. [DOI] [PubMed] [Google Scholar]
- 82.Henderson NC, Arnold TD, Katamura Y, et al. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med. 2013;19:1617–24. doi: 10.1038/nm.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Haque SU, Dashwood MR, Heetun M, et al. Efficacy of the specific endothelin a receptor antagonist zibotentan (ZD4054) in colorectal cancer: a preclinical study. Mol Cancer Ther. 2013;12:1556–67. doi: 10.1158/1535-7163.MCT-12-0975. [DOI] [PubMed] [Google Scholar]
- 84.Quillinan NP, McIntosh D, Vernes J, Haq S, Denton CP. Treatment of diffuse systemic sclerosis with hyperimmune caprine serum (AIMSPRO): a phase II double-blind placebo-controlled trial. Ann Rheum Dis. 2014;73:56–61. doi: 10.1136/annrheumdis-2013-203674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dees C, Akhmetshina A, Zerr P, et al. Platelet-derived serotonin links vascular disease and tissue fibrosis. J Exp Med. 2011;208:961–72. doi: 10.1084/jem.20101629. [DOI] [PMC free article] [PubMed] [Google Scholar]