

Abstract
The bacterial adhesion lectin LecA is an attractive target for interference with the infectivity of its producer P. aeruginosa. Divalent ligands with two terminal galactoside moieties connected by an alternating glucose-triazole spacer were previously shown to be very potent inhibitors. In this study, we chose to prepare a series of derivatives with various new substituents in the spacer in hopes of further enhancing the LecA inhibitory potency of the molecules. Based on the binding mode, modifications were made to the spacer to enable additional spacer–protein interactions. The introduction of positively charged, negatively charged, and also lipophilic functional groups was successful. The compounds were good LecA ligands, but no improved binding was seen, even though altered thermodynamic parameters were observed by isothermal titration calorimetry (ITC).
Keywords: bacterial lectins, carbohydrates, LecA inhibition, molecular modeling, multivalency, virulence factors
Introduction
Enhancing the binding potency of carbohydrate inhibitors of protein–carbohydrate interactions is an important step towards improved medicinal applications.1 This is mostly the case because of the relatively weak interactions between proteins and carbohydrate ligands. Ensuring multivalency of ligands is a proven method to achieve this.2
LecA is an important virulence factor of Pseudomonas aeruginosa and is involved in bacterial adhesion and biofilm formation.3 The protein has recently become a popular target for the design of multivalent inhibitors.4 The tetrameric protein promotes the adhesion of the bacteria to tissue cell surfaces, thus facilitating subsequent steps such as cell invasion and biofilm formation. Inhibiting bacterial adhesion proteins has the potential to become a mild and less resistance-prone method to treat and prevent bacterial infections.5 Two of the four binding sites, with specificity for galactosides, are relatively close together with a separation of about 26 Å.6 This arrangement has led us to design and synthesize divalent galactoside ligands with well-defined and rigid spacers that should allow a chelation-type divalent binding mode.7 Flexible spacers are commonly used as they are forgiving of imperfect design and usually yield sizeable potency enhancements in multivalent systems.7 They are, however, not optimal as there will be a significant entropy loss upon binding and, moreover, achieving selectivity will be less likely. In our search for an optimal spacer we found compound 1 (Figure 1 a) to be a highly potent divalent ligand with nanomolar inhibitory potency.8a The spacer of this structure contains direct linkages between the glucose moieties and the connected triazoles.8 This arrangement leads to a relatively rigid structure, in which rotations of the components can take place, but the overall geometry remains mostly linear.
Figure 1.
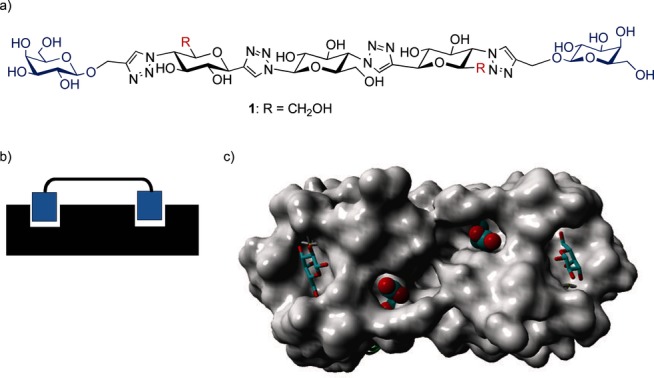
a) Structure of potent divalent LecA inhibitor 1 with the relatively rigid glucose-triazole-based spacer; b) Schematic divalent binding mode of a divalent ligand to two LecA subunits; c) X-ray structure of LecA with bound galactose moieties. The two Asp 47 carboxylates in the spacer path are shown explicitly.6
Most importantly, good solubility in water was observed. We found that three glucose-triazole units was the optimal length for LecA inhibition, while divalent ligands with two and four units showed far inferior inhibition. All data were consistent with a chelating binding mode; especially convincing was the stoichiometry derived from isothermal titration calorimetry (ITC) binding experiments. Furthermore, the short linkage of just a single carbon between the galactoside ligand and the triazole proved to be a major contributing factor to the success of this compound.
In order to further optimize the potency of the compound and to explore the principle of protein–spacer interactions, we now report on our functionalization of the spacer of 1 with various functional groups. The presence of the two carboxylates (Asp 47 in each subunit) is apparent when looking at the path between the two bound galactosides, which the spacer is likely to span on the LecA protein surface when chelating bivalent binding occurs (Figure 1 c). These two carboxylates are likely to be in close proximity to the 6-OH group on the terminal spacer glucoside units, depending on the rotational state of the molecule. This C-6 position can be modified by using the proper protecting group during synthesis.
Molecular modeling was used in order to gauge whether positively charged functional groups at the C-6 position on the terminal spacer glucoside units would be able to interact with the Asp 47 residues. Firstly, ammonium groups were used (as derived from 12, see below). Creating a stabilized conformation with the positive charges in close proximity to the Asp 47 carboxylates was possible, and this orientation was used as the starting point of additional simulations. When running an unrestrained nanosecond molecular dynamics (MD) simulation with explicit water molecules, the stabilized geometry persisted throughout the simulation (see Supporting Information). Especially, the two hydrogen-bonded salt bridges, between the Asp 47 side chains and the ammonium groups of the protonated form of 12, remained within 3 Å and were compatible with a geometry that included two fully bound galactoside ligand units. Such a bound geometry was also possible for a compound that included a pyridinium group (as in 13), but resulted in longer distances between the carboxylate oxygens and the pyridinium nitrogens, due to bigger steric requirements of the pyridinium units. Furthermore, while performing a similar MD simulation, this structure was not maintained, indicating indeed the larger steric requirements of the pyridinium group when facing the protein surface. These experiments lead to the conclusion that the introduction of ammonium groups would be most promising.
We chose to prepare a series of derivatives with various new substituents in the spacer in hopes of further enhancing the LecA inhibitory potency of the molecules. Positively charged substituents were included, as reasoned above. Also, negatively charged and more lipophilic groups were included as these could possibly benefit from interacting with other proximate parts of the protein that would not have been previously obvious.
Results and Discussion
The synthesis of the modified spacers started with two previously prepared building blocks 2 and 3.8 These building blocks were coupled by a double copper(I)-catalyzed azide alkyne cycloaddition (CuAAC) reaction. Next, the two galactosyl axial 4-OH groups of the resulting product 4 were turned into triflates, thus enabling the subsequent substitution by azide with inversion to give 5. The terminal azides were subsequently linked to the protected galactosyl ligand 6 a by CuAAC yielding 7. Selective removal of the tert-butyldimethylsilyl (TBDMS) groups then gave 8 which is ready for further functionalization. The synthetic pathway is summarized in Scheme 1.
Scheme 1.

Reagents and conditions: a) CuSO4⋅5 H2O, NaAsc, DMF with 10 % H2O, 80 °C, 30 min, 85 %; b) 1) Tf2O, 10 % pyridine in CH2Cl2, 0 °C, 3 h, 2) NaN3, acetone/H2O (4:1), rt, o/n, 98 %; c) 6 a, CuSO4⋅5 H2O, NaAsc, DMF with 10 % H2O, 80 °C, 30 min, 64 %; d) p-TsOH, CH3CN/H2O (7:1), rt, 6 h, 83 %.
The two hydroxymethylene groups of compound 8 were oxidized to carboxylic acids using 2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl (TEMPO) (Scheme 2). Removal of the protecting groups gave 9. Reaction of 8 with triflic anhydride in the presence of pyridine followed by Zemplén deprotection gave the bis pyridinium compound 10. Tosylation of the primary hydroxy groups of 8 followed by reaction with sodium azide gave the intermediate 11. Zemplén deprotection of 11, followed by the hydrogenation of the azido groups gave the diamine 12. CuAAC coupling of phenylacetylene to 11 and subsequent Zemplén deprotection gave the bis-triazole 13. All final products were purified by preparative high-performance liquid chromatography (HPLC).
Scheme 2.

Reagents and conditions: a) 1) TEMPO, NaOCl, NaBr, Bu4NBr, NaHCO3/Na2CO3 pH 9.5, 0 °C, 2 h, 2) NaOMe, MeOH, rt, o/n, 38 %; b) 1) Tf2O, 10 % pyridine in CH2Cl2, 0°C→rt, 3 h, 2) NaOMe, MeOH, rt, o/n, 44 %, c) 1) TsCl, DABCO, CH2Cl2, rt, o/n, 2) NaN3, DMF, 95 °C, o/n, 34 %, d) 1) NaOMe, MeOH, rt, o/n, 2) H2, Pd/C, rt, 60 %, e) 1) phenylacetylene, CuSO4⋅5 H2O, NaAsc, DMF with 10 % H2O, 80 °C, 30 min, 2) NaOMe, MeOH, rt, o/n, 24%.
The compounds were tested on an array chip in an assay similar to an enzyme-linked immunosorbent assay (ELISA), as previously reported.8 Fluorescein-labeled LecA was incubated with the inhibitors and exposed to a galactoside-functionalized chip surface. Detection of the fluorescence allowed quantification of the binding. Monovalent ligand 6 b was previously determined to have an IC50 in this assay of 22 μm.8a Clearly all divalent compounds were far more potent and showed major multivalency effects (Table 1). The previous best inhibitor, 1, still remained the most potent compound in the present series with an IC50 in the 2-3 nm range, as before. The pyridinium-functionalized 10 and the phenylacetylene-derived 13 showed only a minor drop in potency, with IC50 values in the 5 nm range. Larger potency drops of about an order of magnitude were observed for both the negatively charged bis-carboxylate 9 and the positively charged bis-amine 12. Subsequently, ITC experiments were conducted, which confirmed the divalent binding mode in all cases, with the stoichiometry n values being close to 0.5. As before8a the dissociation constants (Kd) were somewhat higher than the IC50 values from the chip-based ELISA-like assay. Furthermore the small potency differences of the chip-based assay were not seen in the ITC assay. All compounds showed inhibitory potencies within a narrow range (57–89 nm). Interestingly, when looking at the enthalpic and entropic components of the binding event, the lipophilic and noncharged compound 13, showed enthalpy–entropy compensation, with a lower beneficial binding enthalpy and, at the same time, a lowered entropy loss upon binding. The latter is understandable as an example of the hydrophobic effect where water molecules in an ice-like structure are liberated from the lipophilic surfaces. For this reason some degree of lipophilic association is suggested for 13.
Table 1.
Inhibitory potencies (IC50) of the divalent inhibitors on LecA binding[a] and dissociation constants (Kd), stoichiometry, and thermodynamic binding parameters[b]
| Cmpd | ELISA IC50 [nm] | Kd[d] (ITC) [nm] | n[e] | ΔH [kJ mol−1] | −ΤΔS [kJ mol−1] |
|---|---|---|---|---|---|
| 1 | 1.8 | 57 (±7) | 0.50 | −49.1 | 10.0 |
| 9 | 31 | 68 (±10) | 0.50 | −50.0 | 9.1 |
| 10 | 5.2 | 89 (±7) | 0.51 | −50.1 | 9.9 |
| 12 | 19 | 56 (±3) | 0.49 | −48.7 | 7.4 |
| 13 | 4.9 | 57 (±7)[c] | 0.50 | −44.4 | 3.0 |
[a] Chip-based ELISA-like assay: FITC-labeled LecA (5 μg mL−1) binding to a galactoside functionalized chip surface. [b] Obtained from isothermal titration microcalorimetry (ITC): [LecA]=20–40 μm. [c] 1 % DMSO used. [d] Values reported ± S.D.. [e] Stoichiometry.
Conclusion
New derivatives of a highly potent divalent LecA ligand were prepared. It proved possible to use selectively protected 6-OH groups to build up the ligand in its protected form. Subsequently, the selectively deprotectable silyl groups were removed, and the resulting primary hydroxy groups were converted to carboxylate groups by oxidation, to tosylates and subsequently to azides by substitution, and to pyridiniums via their corresponding triflates. The azides allowed CuAAC coupling and also further reduction to the corresponding amino groups. While it was anticipated that some of these groups would be able to take advantage of additional beneficial interactions with the nearby protein surface, we did not observe this through enhanced inhibitory or binding potencies. This was true even for the positively charged groups that could possibly take advantage of the nearby positioned carboxylate groups of Asp 47. Molecular modeling indicated that these interactions were geometrically possible and could be energetically favorable. However, a possible alternative scenario, where the newly added groups point into the solution and thus away from the protein surface, is apparently more favorable in this case. In future designs, these options should be avoided. Nevertheless, the functionalization of the spacers will also be important in order to fine-tune other properties of this type of ligand, such as toxicity and absorption, distribution, metabolism, and excretion (ADME), which are important for drug development.
Experimental Section
General: Chemicals were obtained from commercial sources and were used without further purification unless noted otherwise. Compounds 2, 3, and 6 were synthesized following literature procedures.7 Solvents were purchased from Biosolve (Valkenswaard, The Netherlands). All moisture- sensitive reactions were performed under an N2 atmosphere. Anhydrous solvents were dried over molecular sieves of 4 Å or 3 Å. Thin-layer chromatography (TLC) was performed on Merck precoated Silica 60 plates. Spots were visualized by UV light and also by 10 % H2SO4 in MeOH. Microwave reactions were carried out in a microwave Initiator system (Biotage, Uppsala, Sweden). The microwave power was limited by temperature control once the desired temperature was reached. Sealed vessels of 2–5 mL and 10–20 mL were used. Analytical HPLC runs were performed on an automated HPLC system (Shimadzu, Kyoto, Japan) with a reversed-phase column (Reprospher 100, C4, 5 μm, 250×4.6 mm, Dr. Maisch GmbH, Ammerbuch-Entringen, Germany) which was equipped with an evaporative light scattering detector (PLELS 1000, Polymer Laboratories, Amherst, USA) and a UV/Vis detector operating at 220 nm and 250 nm. Preparative HPLC runs were performed on an Applied Biosystems (Waltham, USA) workstation. Elution was effected by using a linear gradient of 5 % CH3CN/0.1 % trifluoroacetic acid (TFA) in H2O to 5 % H2O/0.1 % TFA in CH3CN or by a gradient of H2O to 30 % CH3CN in H2O. 1H and 13C NMR spectroscopy was carried on an 400-MR spectrometer (Agilent, Santa Clara, USA) operating at 400 MHz for 1H and 100 MHz for 13C. Heteronuclear single quantum coherence (HSQC) and total correlated spectroscopy (TOCSY) NMR (500 MHz) were performed with an Inova 500 instrument (Varian, Palo Alto, USA). Electrospray ionization mass spectrometry (ESI-MS) experiments were performed with a Shimadzu LCMS QP-8000. High-resolution mass spectrometry (HRMS) analysis was recorded using an ESI-Q-TOF II spectrometer (Bruker, Billerica, USA). The proton numbering scheme of all compounds can be found in the Supporting Information and is used in the assignments of the signals for the NMR spectra below.
Isothermal titration microcalorimetry (ITC): The lectin LecA was obtained from Sigma–Aldrich and was dissolved in buffer (0.1 m Tris-HCl, 6 mm CaCl2, pH 7.5) and degassed. Protein concentration (between 20 and 40 μm depending on the ligand affinity) was checked by measurement of optical density by using a theoretical molar extinction coefficient of 28 000. Carbohydrate ligands were dissolved directly into the same buffer, degassed, and placed in the injection syringe (concentration range: 0.1–0.2 mm). ITC was performed using a MicroCal Auto ITC200 (Malvern, Worcestershire, UK). LecA (0.02–0.04 mm) was placed into the 200 μL sample cell at 25 °C. Titration was performed with injections of carbohydrate ligands (2.5 μL) every 120 s. Data were fitted using the “one-site model” using MicroCal Origin 7 software according to standard procedures. Fitted data yielded the stoichiometry (n), the association constant (Ka), the enthalpy (ΔH) and the entropy of binding. The Kd value was calculated as 1/Ka, and T is 298 K. Each ligand test was performed in duplicate.
LecA inhibiton assay: Lectin LecA was labeled with fluorescein isothiocyanate (FITC) according to a literature procedure.9 Microarray experiments were performed by using a PamChip array run on a PamStation 12 instrument (Pam-Gene, ‘s-Hertogenbosch, The Netherlands). Data were obtained by real-time imaging of the fluorescence signal by a CCD camera. Images were analyzed using BioNavigator 6 software (Pam-Gene). Each array slide contains spots in duplicate. The fluorescence intensities were expressed in arbitrary units, and the relative intensities were the average of the two duplicate spots. Aliquots of a solution of FITC-labeled LecA (5 μg mL−1 for all tested compounds) in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid/bovine serum albumin (HEPES/BSA) buffer (10 mm HEPES, 100 mm NaCl, 0.1 % BSA. pH 7.4), containing different concentrations of the inhibitors were incubated for 1 h at rt and subsequently added to the galactoside-functionalized chip. The binding process was monitored for 2 h, and the end values of the fluorescence detection were taken for the determination of the IC50 by using Prism 5 (Graphpad Software, Inc., La Jolla, USA).
Molecular Modeling: All molecular modeling studies were performed using the molecular graphics, modeling, and simulation program Yasara version 13.9.8 (Yasara Biosciences, Vienna, Austria). The bivalent ligands were first constructed in Yasara as isolated molecules. Subsequently, the complex with LecA was built by superposition of one of the galactose units of the ligand with a bound galactose of one of the subunits of the LecA crystal structure (PDB ID: 1OKO).3a The other galactose unit of the divalent ligand was then pulled into the adjacent galactose binding site of LecA by restraint MD using distance restraints based on the position of the bound galactose present in the X-ray structure with respect to a number of LecA residues. Possible electrostatic interactions between two positively charged amino and pyridinium substitutions in the linker region with two Asp 47s of LecA were investigated in more detail. In order to induce these interactions, the subunits comprising the substituted moieties were first rotated, and subsequently, the nitrogen atoms of respectively the amino and the pyridinium group were restrained to bring them in close proximity to the carbon atoms of the carboxylic acid moieties of Asp 47. After restrained MD, the molecules were subjected to a 1000 ps free MD simulation in water.
General procedure of microwave-assisted click reaction: To a solution of the azide and alkyne compounds in dimethylformamide (DMF) with 10 % H2O, was added CuSO4⋅5 H2O and sodium ascorbate (NaAsc). The mixture was then heated by microwave irradiation at 80 °C for 30 min. When the mixture cooled to rt,0 the copper salts were removed by a resin (Cuprisorb), and the solvents were removed under reduced pressure. The residue was purified by silica gel chromatography to afford the corresponding 1,2,3-triazole product.
Compound 4: A click reaction of a mixture of compound 2 (0.253 g, 0.71 mmol), compound 3 (0.83 g, 1.63 mmol), CuSO4⋅5 H2O (0.06 g, 0.24 mmol), and NaAsc (0.096 g, 0.48 mmol) in DMF (5 mL) containing 10 % H2O was performed following the general procedure described above to afford compound 4 (0.84 g, 85 %); 1H NMR (400 MHz, CDCl3): δ= 8.03–7.89 (m, 5 H, 4×CH benzoyl, H-1), 7.86–7.76 (m, 5 H, 4×CH benzoyl, H-1), 7.52–7.26 (m, 12 H, 12×CH benzoyl), 6.08 (dd, J4,3=J4,5=10 Hz, 1 H, H-4), 6.02–5.90 (m, 2 H, H-4, H-3′), 5.81 (dd, J5′,4′=J5′,6′=9 Hz, 1 H, H-5′), 5.60 (dd, J4′,3′=J4′,5′=9 Hz, 1 H, H-4′), 5.51–5.41 (m, 2 H, 2×H-5), 5.02–4.92 (m, 2 H, 2×H-3), 4.81 (dd, J6′,5′=J6′,7′=10 Hz, 1 H, H-6′), 4.67–4.54 (m, 3 H, H-7′, 2× H-6), 4.16–4.07 (m, 1 H„ H-8′a), 4.07–3.93 (m, 2 H, 2×H-8), 3.91–3.82 (m, 2×H-7), 3.70–3.60 (m, 1 H, H-8′b), 3.57–3.51 (m, 1 H, 6-OH), 3.35–3.29 (1 H, 6-OH), 2.02, 1.65, 1.58 (s, 9 H, 3×CH3, acetyl), 0.90 (s, 18 H, 2×SiC(CH3)3), 0.07 ppm (2 s, 12 H, 2×Si(CH3)2); 13C NMR (100 MHz, CDCl3): δ=69.97, 169.10, 168.95 (3×C=O acetyl), 166.14, 166.05, 165.63, 165.55 (4×C=O benzoyl), 145.61, 145.58 (2×C-2), 133.44–133.21 (CH benzoyl), 129.99–128.41 (CH benzoyl), 129.39–129.20 (C benzoyl) 123.43, 122.13 (2×C-1), 85.55 (C-3′), 77.97 (2×C-7), 75.75 (C-5), 75.48 (C-5), 74.84 (C-7′), 74.03, 73.77 (2×C-3), 72.41 (C-5′), 70.85 (C-4′), 69.94, 69.59 (2×C-4), 68.92, 68.53 (2×C-6), 63.65, 63.13 (2×C-8), 61.61 (C-8′), 59.84 (C-6′), 25.99 (SiC(CH3)3), 20.72, 19.98, 19.89 (3×CH3 acetyl), 18.44 (SiC(CH3)3), −5.31 ppm (Si(CH3)2); MS (ESI) m/z [M+H]+ calcd for C68H84N6O21Si2: 1378.59, found 1378.00; HRMS (Q-TOF) m/z [M+H]+ calcd for C68H84N6O21Si2: 1377.5228, found 1377.5326.
Compound 5: Compound 4 (0.84 g, 0.61 mmol) in anhydrous CH2Cl2 (30 mL) containing pyridine (4.22 mL) was treated with triflic anhydride (12.2 mL of a 1 m solution in CH2Cl2, 12.2 mmol). The mixture was stirred at 0 °C for 3 h, after which cold 1 n KHSO4 (20 mL) was added. The organic layer was washed with cold H2O (2×20 mL) and, once with cold brine (20 mL), dried over Na2SO4, filtered, and concentrated. The residue was used for the next step without further purification. The residue was dissolved in an acetone/H2O mixture (15 mL, 4:1), and NaN3 (0.397 g, 6.1 mmol) was added. The mixture was stirred at rt overnight and diluted with a cold H2O/CH2Cl2 mixture (50 mL, 4:1). The water layer was separated and extracted once with CH2Cl2 (10 mL). The combined organic layers were dried on Na2SO4, filtered, and concentrated to afford compound 5 as a yellowish solid (0.86 g, 0.60 mmol, 98 %); 1H NMR (400 MHz, CDCl3): δ=8.00–7.93 (m, 4 H, 4×CH benzoyl), 7.85–7.72 (m, 5 H, 4×CH benzoyl, H-1), 7.65 (s, 1 H, H-1), 7.58–7.26 (m, 12 H, 12×CH benzoyl), 5.90 (d, J3′,4′=9 Hz, 1 H, H-3′), 5.86–5.65 (m, 3 H, H-4, 2×H-5), 5.57 (m, 2 H, H-4, H-5′), 5.42 (dd, J4′,3′=J4′5′=10 Hz, 1 H, H-4′), 4.97–4.88 (m, 2 H, 2×H-3), 4.81 (dd, J6′,5′=J6′,7′=10 Hz, 1 H, H-6′), 4.61–4.51 (m, 1 H, H-7′), 4.17–4.03 (m, 3 H, H-8′a, 2×H-6), 4.03–3.93 (m, 2 H, 2×H-8), 3.68–3.55 (m, 3 H, H-8′b, 2×H-7), 2.01, 1.69, 1.62 (s, 9 H, 3×CH3, acetyl), 0.95 (s, 18 H, 2×SiC(CH3)3), 0.18–0.04 ppm (m, 12 H, 2×Si(CH3)2); 13C NMR (100 MHz, CDCl3): δ=169.85, 169.01, 168.96 (3×C=O acetyl), 165.85, 165.78, 165.56, 165.47 (4×C=O benzoyl), 145.18 (2×C-2), 133.57–133.39 (CH benzoyl), 129.96–128.44 (CH benzoyl), 129.39–129.20 (C benzoyl) 123.08, 121.61 (2×C-1), 85.64 (C-3′), 79.88, 79.86 (2×C-7), 75.01, 74.96 (2×C-5), 74.69 (C-7′), 73.60, 73.35 (2×C-3), 72.60 (C-4′), 72.17, 72.12 (2×C-4), 70.73 (C-5′), 62.50, 62.40 (2×C-8), 61.50 (C-8′), 60.43 (2×C-6), 59.84 (C-6′), 26.04 (SiC(CH3)3), 20.67, 20.01, 19.94 (3×CH3 acetyl), 18.57 (SiC(CH3)3), −4.97–(−5.26) ppm (Si(CH3)2); MS (ESI) m/z [M+H]+ calcd for C68H82N12O19Si2: 1427.54, found 1427.75; HRMS (Q-TOF) m/z [M+H]+ calcd for C68H82N12O19Si2: 1427.5358, found 1427.5389.
Compound 7: A click reaction of a mixture of compound 5 (0.88 g, 0.62 mmol) and compound 6 a (0.570 g, 1.48 mmol) with CuSO4⋅5 H2O (28 mg, 0.11 mmol) and NaAsc (0.0443 g, 0.22 mmol) in DMF (15 mL) containing 10 % H2O was performed following the general procedure described above to afford compound 7 (0.87 g, 64 %); 1H NMR (400 MHz, CDCl3): δ=7.88 (s, 1 H, H-1), 7.82–7.68 (m, 9 H, 8×CH benzoyl, H-1), 7.68–7.59 (2 s, 2 H, 2×H-1), 7.51–7.39 (m, 4 H, 4×CH benzoyl), 7.36–7.26 (m, 9 H, 8×CH benzoyl), 6.35–6.22 (m, 2 H, 2×H-5), 5.94 (d, J3′,4′=9 Hz, 1 H, H-3′), 5.88–5.73 (m, 2 H, 2×H-4), 5.67–5.52 (m, 2 H, H-5′, H-4′), 5.43–5.33 (m, 2 H, 2×H-13), 5.27–5.07 (m, 6 H, 2×H-3, 2×H-11, 2×H-6), 5.03–4.71 (m, 7 H, 2× H-12, 2×H-9a, H-6′, 2×H-9b), 4.63–4.53 (m, 1 H, H-7′), 4.48–4.37 (m, 4 H, 2×H-7, 2×H-10), 4.29–4.04 (m, 5 H, 2×H-15a, H-8′a, 2×H-15b), 3.94–3.85 (m, 2 H, 2×H-14), 3.80 (d, J8a,8b=12 Hz, 2 H, 2×H-8a), 3.65 (dd, J8′b,8′a=12 Hz, J8′b,7′=3 Hz, 1 H, H-8′b), 3.45–3.32 (m, 2 H, 2× H-8b), 2.20–1.88 (m, 21 H, 7×CH3, acetyl), 1.77–1.63 (m, 12 H, 4×CH3, acetyl), 0.94–0.84 (m, 18 H, 2×SiC(CH3)3), 0.05–(−0.07) ppm (m, 12 H, 2×Si(CH3)2); 13C NMR (100 MHz, CDCl3): δ=170.83–168.96 (C=O acetyl), 165.51 (C=O benzoyl), 165.39 (C=O benzoyl), 165.07 (C=O benzoyl), 144.90 (2×C-2), 143.54 (2×C-2), 133.64, 129.81–128.53 (CH benzoyl), 128.81 (C benzoyl) 123.66, 123.57, 123.14, 121.67 (4×C-1), 99.29, 99.26 (2×C-10), 85.70 (C-3′), 79.40, 79.33 (2×C-7), 75.02 (C-7′), 74.20, 73.85 (2×C-5), 73.67, 73.42 (2×C-3), 72.70 (C-5′), 72.22, 72.15 (2×C-4), 70.99 (2×C-12), 70.78 (2×C-14), 70.73 (C-4′), 68.74 (2×C-11), 67.25 (2×C-13), 61.84 (2×C-9, 2×C-8), 61.43 (2×C-15, C-8′), 60.17, 60.12 (2×C-6), 59.81 (C-6′), 25.96 (SiC(CH3)3), 20.97–19.92 (CH3 acetyl), 18.52 (SiC(CH3)3), −5.11–(−5.34) ppm (Si(CH3)2); MS (ESI) m/z [M+2 H]2+calcd for C102H126N12O39Si2: 1100.89, found 1100.60; HRMS (Q-TOF) m/z [M+2 H]2+ calcd for C102H126N12O39Si2: 1100.3892, found 1100.8980.
Compound 8: Compound 7 (0.87 g, 0.40 mmol) was dissolved in CH3CN/H2O (24 mL, 7:1), and p-TsOH (0.114 g, 0.60 mmol) was added. The mixture was stirred at rt for 6 h and was then diluted with CH2Cl2 (50 mL), which was followed by the addition of 10 % NaHCO3 (20 mL). The organic layer was washed once with brine (20 mL) and dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel chromatography to afford compound 8 (0.65 g, 0.33 mmol, 83 %); 1H NMR (400 MHz, CDCl3): δ=8.22 (s, 1 H, H-1), 8.12 (s, 1 H, H-1), 7.92 (s, 1 H, H-1), 7.87 (s, 1 H, H-1), 7.81 (d, J=8 Hz, 4 H, 4×CH benzoyl), 7.76–7.69 (m, 4 H, 4×CH benzoyl), 7.47–7.35 (m, 4 H, 4×CH benzoyl), 7.31–7.16 (m, 8 H, 8×CH benzoyl), 6.45–6.33 (m, 2 H, 2×H-5), 6.30 (d, J3′,4′=8 Hz, 1 H, H-3′), 6.01–5.88 (m, 2 H, 2×H-4), 5.81 (dd, J5′,4′=J5′,6′=10 Hz, 1 H, H-5′), 5.68 (dd, J4′,3′=J4′,5′=10 Hz 1 H, H-4′), 5.53–5.37 (m, 4 H, 2× H-13, 2×H-6), 5.32 (d, J3,4=12 Hz, 2 H, 2×H-3), 5.22–5.09 (m, 3 H, 2×H-11, H-6′), 5.04–4.80 (m, 6 H, 2×H-12, 2×H-9a, 2×H-9b), 4.73–4.65 (m, 1 H, H-7′), 4.51–4.40 (m, 4 H, 2×H-7, 2×H-10), 4.33–4.23 (m, 2 H, 2×H-15a), 4.17–4.07 (m, 2 H, 2×H-15b), 4.04–3.93 (m, 3 H, 2×H-14, H-8′a), 3.80 (d, J8a,8b=12 Hz, 2 H, 2×H-8a), 3.56 (dd, J8′b,8′a=12 Hz, J8′b,7′=4 Hz, 1 H, H-8′b), 3.29 (d, J8b,8a=12 Hz, 2 H, 2×H-8b), 2.20–1.93 (m, 21 H, 7×CH3, acetyl), 1.86–1.48 ppm (m, 12 H, 4×CH3, acetyl); 13C NMR (100 MHz, CDCl3): δ=170.88–169.10 (C=O acetyl), 165.51 (C=O benzoyl), 165.26 (C=O benzoyl), 165.11 (C=O benzoyl), 144.59, 144.40, 143.75, 143.71 (4×C-2), 133.60, 129.91–128.44 (CH benzoyl), 128.77, 128.37 (C benzoyl) 123.79 (2×C-1), 123.63 (2×C-1), 99.20, 99.10 (2×C-10), 85.29 (C-3′), 79.35, 79.15 (2×C-7), 74.85 (C-7′), 73.99 (2×C-5), 73.68, 73.33 (2×C-3), 72.72 (C-5′), 72.12 (2×C-4), 70.94 (2×C-12), 70.83 (2×C-14), 70.81 (C-4′), 68.84 (2×C-11), 67.27 (2×C-13), 61.64 (2×C-9), 61.45 (2×C-15), 61.31 (C-8′), 60.64, 60.56 (2×C-8), 59.92, 59.88 (2×C-6), 59.84 (C-6′), 20.96–19.83 ppm (CH3 acetyl); HRMS (Q-TOF) m/z [M+2 H]2+ calcd for C90H98N12O39: 986.3027, found 986.8078.
Compound 9: A solution of compound 8 (51.6 mg, 0.026 mmol) and TEMPO (0.164 mg, 1.05 μmol) in CH2Cl2 (1 mL) was added to a solution of NaBr (1.1 mg, 10.5 μmol) and Bu4NBr (3.4 mg, 10.5 μmol) in NaHCO3/Na2CO3 buffer (1 mL, pH was adjusted to 9.5 with saturated Na2CO3). The mixture was cooled to 0 °C, and NaOCl (6–14 %, 105 μL) was added dropwise. The mixture was stirred vigorously at 0 °C for 2 h and then quenched by the addition of Na2S2O3 (sat., 0.5 mL), after which H2O (1.5 mL) and CH2Cl2 (4 mL) were added, and the pH was adjusted to pH 3 with aqueous HCl (6 n). The organic phase was washed once with brine (1 mL), dried over Na2SO4, and concentrated in vacuo. The resulting oxidized compound was exposed to Zémplén conditions, followed by H+ resin, and was concentrated. The residue was subjected to preparative HPLC purification, which gave compound 9 (11.1 mg, 0.010 mmol, 38 %); 1H NMR (400 MHz, D2O): δ=8.56 (s, 1 H, H-1), 8.43 (s, 1 H, H-1), 8.26 (s, 2 H, 2×H-1), 6.10 (d, J3′,4′=12 Hz, 1 H, H-3′), 5.06 (d, J9a,9b=12 Hz, 2 H, 2×H-9a), 5.01–4.91 (m, 5 H, 2×H-9b, H-6′, 2×H-3), 4.89–4.72 (m, 4 H, 2×H-7, 2×H-6), 4.58–4.41 (m, 6 H, H-7′, 2×H-10, H-5′, 2×H-5), 4.30 (dd, J4′,3′=J4′,5′=9.2 Hz, 1 H, H-4′), 4.08 (dd, J4,3=J4,5=9.2 Hz, 2 H, 2×H-4), 3.97–3.92 (m, 2 H, 2×H-13), 3.88–3.70 (m, 6 H, 2×H-15ab, 2×H-14), 3.69–3.61 (m, 3 H, 2×H-12, H-8′a), 3.56 (dd, J11,10=J11,12=8 Hz, 2 H, 2×H-11), 3.40 ppm (dd, J8′b,8′a=12 Hz, J8′b,7′=4 Hz, 1 H, H-8′b); 13C NMR (100 MHz, D2O): δ=172.26 (2×C-8), 145.12 (C-2), 145.01 (C-2), 144.38 (2×C-2), 126.65 (2×C-1), 126.51 (C-1), 125.47 (C-1), 102.56 (2×C-10), 88.28 (C-3′), 78.82 (2×C-7), 77.71 (C-7′), 75.95 (2×C-14), 75.04 (C-5), 75.00 (C-5), 74.40 (C-3), 74.38 (C-3), 74.28 (C-5′), 73.64 (C-4), 73.59 (C-4), 73.42 (2×C-12), 73.30 (C-4′), 71.39 (2×C-11), 69.33 (2×C-13), 64.60 (2× C-6), 62.41 (2×C-9), 62.33 (C-6′), 61.68 (2×C-15), 60.32 ppm (C-8′); MS (ESI) m/z [M+H]+ calcd for C40H56N12O26: 1121.34, found 1121.05; HRMS (Q-TOF) m/z [M+H]+ calcd for C40H56N12O26: 1121.3429, found 1121.3465, [M+Na]+ 1143.3282.
Compound 10: Compound 8 (49.2 mg, 0.025 mmol) was dissolved in dry CH2Cl2 (2 mL) with pyridine (200 μL). The mixture was cooled down to 0 °C, after which triflic anhydride (203 μL, 0.203 mmol) was added dropwise to the above solution. The reaction was allowed to warm up to rt and stirred for 3 h after which 1 n KHSO4 (2 mL) and CH2Cl2 (15 mL) were added. The organic layer was washed once with H2O (5 mL) and once with brine (5 mL), dried on Na2SO4, filtered, and concentrated. The residue was purified by preparative HPLC to afford the corresponding pyridinium compound. The resulting material was then treated with 0.5 m NaOMe in MeOH (5 mL), stirred at rt overnight, after which 1 n HCl was added to adjust to pH≈6, and the solvents were evaporated in vacuo. The residue was purified by preparative HPLC, which gave compound 10 (13.3 mg, 0.011 mmol, 44 %); 1H NMR (400 MHz, D2O): δ=8.69 (d, J16,17=8 Hz, 4 H, 4×H-16), 8.56 (dd, J18,17=J18,17=8 Hz, 2 H, 2×H-18), 8.42 (s, 1 H, H-1), 8.38 (s, 2 H, 2×H-1), 8.28 (s, 2 H, H-1), 8.05–7.98 (m, 4 H, 4×H-17), 6.06 (d, J3′,4′=8 Hz, 1 H, H-3′), 5.12 (d, J9a,9b=12 Hz, 2 H, 2×H-9a), 5.00–4.92 (m, 3 H, 2×H-9b, H-6′), 4.92–4.73 (m, 8 H, 2×H-7, 2×H-6, 2×H-3, 2×H-8a), 4.62–4.42 (m, 6 H, 2×H-10, 2×H-8b, H-7′, H-5′), 4.34–4.23 (m, 3 H, 2×H-5, H-4′), 4.07–3.98 (m, 2 H, 2×H-4), 3.98–3.93 (m, 2 H, 2×H-13), 3.88–3.73 (m, 6 H, 2×H-15ab, 2×H-14), 3.71–3.63 (m, 3 H, 2×H-12, H-8′a), 3.63–3.54 (m, 2 H, 2×H-11), 3.35 ppm (dd, J8′b,8′a=12 Hz, J8′b,7′=4 Hz, 1 H, H-8′b); 13C NMR (100 MHz, D2O): δ=146.77 (2× C-18), 145.32 (4×C-16) 144.63 (2×C-2), 144.37 (C-2), 144.23 (C-2), 128.21 (4×C-17), 125.96 (2×C-1), 125.56 (C-1), 124.75 (C-1), 102.42 (2×C-10), 87.61 (C-3′), 77.08 (C-7′), 75.87 (2×C-7), 75.49 (2×C-14), 74.68(2×C-5), 73.84 (2×C-3), 73.73 (C-5′), 72.96(2×C-4), 72.89 (2×C-12), 72.72 (C-4′), 70.84 (2×C-11), 68.76 (2×C-13), 63.61 (2×C-6), 62.07 (2×C-9), 61.71 (C-6′), 61.43 (2×C-8), 61.20 (2×C-15), 59.76 ppm (C-8′); MS (ESI) m/z [M]2+ calcd for C50H68N14O22: 608.23, found 608.30; HRMS (Q-TOF) m/z [M]2+ calcd for C50H68N14O222+: 608.2311, found 608.2356.
Compound 11: Compound 8 (246 mg, 0.125 mmol) was dissolved in dry CH2Cl2 (5 mL), and tosyl chloride (238.4 mg, 1.25 mmol) and 1,4-diazabicyclo[2.2.2]octane (DABCO) (38 mg, 0.34 mmol) were added. The mixture was stirred at rt overnight after which the solvent was removed. The residue was redissolved in CH2Cl2 (15 mL) and washed once with H2O (5 mL) and once with brine (5 mL), dried on Na2SO4, filtered, and concentrated. The resulting material was purified by silica gel chromatography to give the tosylated compound (220 mg, 77.2 %). The residue was then dissolved in dry DMF (8 mL) to which NaN3 (33.6 mg, 0.52 mmol) was added. The mixture was stirred at 95 °C overnight after which the solvent was removed. The residue was dissolved in CH2Cl2 (15 mL), washed once with 1 n KHSO4 (5 mL) and once with H2O (5 mL), dried on Na2SO4, filtered, and concentrated. The residue was purified by silica gel chromatography to afford compound 11 (86.8 mg, 0.043 mmol, 34 %); 1H NMR (400 MHz, CDCl3): δ=7.95 (s, 1 H, H-1), 7.82–7.765 (m, 11 H, 4×CH benzoyl, 3×H-1), 7.51–7.39 (m, 4 H, 4×CH benzoyl), 7.36–7.22 (m, 8 H, 8×CH benzoyl), 6.36–6.23 (m, 2 H, 2×H-5), 5.98 (d, J3′,4′=8 Hz, 1 H, H-3′), 5.92–5.76 (m, 2 H, 2×H-4), 5.65 (t, J5′,4′=J5′,6′=8 Hz, 1 H, H-5′), 5.57 (t, J4′,3′=J4′,5′=8 Hz, 1 H, H-4′), 5.43–5.35 (m, 2 H, 2×H-13), 5.29 (d, J3,4=8 Hz, 2 H, 2×H-3), 5.22–5.09 (m, 4 H, 2×H-11, 2×H-6), 5.02–4.83 (m, 5 H, 2×H-12, 2×H-9a, H-6′), 4.77 (d, J9b,9a=12 Hz, 2 H, 2×H-9b), 4.71–4.56 (m, 3 H, 2×H-7, H-7′), 4.44 (d, J10,11=8 Hz, 2 H, 2×H-10), 4.28–4.18 (m, 2 H, 2×H-15a), 4.18–4.05 (m, 3 H, 2×H-15b, H-8′a), 3.94–3.86 (m, 2 H, 2×H-14), 3.72–3.62 (m, 1 H, H-8′b), 3.56 (t, J8a,8b=12 Hz, 2 H, 2× H-8a), 2.95, 2.88 (2×dd, J8b,8a=12 Hz, J8b,7=4 Hz, 2 H, 2×H-8b), 2.21–1.92 (21 H, 7×CH3, acetyl), 1.83–1.59 ppm (12 H, 4×CH3, acetyl); 13C NMR (100 MHz, CDCl3): δ=170.88–169.06 (C=O acetyl), 165.36 (C=O benzoyl), 165.28 (C=O benzoyl), 164.99 (C=O benzoyl), 144.72, 144.65, 144.25, 144.24 (4×C-2), 133.75–133.54, 129.85–128.49 (CH benzoyl), 128.72, 128.29, 128.26 (C benzoyl) 123.49, 123.40, 123.30 (3×C-1), 121.72 (2×C-1), 99.66, 99.60 (2×C-10), 85.68 (C-3′), 77.93, 77.72 (2×C-7), 75.00 (C-7′), 73.76, 73.71 (2×C-5), 73.50, 73.28 (2×C-3), 72.66 (C-5′), 72.07, 72.03 (2×C-4), 70.91 (2× C-12, C-4′), 70.85 (2×C-14), 70.81 (C-4′), 68.78 (2×C-11), 67.23 (2× C-13), 62.17, 62.13 (2×C-9), 61.43 (2×C-15, C-8′), 60.81 (2×C-6), 59.72 (C-6′), 50.58, 50.37 (2×C-8), 20.94–19.92 ppm (CH3 acetyl); MS (ESI) m/z calcd for C90H96N18O37 (M+2 H)2+ 1011.91, found 1012.35; HRMS (Q-TOF) m/z [M+2 H]2+ calcd for C90H96N18O37: 1011.3092, found 1011.3091, [M+H+Na]2+ 1022.8011, [M+2Na]2+ 1033.2923.
Compound 12: Compound 11 (22.6 mg, 0.020 mmol) was treated with 0.5 m NaOMe in MeOH (2.5 mL) at rt overnight and briefly with H+ resin. The residue was concentrated and dissolved in H2O (2 mL), and Pd/C (15 mg, 10 % Pd) was added. The pH was adjusted to pH 1 by 6 n HCl, and the mixture was stirred at rt under an H2 atmosphere until the hydrogenation was complete. After that, the reaction mixture was filtered through celite. The filtrate was concentrated under reduced pressure and subjected to preparative HPLC purification, which gave compound 12 (12.9 mg, 0.012 mmol, 60 %); 1H NMR (400 MHz, D2O): δ=8.56 (s, 1 H, H-1), 8.43 (s, 1 H, H-1), 8.33 (s, 2 H, 2×H-1), 6.13 (d, J3′,4′=8 Hz, 1 H, H-3′), 5.09 (d, J9a,9b=10 Hz, 2 H, 2×H-9a), 5.03–4.89 (m, 5 H, 2×H-3, H-6′, 2×H-9b), 4.89–4.70 (m, 2 H, 2×H-6), 4.66–4.47 (m, 6 H, 2×H-7, 2×H-10, H-7′, H-5′), 4.41–4.28 (m, 3 H, 2×H-5, H-4′), 4.08–3.99 (m, 2 H, 2×H-4), 3.99–3.93 (m, 2 H, 2×H-13), 3.89–3.73 (m, 6 H, 2×H-15ab, 2×H-14), 3.71–3.64 (m, 3 H, 2×H-12, H-8′a), 3.57 (dd, J11,10=J11,12=8 Hz, 2 H, 2×H-11), 3.40 (dd, J8′b,8′a=12 Hz, J8′b,7′=4 Hz, 1 H, H-8′b), 3.24–3.11 (m, 2 H, 2×H-8a), 2.86 ppm (d, J8b,8a=12 Hz, 2 H, 2×H-8b); 13C NMR (100 MHz, D2O): δ=144.94, 144.82 (2×C-2), 144.69 (2×C-2), 126.18 (2×C-1), 126.13 (C-1), 125.07 (C-1), 102.63 (2×C-10), 87.97 (C-3′), 77.46 (C-7′), 75.76 (2×C-14), 74.90 (2×C-5), 74.51 (2×C-7), 74.33 (2×C-3), 74.07 (C-5′), 73.45 (2×C-4), 73.18 (2×C-12), 73.13 (C-4′), 71.14 (2×C-11), 69.09 (2×C-13), 64.27 (2×C-6), 62.39 (2×C-9), 62.08 (C-6′), 61.48 (2×C-15), 60.12 (C-8′), 40.59 ppm (2×C-8); MS (ESI) m/z [M+2 H]2+ calcd for C40H62N14O22: 546.21, found 546.90; HRMS (Q-TOF) m/z [M+H]+ calcd for C40H62N14O22: 1091.4163, found 1091.4282, [M+Na]+ 1113.4132.
Compound 13: A click reaction of a mixture of compound 11 (61.8 mg, 0.031 mmol), phenylacetylene (11.1 mg, 0.109 mmol), NaAsc (6.5 mg, 0.033 mmol), and CuSO4⋅5 H2O (4.1 mg, 0.016 mmol) was performed following the general procedure described above to afford the coupling product compound (54 mg, 80 %), which was then treated with NaOMe in MeOH (0.5 m, 5 mL). The mixture was stirred at rt overnight, treated with H+ resin, concentrated, and subjected to preparative HPLC purification, which gave compound 13 (9.8 mg, 7.3 μmol, 24 %); 1H NMR (400 MHz, D2O with 30 % CD3CN): δ=8.62 (s, 1 H, H-1), 8.50, 8.49 (2 s, 2 H, 2×H-1), 8.46 (s, 1 H, H-1), 8.42, 8.41 (2 s, 2 H, 2×H-1), 8.05–7.98 (m, 4 H, 4×C-17), 7.75 (t, J18,17=J18,19=8 Hz, 4 H, 4×C-18), 7.71–7.62 (m, 2 H, 2×C-19), 6.21 (d, J3′4′=9 Hz, 1 H, H-3′), 5.27 (d, J9a,9b=12 Hz, 2 H, 2×H-9a), 5.16–4.99 (m, 7 H, 2×H-9b, 2×H-3, H-6′, 2×H-7), 4.92–4.83 (m, 2 H, 2×H-6), 4.83–4.70 (m, 4 H, 2×H-8a, 2×H-10), 4.70–4.58 (m, 4 H, 2×H-8b, H-5′, H-7′), 4.57–4.48 (m, 2 H, 2×H-5), 4.43 (t, J4′,3′=J4′,5′=8 Hz, 1 H, H-4′), 4.21–4.11 (m, 4 H, 2×H-4, 2×H-13), 4.08–3.90 (m, 6 H, 2×H-15ab, 2×H-14), 3.89–3.82 (m, 2 H, 2×H-12), 3.82–3.73 (m, 3 H, 2×H-11, H-8′a), 3.46 ppm (dd, J8′b,8′a=13 Hz, J8′b,7′=4 Hz, 1 H, H-8′b); 13C NMR (100 MHz, D2O with 30 % CD3CN): δ=147.90 (4×C-2), 145.44 (2×C-2), 130.82–129.48 (4×C-18, 2×C-19), 130.00 (2×C-16), 126.40 (2×C-1), 126.21 (4×C-17), 125.78 (C-1), 124.76 (C-1), 123.66 (2×C-1), 102.73 (2×C-10), 88.11 (C-3′), 77.554 (C-7′), 76.27 (2×C-7), 75.81 (2×C-14), 75.31 (2×C-5), 74.50 (2×C-3), 74.20 (C-5′), 73.62 (2×C-4), 73.38 (2×C-12), 73.25 (C-4′), 71.29 (2×C-11), 69.18 (2× C-13), 64.29 (2×C-6), 62.37 (2×C-9), 62.07 (C-6′), 61.55 (2×C-15), 60.22 (C-8′), 51.44 ppm (2×C-8); MS (ESI) m/z [M+2 H]2+ calcd for C56H70N18O22: 674.63, found 674.95; HRMS (Q-TOF) m/z [M+H]+ calcd for C56H70N18O22: 1348.2624, found 1348.4964.
Acknowledgments
This research was supported by the Dutch Technology Foundation STW, Applied Science Division of the Netherlands Organization for Science Research (NWO), the Technology Program of the Ministry of Economic Affairs (Netherlands), and by the European Cooperation in Science and Technology (COST) action CM1102 MultiGlycoNano.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
References
- 1a.Hudak JE, Bertozzi CR. Chem. Biol. 2014;21:16–37. doi: 10.1016/j.chembiol.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b.Ernst B, Magnani JL. Nat. Rev. Drug. Discov. 2009;8:661–677. doi: 10.1038/nrd2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2a.Dam TK, Gerken TA, Brewer CF. Biochemistry. 2009;48:3822–3827. doi: 10.1021/bi9002919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b.Gouin SG. Chem. Eur. J. 2014;20:11616–11628. doi: 10.1002/chem.201402537. [DOI] [PubMed] [Google Scholar]
- 2c.Jayaraman N. Chem. Soc. Rev. 2009;38:3463–3483. doi: 10.1039/b815961k. [DOI] [PubMed] [Google Scholar]
- 2d.Pieters RJ. Org. Biomol. Chem. 2009;7:2013–2025. doi: 10.1039/b901828j. [DOI] [PubMed] [Google Scholar]
- 2e.Gingras M, Chabre YM, Roy M, Roy R. Chem. Soc. Rev. 2013;42:4823–4841. doi: 10.1039/c3cs60090d. [DOI] [PubMed] [Google Scholar]
- 2f.Levine PM, Carberry TP, Holub JM, Kirshenbaum K. MedChemComm. 2013;4:493–509. [Google Scholar]
- 2g.Kiessling LL, Grim JC. Chem. Soc. Rev. 2013;42:4476–4491. doi: 10.1039/c3cs60097a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2h.Galan MC, Dumy P, Renaudet O. Chem. Soc. Rev. 2013;42:4599–4612. doi: 10.1039/c2cs35413f. [DOI] [PubMed] [Google Scholar]
- 3a.Imberty A, Wimmerová M, Mitchell EP, Gilboa-Garber N. Microbes Infect. 2004;6:221–228. doi: 10.1016/j.micinf.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 3b.Cioci G, Mitchell EP, Gautier C, Wimmerová M, Sudakevitz D, Pérez S, Gilboa-Garber N, Imberty A. FEBS Lett. 2003;555:297–301. doi: 10.1016/s0014-5793(03)01249-3. [DOI] [PubMed] [Google Scholar]
- 4a.Gening ML, Titov DV, Cecioni S, Audfray A, Gerbst AG, Tsvetkov YE, Krylov VB, Imberty A, Nifantiev NE, Vidal S. Chem. Eur. J. 2013;19:9272–9285. doi: 10.1002/chem.201300135. [DOI] [PubMed] [Google Scholar]
- 4b.Cecioni S, Praly J, Matthews SE, Wimmerova M, Imberty A, Vidal S. Chem. Eur. J. 2012;18:6250–6263. doi: 10.1002/chem.201200010. [DOI] [PubMed] [Google Scholar]
- 4c.Gerland B, Goudot A, Pourceau G, Meyer A, Vidal S, Souteyrand E, Vasseur J, Chevolot Y, Morvan F. J. Org. Chem. 2012;77:7620–7626. doi: 10.1021/jo300826u. [DOI] [PubMed] [Google Scholar]
- 4d.Cecioni S, Faure S, Darbost U, Bonnamour I, Parrot-Lopez H, Roy O, Taillefumier C, Wimmerova M, Praly J, Imberty A, Vidal S. Chem. Eur. J. 2011;17:2146–2159. doi: 10.1002/chem.201002635. [DOI] [PubMed] [Google Scholar]
- 4e.Cecioni S, Oerthel V, Iehl J, Holler M, Goyard D, Praly J, Imberty A, Nierengarten J, Vidal S. Chem. Eur. J. 2011;17:3252–3261. doi: 10.1002/chem.201003258. [DOI] [PubMed] [Google Scholar]
- 4f.Gerland B, Goudot A, Ligeour C, Pourceau G, Meyer A, Vidal S, Gehin T, Vidal O, Souteyrand E, Vasseur J, Chevolot Y, Morvan F. Bioconjugate Chem. 2014;25:379–392. doi: 10.1021/bc4005365. [DOI] [PubMed] [Google Scholar]
- 4g.Novoa A, Eierhoff T, Topin J, Varrot A, Barluenga S, Imberty A, Römer W, Winssinger N. Angew. Chem. Int. Ed. 2014;53:8885–8889. doi: 10.1002/anie.201402831. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014;126 [Google Scholar]
- 4h.Kadam RU, Bergmann M, Garg D, Gabrieli G, Stocker A, Darbre T, Reymond J-L. Chem. Eur. J. 2013;19:17054–17063. doi: 10.1002/chem.201302587. [DOI] [PubMed] [Google Scholar]
- 4i.Kadam RU, Garg D, Schwartz J, Visini R, Sattler M, Stocker A, Darbre T, Reymond J-L. ACS Chem. Biol. 2013;8:1925–1930. doi: 10.1021/cb400303w. [DOI] [PubMed] [Google Scholar]
- 5a.Pieters RJ. Med. Res. Rev. 2007;27:796–816. doi: 10.1002/med.20089. [DOI] [PubMed] [Google Scholar]
- 5b.Bernardi A, Jiménez-Barbero J, Casnati A, De Castro C, Darbre T, Fieschi F, Finne J, Funken H, Jaeger K, Lahmann M, Lindhorst TK, Marradi M, Messner P, Molinaro A, Murphy PV, Nativi C, Oscarson S, Penadés S, Peri F, Pieters RJ, Renaudet O, Reymond J, Richichi B, Rojo J, Sansone F, Schäffer C, Turnbull WB, Velasco-Torrijos T, Vidal S, Vincent S, Wennekes T, Zuilhof H, Imberty A. Chem. Soc. Rev. 2013;42:4709–4727. doi: 10.1039/c2cs35408j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c.Pieters RJ. Adv. Exp. Med. Biol. 2011;715:227–240. doi: 10.1007/978-94-007-0940-9_14. [DOI] [PubMed] [Google Scholar]
- 5d.Pera NP, Pieters RJ. MedChemComm. 2014;5:1027–1035. [Google Scholar]
- Measured between the two anomeric oxygens of the bound galactose units of the X-ray structure with PDB ID: 1OKO (see Ref. 3 a)
- Wittmann V, Pieters RJ. Chem. Soc. Rev. 2013;42:4492–4503. doi: 10.1039/c3cs60089k. [DOI] [PubMed] [Google Scholar]
- 8a.Pertici F, de Mol NJ, Kemmink J, Pieters RJ. Chem. Eur. J. 2013;19:16923–16927. doi: 10.1002/chem.201303463. [DOI] [PubMed] [Google Scholar]
- 8b.Pertici F, Pieters RJ. Chem. Commun. 2012;48:4008–4010. doi: 10.1039/c2cc30234a. [DOI] [PubMed] [Google Scholar]
- Goding JW. J. Immunol. Methods. 1976;13:215–226. doi: 10.1016/0022-1759(76)90068-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary