

Abstract
Recently a novel method for the preparation of 18F-labeled arenes via oxidative [18F]fluorination of easily accessible and sufficiently stable nickel complexes with [18F]fluoride under exceptionally mild reaction conditions was published. The suitability of this procedure for the routine preparation of clinically relevant positron emission tomography (PET) tracers, 6-[18F]fluorodopamine (6-[18F]FDA), 6-[18F]fluoro-l-DOPA (6-[18F]FDOPA) and 6-[18F]fluoro-m-tyrosine (6-[18F]FMT), was evaluated. The originally published base-free method was inoperative. However, a “low base” protocol afforded protected radiolabeled intermediates in radiochemical conversions (RCCs) of 5–18 %. The subsequent deprotection step proceeded almost quantitatively (>95 %). The simple one-pot two-step procedure allowed the preparation of clinical doses of 6-[18F]FDA and 6-[18F]FDOPA within 50 min (12 and 7 % radiochemical yield, respectively). In an unilateral rat model of Parkinsons disease, 6-[18F]FDOPA with high specific activity (175 GBq μmol−1) prepared using the described nickel-mediated radiofluorination was compared to 6-[18F]FDOPA with low specific activity (30 MBq μmol−1) produced via conventional electrophilic radiofluorination. Unexpectedly both tracer variants displayed very similar in vivo properties with respect to signal-to-noise ratio and brain distribution, and consequently, the quality of the obtained PET images was almost identical.
Keywords: [18F]fluoride, nucleophilic aromatic substitution, positron emission tomography (PET), radiopharmaceuticals, radiosynthesis
Introduction
Radiofluorinated aromatic amines and amino acids are important diagnostic tools in modern positron emission tomography (PET) imaging (Figure 1). 6-[18F]Fluoro-3,4-l-dihydroxyphenylalanine (6-[18F]FDOPA, [18F]1 a) is applied in clinical practice as a biomarker of catecholamine synthesis, storage and metabolism, and enables visualization of neuroendocrine tumors as well as the measurement of integrity and function of the nigrostriatal dopaminergic system.1 6-[18F]Fluoro-l-m-tyrosine (6-[18F]FMT, [18F]1 b), a close structural and functional analogue of 6-[18F]FDOPA with improved biodistribution properties, is also used in the clinic for the same purposes.2
Figure 1.
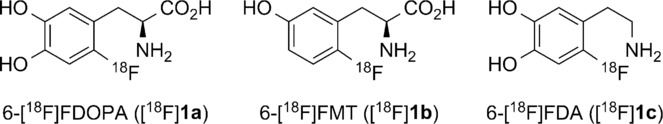
Structures of 6-[18F]FDOPA ([18F]1 a), 6-[18F]FMT ([18F]1 b), and 6-[18F]FDA ([18F]1 c).
In addition to 6-[18F]FDOPA and 6-[18F]FMT, 6-[18F]FDA ([18F]1 c) has also gained importance for the detection of neuroendocrine tumors such as pheochromocytomas and paragangliomas.3 However, until quite recently, widespread application of these compounds has been hampered by a paucity of effective production routes, since incorporation of [18F]fluoride into electron-rich aromatic systems is challenging. Electrophilic fluorodestannylation using [18F]F2 or [18F]CH3COOF is routinely used for the preparation of these tracers.4 However, this method suffers from several disadvantages. [18F]F2 gas target handling is difficult owing to the toxicity and corrosive character of fluorine. Moreover, [19F]F2 has to be applied as a carrier gas for 18F to compete with surface adsorption enabling complete recovery of radioactivity from the target. As a consequence, specific activity (SA) of [18F]F2 does not exceed 350–500 MBq μmol−1.5 Accordingly, production of tracers with high SA via electrophilic radiofluorination is impossible. In order to obtain radiotracers with high SA and avoid F2 target gas handling, radiosyntheses should start from [18F]fluoride. Wagner et al.6 proposed a synthesis route for 6-[18F]FDOPA via isotopic 18/19F exchange using a formyl-activated precursor followed by Baeyer–Villiger oxidation and final hydrolysis of the radiolabeled intermediate. 6-[18F]FMT and several other aromatic amino acids were also successfully prepared using this synthetic route.7
Various multistep radiosyntheses of no-carrier-added (n.c.a.) 6-[18F]FDOPA using nucleophilic [18F]fluoride have been reported. All these procedures start with the preparation of a suitably protected 6-[18F]fluoro-3,4-dihydroxybenzaldehyde (usually, 6-[18F]fluoroveratraldehyde).8 The latter is converted into the corresponding benzyl bromide or iodide, which is subsequently used for the alkylation of the chiral glycine equivalent or achiral glycine derivative with or without a chiral phase transfer catalyst.9 Finally, acidolytic cleavage of protecting groups (typically under harsh reaction conditions) followed by HPLC or if necessary chiral HPLC purification is carried out. 6-[18F]FDOPA is obtained in moderate radiochemical yields (RCYs), with SA >37 GBq μmol−1 and high enantiomeric purity. Several procedures for the preparation of 6-[18F]FDA from [18F]fluoride have also been reported.4,10 Unfortunately, similarly to those for 6-[18F]FDOPA and 6-[18F]FMT, the majority of these syntheses consist of numerous laborious reaction and operation steps, and are therefore difficult to implement in routine radiopharmaceutical production. Accordingly, convenient preparation procedures for the preparation of [18F]1 a–c via operationally simple nucleophilic 18F-labeling of electron-rich arenes are highly sought after.
Recently Lee et al. reported a radiofluorination procedure using a palladium-based fluoride-derived electrophilic radiofluorination reagent for the synthesis of 18F-labeled aromatic compounds (Scheme 1 A).11a It comprises the reaction of a radiofluorinated PdIV complex prepared in advance with an appropriate arylpalladium(II) complex yielding, after reductive elimination, the corresponding 18F-labeled arene. A further development of this method involves radiolabeling of an arylnickel(II) complex with a radiofluorination agent generated in situ from a hypervalent iodine oxidant [bis(onio)-substituted aryliodine(III), 2] and [18F]fluoride (Scheme 1 B).11b According to the latter procedure, radiolabeling was carried out in the presence of only 1 mg of the corresponding nickel complex at room temperature under ambient atmosphere within less than 1 min to yield radiolabeled arenes with >37 GBq μmol−1 SA in fair to moderate radiochemical conversions (RCCs).12 According to this publication, neither base nor azeotropic drying were necessary. This method was applied to prepare protected 6-[18F]FDOPA ([18F]3 a) from the corresponding nickel complex (4 a) in 15 % RCC (Scheme 1 B). The exceptional operational simplicity, very mild reaction conditions and short reaction time prompted us to study the applicability of this method for the preparation of clinically relevant doses of [18F]1 a–c. Finally, high and low SA preparations of 6-[18F]FDOPA obtained via nickel-mediated and conventional electrophilic radiofluorination, respectively, were compared in a rat model of hemi-Parkinsons disease.
Scheme 1.
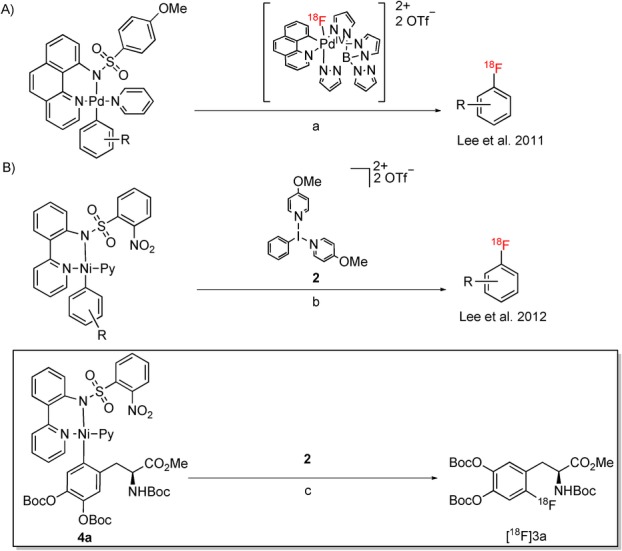
Palladium- and nickel-mediated preparation of [18F]fluoroarenes according to Lee et al. (A and B, respectively).11 Reagents and conditions: a) acetone, 85 °C, 10 min; b) aq [18F]fluoride, 18-crown-6, MeCN, RT, <1 min; c) aq [18F]fluoride, 18-crown-6, MeCN, RT, <1 min, RCC=15±7 %; RCC=radiochemical conversion.12
Results and Discussion
At the outset, we tried to prepare protected 6-[18F]FDA ([18F]1 c) from corresponding nickel complex 4 c (Scheme 2). Unexpectedly, under base-free conditions originally reported by Lee et al.,11b no product formation was observed; only [18F]F− was detected in the reaction mixture. Similarly, if azeotropically dried [18F]KF/18-crown-6 prepared according to the conventional nucleophilic radiofluorination protocol11b using potassium carbonate (2.5–3.2 mg) was applied, no 18F-incorporation took place. We assumed that oxidant 2 is prone to decomposition under strongly basic conditions. Consequently, the applicability of our “low base” protocol initially developed for copper-mediated radiofluorination was tested.13
Figure 2.
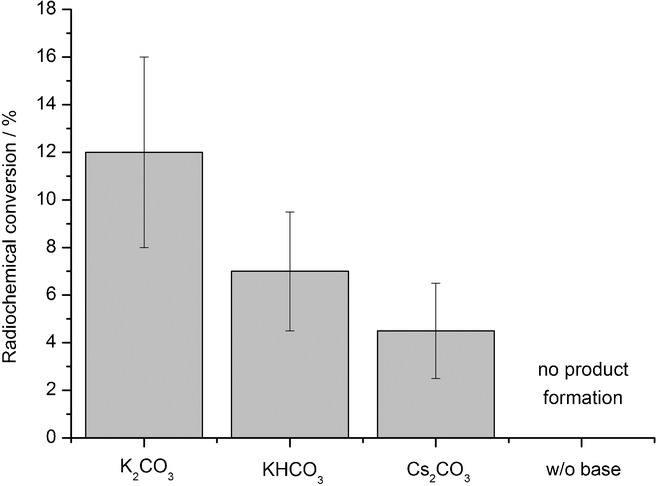
Influence of base on radiochemical conversion (RCC) of [18F]3 c. The appropriate [18F]fluoride salt/18-crown-6 complex (50–500 MBq) was prepared using the corresponding base (1.16 μmol) in MeOH (200 μL). MeOH was evaporated, and the residue taken up in MeCN (900 μL). The solution was added to nickel complex 4 c (5 μmol) and 2 (1 equiv), and the reaction mixture was stirred for 5 min at RT. Thereafter, water (5 mL) was added, the mixture was vigorously stirred for 1 min and then analyzed by radio-HPLC. Values represent the mean± standard deviation (SD) of at least three experiments.
Scheme 2.
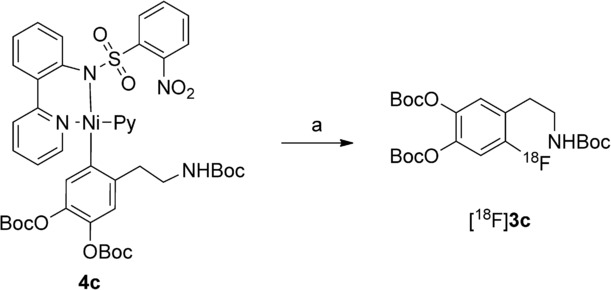
Radiosynthesis of protected [18F]FDA ([18F]3 c). Reagents and conditions: a) [18F]fluoride, base, 18-crown-6, oxidant 2, MeCN, RT, 1—20 min.
18F− was trapped on an anion-exchange resin and then eluted with a methanolic solution of potassium carbonate into a reaction vial containing 18-crown-6. A small amount of potassium carbonate (0.16 mg) was sufficient to recover 18F− quantitatively (>98 %). Since low-boiling methanol could be completely removed at 70–80 °C within 2–3 min, the time of drying of [18F]KF/18-crown-6 complex was significantly decreased. The residue was taken up in acetonitrile and added to nickel complex 4 c and oxidant 2. The resulting mixture was stirred at ambient temperature under inert conditions. Application of 4 c (1 mg) according to the literature11b afforded 18F-labeled protected 6-FDA [18F]3 c in only 1–4 % RCC after 1 min. In contrast, if a greater amount of nickel complex 4 c (5–10 mg) was utilized, [18F]3 c was formed in 12±4 % RCC within 5 min. At elevated temperatures (>30 °C), no 18F incorporation was observed. Use of other bases such as cesium carbonate and potassium bicarbonate also provided [18F]3 c, but in lower RCCs (5±2 and 7±3 %, respectively) (Figure 2).
In contrast to the literature,11b formation of [18F]3 c took place only in the presence of a base. This finding prompted us to optimize this radiolabeling procedure using nickel complex 4 c as a model substrate with respect to other reaction parameters such as oxidant/precursor ratio, reaction time and solvent.
The oxidant/precursor ratio strongly affected the formation of [18F]3 c (Figure 3). Application of an excess of radiolabeling precursor 4 c afforded only traces of [18F]3 c. If equimolar amounts of oxidizing agent 2 and nickel complex 4 c were used, the RCC value of [18F]3 c amounted to 6±3 %. If the oxidant to nickel complex molar ratio was 1.3, a maximal RCC value of 18±10 % was obtained. Above a ratio of 1.5, a considerable decrease in RCC values was observed.
Figure 3.
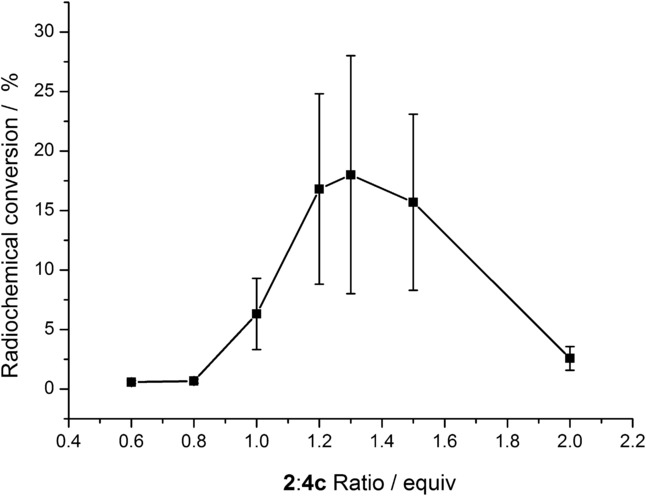
Radiochemical conversion (RCC) of [18F]3 c as a function of the oxidant/precursor ratio. A solution of [18F]KF/18-crown-6 (50–500 MBq) in MeCN (900 μL) was added to nickel complex 4 c (5 μmol) and 2 (0.6–2.0 equiv), and the reaction mixture was stirred for 5 min at RT. Thereafter, water (5 mL) was added, the mixture was vigorously stirred for 1 min and then analyzed by radio-HPLC. Values represent the mean± standard deviation (SD) of at least three experiments.
Radiofluorination of 4 c was tested in different solvents (Figure 4). The highest RCC values were achieved in anhydrous acetonitrile. In contrast to literature reports,11b the presence of a low amount of water (1 %) in the reaction mixture caused a significant decrease in RCC values. In sulfolane, diglyme and dimethylformamide (DMF), formation of [18F]3 c was significantly lower in comparison to that observed in acetonitrile. Dependency of RCC of 4 c on reaction time was also determined (Figure 5). Under optimized conditions, reaction times of 1 and 5 min afforded [18F]3 c in RCCs of 7±3 % and 18±10 %, respectively. A further extension of the reaction time did not increase the RCC.
Figure 4.
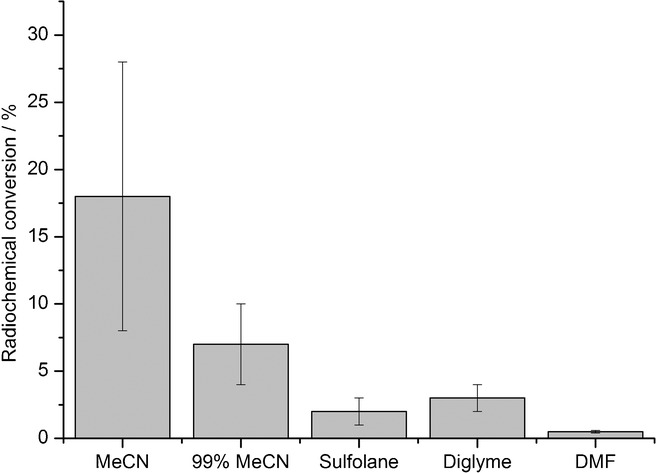
Radiochemical conversion (RCC) of [18F]3 c in different solvents. A solution of [18F]KF/18-crown-6 (50–500 MBq) in the corresponding solvent (900 μL) was added to nickel complex 4 c (5 μmol) and 2 (6.5 μmol), and the reaction mixture stirred for 5 min at RT. Thereafter, water (5 mL) was added, the mixture was vigorously stirred for 1 min and then analyzed by radio-HPLC. Values represent the mean± standard deviation (SD) of at least three experiments.
Figure 5.
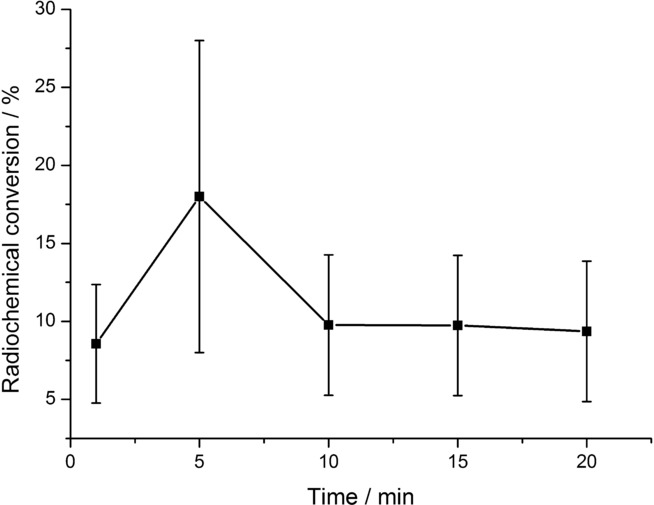
Dependence of radiochemical conversion (RCC) of [18F]4 c on reaction time. A solution of [18F]KF/18-crown-6 (50–500 MBq) in MeCN (900 μL) was added to nickel complex 4 c (5 μmol) and 2 (1.3 equiv), and the mixture was stirred for the corresponding time at RT. Thereafter, water (5 mL) was added, the mixture was vigorously stirred for 1 min and then analyzed by radio-HPLC. Values represent the mean± standard deviation (SD) of at least three experiments.
During our experiments, we noticed the extreme moisture sensitivity of the oxidizing agent 2.14 This was, for example, the reason for the high statistical deviations of RCC values that were observed in the optimization experiments. In an attempt to overcome this problem, we prepared several hypervalent iodine compounds, such as 5,15 and iodosylarenes 6 a,b16 as less moisture-sensitive alternatives to 2 (Figure 6). However, application of these compounds as oxidants did not afford incorporation of 18F.
Figure 6.
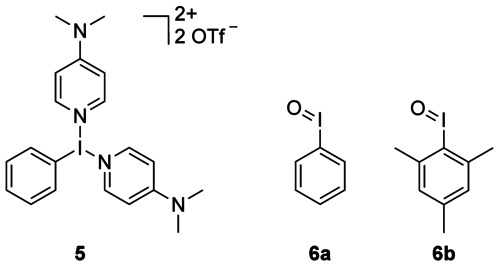
Hypervalent iodine oxidants tested as possible alternatives to 2.
With optimized reaction conditions for radiolabeling of 4 c in hand, we tested 18F-radiofluorination of nickel complexes 4 a11b and 4 b. Accordingly, protected derivatives of 6-[18F]FDOPA and 6-[18F]FMT, [18F]3 a and [18F]3 b, were prepared in 13±2 % and 9±1 % RCC, respectively. The next preparation step should comprise the deprotection of the corresponding 18F-labeled intermediates, [18F]3 a–c, to the desired PET tracers. Initially, trifluoroacetic acid (TFA) at 80 and 130 °C was tested as a deprotection agent. Under these conditions, complete decomposition of radiolabeled intermediates was observed with 18F− as the only detectable radioactive product. In contrast, quantitative deprotection of [18F]3 c was accomplished using 12 m aq HCl at 130 °C after just 5 min. In the case of [18F]3 a and [18F]3 b, cleavage of the methyl ester group required a slightly extended deprotection time of 10 min.
Once the protocol for the radiosynthesis of [18F]3 a–c via nickel-mediated radiofluorination had been established, we tried to implement a scale-up synthesis procedure to obtain the corresponding PET tracers in amounts sufficient for biological evaluation. After accomplishment of the radiofluorination step, acetonitrile was evaporated under a gentle stream of argon, and the residue was redissolved in 12 m aq HCl. After hydrolysis had been completed, the bulk of HCl was removed by coevaporation with acetone. The crude products were purified by HPLC to give the corresponding PET tracers as ready-to-use solutions in a 4 % ethanolic phosphate buffer. Application of a one-pot protocol allowed us to minimize loss of radioactivity due to surface adsorption and to decrease radiosynthesis time significantly.
Using this procedure, [18F]1 a, [18F]1 b17 and [18F]1 c were isolated in RCYs of 7±1, 5±1 and 12±2 %, respectively, with excellent radiochemical and chemical purity (Scheme 3). Specific activities of [18F]1 a and [18F]1 c were determined to 175 and 60 GBq μmol−1, respectively.18
Scheme 3.
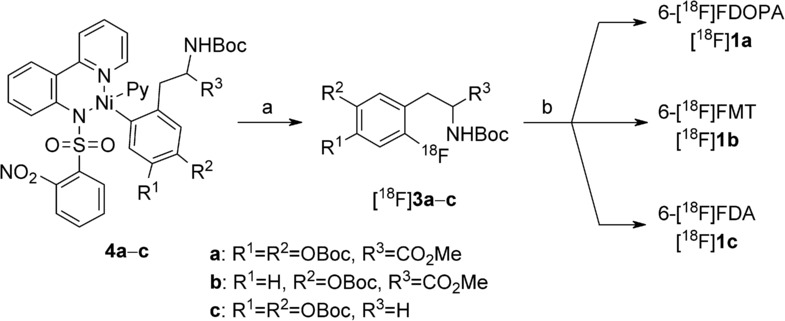
Preparation of 6-[18F]FDOPA ([18F]1 a), 6-[18F]FMT ([18F]1 b) and 6-[18F]FDOPA ([18F]1 c) via radiofluorination of nickel complexes 4 a–c. Reagents and conditions: a) [18F]KF/18-crown-6, oxidant 2, MeCN, RT, 5 min; b) 12 m aq HCl, 130 °C, 5–10 min. [18F]1 a, RCY=7 %, 220 MBq from 6.3 GBq 18F−, SA=175 GBq μmol−1; [18F]1 b, RCY=5 %; [18F]1 c, RCY=12 %, 250 MBq from 4 GBq 18F−, SA=60 GBq μmol−1; RCY=radiochemical yield;12 SA=specific activity.
Finally, we studied, whether the specific activity of 6-[18F]FDOPA affects PET imaging of dopaminergic activity in brain. For this purpose, n.c.a. 6-[18F]FDOPA produced via nickel-mediated radiofluorination and carried-added (c.a.) 6-[18F]FDOPA prepared via conventional electrophilic radiofluorination were compared in an unilateral rat model of hemi-Parkinsons disease.19 Accordingly, loss of dopaminergic midbrain neurons was induced by stereotaxic injection of neurotoxic 6-hydroxydopamine (6-OHDA) into the medial forebrain bundle.20 Six weeks after 6-OHDA injection, rats pretreated with carbidopa were injected into the lateral tail vein with 56–74 MBq of n.c.a 6-[18F]FDOPA and measured. Eight weeks after 6-OHDA injection, the same animals were measured using c.a 6-[18F]FDOPA (67–72 MBq) (Figure 7).
Figure 7.
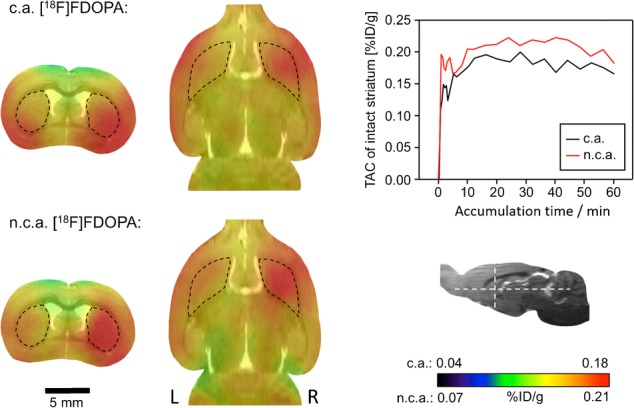
No-carrier-added (n.c.a.) versus carrier-added (c.a.) 6-[18F]FDOPA: magnetic resonance/positron emission tomography (MR/PET) imaging in a rat model of Parkinsons disease. Two transverse (left column) and two horizontal images (middle column) from the same animal with left side lesion induced by the injection of 6-hydroxydopamine (6-OHDA) are shown. Left and right striatum are indicated by dashed black outlines. 6-OHDA-Induced lesion is visible as a reduction of the PET signal in the left striatum. Section levels are indicated by white dashed lines in the insert (bottom right). The time activity curves (TAC; top right) for both images were taken from the intact right striatum.
Specific activity of the c.a. tracer (30 MBq μmol−1) was more than three orders of magnitude lower than that of the n.c.a. compound. Both tracer variants were taken up by the intact striatum with similar kinetics (time activity curves in Figure 7). Mean striatal standardized uptake values (SUVs) (%ID/g) were 0.22 and 0.21 for n.c.a. and c.a. 6-[18F]FDOPA, respectively. In addition, the striatum-to-cerebellum ratio was determined to be independent of the specific activity of the tracer. In both cases, the dopaminergic lesion was clearly visible with an identical ipsi-to-contralateral striatal ratio.
This somewhat unexpected result could be explained by the high capacity and/or high competitiveness of the main biochemical processes responsible for the accumulation of 6-[18F]FDOPA and its metabolites in dopaminergic neurons. In this case, competition between 6-[18F]FDOPA and 6-FDOPA should be negligible compared with that between 6-[18F]FDOPA and other competitors.
6-[18F]FDOPA metabolism in brain comprises five main steps. Initially, the radiotracer is taken up across the blood–brain barrier (BBB) mainly via l-type amino acid transporter (LAT-1).21 LAT-1 is also responsible for brain uptake of proteinogenic neutral amino acids like Phe, Tyr, Val, Ile, Leu, and Trp.22 Therefore, the competition between 6-[18F]FDOPA and other amino acids is significantly higher relative to that with 6-FDOPA. In striatal neurons, 6-[18F]FDOPA is decarboxylated into 6-[18F]FDA.23 This step is catalyzed by aromatic l-amino acid decarboxylase (AADC) with very broad substrate specificity and high capacity.24 6-[18F]FDA is transported into the vesicles by the vesicular monoamine transporter type 2 (VMAT2).23 Since VMAT2 is responsible for detoxification of toxic amines like 6-FDA,25 its transport capacity should be high. Subsequently, stored 6-[18F]FDA is transported to the synaptic cleft by exocytosis and taken up there again by dopamine transporters (DAT). Finally, cytosolic 6-[18F]FDA is metabolized to 6-[18F]fluoro-3,4-dihydroxyphenylacetic acid (6-[18F]FDOPAC), 6-[18F]fluoro-3-methoxytyramine (6-[18F]FMTA) and 6-[18F]fluorohomovanilic acid (6-[18F]FHVA) by monoamine oxidase (MAO) and catechol-O-methyltransferase (COMT).26 These enzymes also exhibit broad substrate specificity and high capacity. The acidic metabolites, 6-[18F]FDOPAC and 6-[18F]FHVA, are rapidly cleared from brain.27 Vesicular stored 6-[18F]FDA as well as its cytosolic and extracellular metabolites are responsible for more than 90 % of the striatal PET signal generated by 6-[18F]FDOPA in brain at 10–90 min p.i.26
Conclusions
Nickel-mediated radiofluorination under optimized “low base” conditions enabled fast access to n.c.a. 6-[18F]FDA, 6-[18F]FDOPA, and 6-[18F]FMT via a one-pot two-step procedure. Owing to the simplicity, this procedure should be well suited for automation given that a less moisture-sensitive alternative for oxidant 2 will be found. Furthermore, it was shown that, in a Parkinsons rat model, biodistribution and, consequently, imaging properties of 6-[18F]FDOPA were independent from specific activity.28
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
References
- 1a.Juhasz C, Dwivedi S, Kamson DO, Michelhaugh SK, Mittal S. Mol. Imaging. 2014;13:1–16. doi: 10.2310/7290.2014.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b.Varrone A, Halldin C. J. Nucl. Med. 2010;51:1331–1334. doi: 10.2967/jnumed.109.065656. [DOI] [PubMed] [Google Scholar]
- 1c.Politis M. Nat. Rev. Neurol. 2014;10:708–722. doi: 10.1038/nrneurol.2014.205. [DOI] [PubMed] [Google Scholar]
- 1d.Berti V, Pupi A, Mosconi L. Ann. N. Y. Acad. Sci. 2011;1228:93–108. doi: 10.1111/j.1749-6632.2011.06025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1e.Cropley VL, Fujita M, Innis RB, Nathan PJ. Biol. Psychiatry. 2006;59:898–907. doi: 10.1016/j.biopsych.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Kaira K, Oriuchi N, Shimizu K, Tominaga H, Yanagitani N, Sunaga N, Ishizuka T, Kanai Y, Mori M, Endo K. J. Nucl. Med. 2009;50:1770–1776. doi: 10.2967/jnumed.109.066837. [DOI] [PubMed] [Google Scholar]
- 3a.Timmers HJ, Eisenhofer G, Carrasquillo JA, Chen CC, Whatley M, Ling A, Adams KT, Pacak K. Clin. Endocrinol. 2009;71:11–17. doi: 10.1111/j.1365-2265.2008.03496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b.Taieb D, Neumann H, Rubello D, Al-Nahhas A, Guillet B, Hindie E. J. Nucl. Med. 2012;53:264–274. doi: 10.2967/jnumed.111.098152. [DOI] [PubMed] [Google Scholar]
- Pretze M, Wängler C, Wängler B. BioMed Res. Int. 2014;2014:674063. doi: 10.1155/2014/674063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess E, Blessing G, Coenen HH, Qaim SM. Appl. Radiat. Isot. 2000;52:1431–1440. doi: 10.1016/s0969-8043(99)00248-1. [DOI] [PubMed] [Google Scholar]
- Wagner FM, Ermert J, Coenen HH. J. Nucl. Med. 2009;50:1724–1729. doi: 10.2967/jnumed.109.063297. [DOI] [PubMed] [Google Scholar]
- 7a.Castillo Meleán J. PhD Thesis, Universität zu Köln, Köln (Germany) 2011.
- 7b.Castillo Meleán J, Ermert J, Coenen HH. Org. Biomol. Chem. 2011;9:765–769. doi: 10.1039/c0ob00440e. [DOI] [PubMed] [Google Scholar]
- Wadsak W, Wirl-Sagadin B, Mitterhauser M, Mien L-K, Ettlinger DE, Keppler BK, Dudczak R, Kletter K. Appl. Radiat. Isot. 2006;64:355–359. doi: 10.1016/j.apradiso.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 9a.Lemaire C, Damhaut P, Plenevaux A, Comar D. J. Nucl. Med. 1994;35:1996–2002. [PubMed] [Google Scholar]
- 9b.Shen B, Ehrlichmann W, Uebele M, Machulla HJ, Reischl G. Appl. Radiat. Isot. 2009;67:1650–1653. doi: 10.1016/j.apradiso.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 10a.Vavere A, Butch E, Shulkin B, Snyder S. J. Nucl. Med. 2014;55 (Suppl. 1):1410. [Google Scholar]
- 10b.Ding YS, Fowler JS, Gatley SJ, Dewey SL, Wolf AP, Schlyer DJ. J. Med. Chem. 1991;34:861–863. doi: 10.1021/jm00106a055. [DOI] [PubMed] [Google Scholar]
- 11a.Lee E, Kamlet AS, Powers DC, Neumann CN, Boursalian GB, Furuya T, Choi DC, Hooker JM, Ritter T. Science. 2011;334:639–642. doi: 10.1126/science.1212625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b.Lee E, Hooker JM, Ritter T. J. Am. Chem. Soc. 2012;134:17456–17458. doi: 10.1021/ja3084797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radiochemical conversion (RCC), determined by radio-HPLC, refers to the amount of radiofluoride reacted with the labeling precursor to give the desired 18F-labeled compound. Radiochemical yield (RCY) refers to the isolated yield of the radiochemically and chemically pure radiolabeled compound; all RCYs reported here are corrected for decay
- Zlatopolskiy BD, Zischler J, Krapf P, Zarrad F, Urusova EA, Kordys E, Endepols H, Neumaier B. Chem. Eur. J. 2015;21:5972–5979. doi: 10.1002/chem.201405586. [DOI] [PubMed] [Google Scholar]
- To avoid decomposition, compound 2should be handled and stored (for not longer than two months) in a glove box under strict exclusion of moisture
- Weiss R, Seubert J. Angew. Chem. Int. Ed. Engl. 1994;33:891–893. [Google Scholar]; Angew. Chem. 1994;106 [Google Scholar]
- Huang X, Liu W, Ren H, Neelamegam R, Hooker JM, Groves JT. J. Am. Chem. Soc. 2014;136:6842–6845. doi: 10.1021/ja5039819. [DOI] [PubMed] [Google Scholar]
- In this work, racemic 6-[18m18F]fluoro- -tyrosine (6-[ F]FMT) was prepared
- Complete experimental details for the preparation of the reference compounds and precursors for radiofluorination, as well as radiochemical and biological experiments, together with HPLC chromatograms, NMR spectra, and details of the specific activity calculations are available on the WWW underhttp://dx.doi.org/10.1002/open.201500056
- Sioka C, Fotopoulos A, Kyritsis AP. Eur. J. Nucl. Med. Mol. Imaging. 2010;37:1594–1603. doi: 10.1007/s00259-009-1357-9. [DOI] [PubMed] [Google Scholar]
- Kyono K, Takashima T, Katayama Y, Kawasaki T, Zochi R, Gouda M, Kuwahara Y, Takahashi K, Wada Y, Onoe H, Watanabe Y. EJNMMI research. 2011;1:25. doi: 10.1186/2191-219X-1-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Amo EM, Urtti A, Yliperttula M. Eur. J. Pharm. Sci. 2008;35:161–174. doi: 10.1016/j.ejps.2008.06.015. [DOI] [PubMed] [Google Scholar]
- Boado RJ, Li JY, Nagaya M, Zhang C, Pardridge WM. Proc. Natl. Acad. Sci. USA. 1999;96:12079–12084. doi: 10.1073/pnas.96.21.12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara K, Watabe H, Kumakura Y, Hayashi T, Endres CJ, Minato K, Iida H. Synapse. 2011;65:751–762. doi: 10.1002/syn.20899. [DOI] [PubMed] [Google Scholar]
- Brown WD, Taylor MD, Roberts AD, Oakes TR, Schueller MJ, Holden JE, Malischke LM, DeJesus OT, Nickles RJ. Neurology. 1999;53:1212–1218. doi: 10.1212/wnl.53.6.1212. [DOI] [PubMed] [Google Scholar]
- De La Fuente-Fernández R, Furtado S, Guttman M, Furukawa Y, Lee CS, Calne DB, Ruth TJ, Stoessl AJ. Synapse. 2003;49:20–28. doi: 10.1002/syn.10199. [DOI] [PubMed] [Google Scholar]
- DeJesus OT, Haaparanta M, Solin O, Nickles RJ. Brain Res. 2000;877:31–36. doi: 10.1016/s0006-8993(00)02649-4. [DOI] [PubMed] [Google Scholar]
- Kumakura Y, Cumming P. Neuroscientist. 2009;15:635–650. doi: 10.1177/1073858409338217. [DOI] [PubMed] [Google Scholar]
- While this article was in preparation, Kuik et al. reported that no-carrier-added (n.c.a.) and carried-added (c.a.) [18F]FDOPA offer equal imaging quality in visualization of xenografted neuroendocrine tumors in mice. [Google Scholar]
- Kuik WJ, Kema IP, Brouwers AH, Zijlma R, Neumann KD, Dierckx RA, DiMagno SG, Elsinga PH. J. Nucl. Med. 2015;56:106–112. doi: 10.2967/jnumed.114.145730. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary