

Significance
Most enzymatic reactions involve hydrogen or proton transfer among the enzyme, substrate, and water at physiological pH. Thus, enzyme catalysis cannot be fully understood without accurate mapping of hydrogen atom positions in these macromolecular catalysts. Direct information on the location of hydrogen atoms can be obtained using neutron crystallography. We used neutron crystallography and biomolecular simulation to characterize the initial stage of the glycoside hydrolysis reaction catalyzed by a family 11 glycoside hydrolase. We provide evidence that the catalytic glutamate residue alternates between two conformations bearing different basicities, first to obtain a proton from the bulk solvent, and then to deliver it to the glycosidic oxygen to initiate the hydrolysis reaction.
Keywords: glycoside hydrolase, protonation state, macromolecular neutron crystallography, xylanase, molecular simulations
Abstract
Glycoside hydrolase (GH) enzymes apply acid/base chemistry to catalyze the decomposition of complex carbohydrates. These ubiquitous enzymes accept protons from solvent and donate them to substrates at close to neutral pH by modulating the pKa values of key side chains during catalysis. However, it is not known how the catalytic acid residue acquires a proton and transfers it efficiently to the substrate. To better understand GH chemistry, we used macromolecular neutron crystallography to directly determine protonation and ionization states of the active site residues of a family 11 GH at multiple pD (pD = pH + 0.4) values. The general acid glutamate (Glu) cycles between two conformations, upward and downward, but is protonated only in the downward orientation. We performed continuum electrostatics calculations to estimate the pKa values of the catalytic Glu residues in both the apo- and substrate-bound states of the enzyme. The calculated pKa of the Glu increases substantially when the side chain moves down. The energy barrier required to rotate the catalytic Glu residue back to the upward conformation, where it can protonate the glycosidic oxygen of the substrate, is 4.3 kcal/mol according to free energy simulations. These findings shed light on the initial stage of the glycoside hydrolysis reaction in which molecular motion enables the general acid catalyst to obtain a proton from the bulk solvent and deliver it to the glycosidic oxygen.
Glycoside hydrolases (GHs) catalyze the cleavage of the glycosidic bond, one of the most stable linkages in biological macromolecules, with rate enhancements on the order of 1017 to 1018 relative to the uncatalyzed reaction in neutral solution (1), placing them among the most efficient biochemical catalysts known. GHs naturally decompose billions of tons of organic matter worldwide annually, and they produce commercially valuable chemicals from lignocellulosic plant biomass (2, 3). They are also important targets for the development of therapeutic agents against bacterial and viral infections, diabetes, and other diseases (4, 5). Understanding their catalytic mechanisms and ligand binding characteristics requires knowledge of the protonation states and hydrogen bonding at the atomic level, which is of paramount importance for protein engineering and drug design (6).
Glycosides are acetals that are spontaneously cleaved through a specific acid hydrolysis mechanism involving protonation of the glycosidic oxygen of the scissile C–O bond by H3O+ to generate an oxocarbenium cation intermediate, followed by the nucleophilic attack of a water molecule that leads to a racemic hemiacetal product (7). GH enzymes enhance the rate of this slow hydrolysis reaction using a general acid, usually the carboxylic acid of a Glu residue, which donates a proton to the leaving group oxygen in the rate-limiting step (8–10). Two models for the catalytic mechanism of β-glycoside hydrolases have been proposed: (i) a double displacement pathway first proposed by Koshland, essentially proceeding as an SN2 reaction (11, 12) and (ii) an oxocarbenium ion intermediate pathway originally proposed by Phillips (13), occurring as an SN1 reaction (Fig. S1) (14, 15). The Koshland mechanism suggests a glycosylation step, in which a covalent glycosyl-enzyme intermediate bearing inverted stereochemistry at the anomeric C1 is formed, followed by the deglycosylation step, in which the latter is hydrolyzed by an incoming H2O activated by the first Glu residue and switched its role to act as a general base. This canonical double-displacement mechanism has been challenged by proponents of the oxocarbenium ion intermediate mechanism, in which the general acid-assisted departure of an aglycon leaving group leads to a short-lived, planar carbocation on C1 of the glycon, which is then hydrated by an incoming H2O with an estimated rate constant of 1012 s−1 (16). Each mechanism draws support from different types of experimental and computational evidence (17–22), with the double-displacement mechanism being generally accepted.
Fig. S1.

Proposed mechanisms for glycoside hydrolysis catalyzed by retaining GH enzymes. (A) Double displacement mechanism. (B) Oxocarbenium ion intermediate mechanism.
Two catalytic Glu residues, one serving as an acid/base catalyst and the other as a nucleophile, are nearly ubiquitous in GH enzymes (23). Such a scenario necessitates the former being a weaker acid than the latter and implies that, in the reactant state, the general acid must be protonated and neutral (i.e., -COOH), with a pKa greater than 6, and the nucleophile must be deprotonated and negatively charged (i.e., -COO−), with a pKa less than 5 (24). However, the protonation states of the key residues in the active site as well as the distribution of hydrogen bonds have not been directly observed in GH enzymes. Thus, the changes in protonation predicted by the reaction mechanism have been inferred only from indirect measurements. H atoms are difficult to visualize by X-ray crystallography, even in structures determined at sub-ångström resolution. Whereas X-rays are scattered from electron density, neutrons are scattered by atomic nuclei through the strong force, which is independent of atomic number. The smallest atom, H, and its heavy isotope deuterium (D) scatter neutrons as well as other atoms constituting biological molecules. As a result, locating H and D atoms in macromolecular neutron structures is straightforward even at medium resolutions of 2.0–2.5 Å (25).
Family 11 GH (GH11) enzymes are small (∼20-kDa) proteins called xylanases, with a conserved jelly-roll fold, that catalyze the hydrolysis of the most abundant component of hemicellulose, xylan, into smaller oligosaccharides (26–29). The active-site cavity of xylanase, XynII, from Trichoderma reesei adopts a half-pipe shape and is sculpted by an extensive β-sheet that twists on itself (Fig. S2). On the basis of previous X-ray structures (30), the active site cleft has six subsites that bind the xylose units of xylan, spanning positions −3 to +3, with hydrolysis occurring between subsites −1 and +1 (31). Two catalytic Glu residues, Glu177 and Glu86, face each other from opposite sides of the cleft, and their side chains are strategically positioned within about 6 Å from each other (32). Glu177 is believed to serve as the acid/base catalyst and must be protonated before the reaction whereas Glu86 is the nucleophile and must be deprotonated.
Fig. S2.

The jelly-roll fold of GH11 enzymes in diagram representation. The catalytic residues Glu177 and Glu86 are shown as sticks. The xylohexaose substrate is docked to the active site of native XynII (pD6.2) by superimposing with the pseudo-Michaelis complex structure E177Q-X6 reported previously (33). Glu177 is in the upward conformation, poised to attack the glycosidic oxygen.
To directly determine protonation states in a GH, and specifically to locate the H atom on carboxylic acid of the catalytic Glu, we obtained five neutron structures of WT and mutant (N44D) XynII at high resolutions of 1.7–2.0 Å in the apo- and 2-(N-morpholino)ethanesulfonic acid (Mes)-bound forms. The N44D mutant displays altered pKa values of key residues in the active site. These structures were obtained from H/D exchanged crystals formed at pD values from 4.8 to 8.9, spanning the range of xylanase activity and the pKa values of key residues (pD = pH + 0.4; see SI Methods). To provide additional insight into active site protonation states, selected structures were further characterized with pKa calculations and free energy simulations.
SI Methods
Protein Expression and Purification.
The native Trichoderma longibrachiatum XynII was purchased from Hampton Research as a 36 mg/mL solution in 0.18 M Na/K phosphate and 43% (vol/vol) glycerol. This protein shares the same amino acid sequence as its Trichoderma reesei orthology, and the two XynII enzymes are considered to be equivalent. For crystallization trials at specific pH, the sample was run twice on a desalting column (HiPrep 26/10; GE Healthcare) with 0.1 M sodium cacodylate-HCl (pH 5.8), 0.1 M Mes-NaOH (pH 6.0), or 0.1 M Tris⋅HCl (pH 8.5) buffers. The protein samples were concentrated to 70 mg/mL (pH 5.8–6.0) or 20 mg/mL (pH 8.5). The procedures for site-directed mutagenesis, protein expression, and purification to obtain the WT recombinant XynII, N44D, and N44E variants were reported previously (44). The proteins were concentrated to 30–40 mg/mL in 0.1 M Mes-NaOH (pH 6.0) or 0.1 M sodium citrate-HCl (pH 4.6).
Crystallization.
pH values were measured directly in the crystallization drops for all crystals chosen for neutron crystallography, and H2O was exchanged with D2O in capillaries (pD = pH + 0.4). To obtain native XynII crystals for neutron diffraction at different pH values, the following crystallization conditions and manipulations were used. Crystals of pD6.2 grew in microbridges (Hampton Research) with 0.1 M sodium cacodylate-HCl (pH 5.8), 0.2 M NaI, and 5–7% (wt/vol) PEG8000 at 18 °C. Crystals of N44D grew in the similar conditions, except sodium cacodylate was replaced with Mes-NaOH. Crystals of pD8.9 grew in nine-well glass plates using the sandwich box setups (Hampton Research) with 0.1 M Tris⋅HCl (pH 8.5), 0.2 M NaI, and 10% (wt/vol) PEG8000 at 18 °C. Crystals of WT-Mes were obtained using a similar technique with 0.1 M Mes-NaOH (pH 6.0), 0.2 M NaI, and 14% (wt/vol) PEG8000. To ensure full occupancy of Mes in the enzyme active site, the crystallization drops were injected with 1.5 M Mes (pH 5.5) to reach the final buffer concentration of 0.7 M and final pH of 5.7. To grow neutron quality crystals, the temperature was slowly decreased from 18 °C to 6 °C over the course of 2 mo. The low pH crystals of pD4.8 were obtained differently. First, neutron quality crystals were grown at pH 5.8 in sodium cacodylate. Next, the pH in the crystallization drops was changed by removing the initial well solution and replacing it three times with 0.1 M NaOAc (pH 5.5, 5.0 and 4.4), 0.2 M NaI, and PEG8000 over the period of several days. The pH in the crystallization drop was measured after the last buffer change to give the same pH of 4.4 as in the well solution. Crystal of recombinant WT XynII at pH 4.0 grew using the hanging drop method in 20% (wt/vol) PEG8000, 0.2 M NaI, 0.1 M NaOAc at pH 4.0. Crystals of N44D at pH 4.6 grew in the similar conditions [22% (wt/vol) PEG8000, 0.2 M NaI, 0.1 M NaOAc at pH 4.6]. Crystals of N44E I in complex with Mes were obtained in hanging drops using 16% (wt/vol) PEG8000, 0.2 M NaI, 0.1 M Mes-NaOH at pH 6.0.
X-Ray Crystallography.
Room temperature X-ray crystallographic data sets were collected from the crystals chosen for neutron diffraction. All these datasets were collected on a Rigaku HighFlux HomeLab instrument equipped with a MicroMax-007 HF X-ray generator and Osmic VariMax optics. The diffraction images were obtained using an R-Axis IV++ image plate detector. Low-temperature X-ray diffraction datasets were collected on beamline ID19 at 100 K at the Advanced Photon Source (Argonne, IL). Diffraction data were collected, integrated, and scaled using the HKL3000 software suite (45). Structures were solved by the molecular replacement method using phasing information from the XynII structure with PDB ID code 2DFB. Room-temperature X-ray structures were refined using SHELX-97 (46) whereas 100 K structures were refined with PHENIX (47). Joint X-ray/neutron refinement was performed on isomorphous diffraction data sets to reduce errors; thus, the X-ray data for pD4.8, pD6.2, pD8.9, WT-Mes, and N44D complexes were collected from crystals that had been taken from the same crystallization drops that provided the larger crystals for neutron data and were exchanged with D2O in a similar way as was done for neutron studies. A summary of the crystallographic data is given in Tables S1 and S2.
Table S1.
Room temperature neutron crystallographic data collection and joint XN refinement statistics
| Data and refinement | pD8.9 | pD6.2 | pD4.8 | WT-Mes | N44D |
| PDB ID code 4S2H | PDB ID code 4S2G | PDB ID code 4S2F | PDB ID code 4S2D | PDB ID code 4XPV | |
| Data collection | |||||
| Beamline/facility | PCS/LANSCE | BioDiff/FRM II | MaNDi/SNS | PCS/LANSCE | PCS/LANSCE |
| Space group | P212121 | P212121 | P212121 | P212121 | P212121 |
| Cell dimensions | |||||
| a, b, c, Å | 49.61, 60.03, 70.55 | 49.51, 59.87, 70.43 | 49.96, 59.62, 70.59 | 49.17, 60.21, 70.43 | 49.19, 60.29, 70.52 |
| α, β, γ, ° | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution, Å | 22.86–1.70 (1.79–1.70)* | 50.00–2.00 (2.07–2.00) | 50.00–2.00 (2.10–2.00) | 22.85–2.00 (2.11–2.00) | 33.50–2.00 (2.11–2.00) |
| No. reflections measured | 65,131 (4,655) | 67,029 | N/A | 40,746 (3069) | 39,757 (3077) |
| No. reflections unique | 20,012 (2,359) | 14,150 (1,307) | 13,899 (1,721) | 12,398 (1501) | 11,878 (1462) |
| Rmerge | 0.237 (0.364) | 0.097 (0.327) | 0.253 (0.271) | 0.223 (0.369) | 0.229 (0.381) |
| I/σI | 5.1 (1.7) | 11.2 (3.1) | 6.2 (3.2) | 5.0 (1.6) | 4.9 (1.7) |
| Completeness, % | 85.3 (70.7) | 96.2 (91.0) | 90.6 (84.2) | 85.5 (72.7) | 82.5 (71.2) |
| Redundancy | 3.3 (2.0) | 4.7 (3.3) | 4.8 (2.7) | 3.3 (2.0) | 3.3 (2.1) |
| Joint XN Refinement | |||||
| Resolution (neutron), Å | 20–1.70 | 40–2.00 | 40–2.00 | 20–2.00 | 20–2.00 |
| Resolution (X-ray), Å | 20–1.60 | 40–1.60 | 40–1.70 | 20–1.60 | 20–1.70 |
| Data rejection criteria | No observation & |F|=0 | No observation & |F|=0 | No observation & |F|=0 | No observation & |F|=0 | No observation & |F|=0 |
| Sigma cutoff | 2.5 | 2.0 | 3.0 | 2.2 | |
| No. reflections (neutron) | 17,268 | 12,923 | 13,048 | 11,689 | 11,552 |
| No. reflections (X-ray) | 26,410 | 25,375 | 19,665 | 26,870 | 22,806 |
| Rwork/Rfree (neutron) | 0.261/0.268 | 0.164/0.183 | 0.262/0.305 | 0.244/0.279 | 0.264/0.304 |
| Rwork/Rfree (X-ray) | 0.199/0.210 | 0.203/0.213 | 0.199/0.212 | 0.192/0.196 | 0.133/0.157 |
| No. atoms | |||||
| Protein including H and D | 2,832 | 2,832 | 2,834 | 2,833 | 3,162 |
| Iodide | 1 | 1 | 1 | 1 | 3 |
| Ligand | N/A | N/A | N/A | 25 (Mes) | N/A |
| Water | 510 (170 D2Os) | 444 (148 D2Os) | 438 (146 D2Os) | 453 (151 D2Os) | 519 (173 D2Os) |
| B-factors | |||||
| Protein | 22.9 | 33.8 | 26.5 | 19.7 | 16.9 |
| Iodide | 20.7 | 32.7 | 24.2 | 18.2 | 33.3 |
| Ligand | N/A | N/A | N/A | 23.1 (Mes) | N/A |
| Water | 46.0 | 55.8 | 44.9 | 42.0 | 27.8 |
| rms deviations | |||||
| Bond lengths, Å | 0.006 | 0.006 | 0.007 | 0.008 | 0.014 |
| Bond angles, ° | 1.176 | 1.359 | 1.225 | 1.073 | 1.200 |
Values in parentheses are for highest resolution shell. Data were collected from one crystal for each structure.
Table S2.
100 K X-ray crystallographic data collection and refinement statistics
| Data and refinement | WT pH 4.0 | N44D pH 4.6 | N44E-Mes pH6.0 |
| PDB ID 4XQD | PDB ID 4XQ4 | PDB ID 4XQW | |
| Data collection | |||
| Space group | C2 | C2 | P212121 |
| Cell dimensions | |||
| a, b, c, Å | 80.86, 69.63, 78.19 | 80.94, 69.75, 78.28 | 47.99, 59.63, 69.55 |
| α, β, γ, ° | 90, 117.5, 90 | 90, 117.5, 90 | 90, 90, 90 |
| Resolution, Å | 20.00–1.50 (1.53–1.50)* | 27.20–1.22 (1.29–1.22) | 50.00–1.50 (1.53–1.50) |
| No. reflections unique | 56,145 (2,752) | 114,048 (16,604) | 31,754 (1,494) |
| Rmerge | 0.098 (0.380) | 0.113 (0.421) | 0.066 (0.427) |
| I/σ(I) | 7.5 (2.1) | 4.7 (1.8) | 17.0 (3.1) |
| Completeness, % | 91.2 (88.9) | 99.6 (99.7) | 96.5 (92.7) |
| Redundancy | 2.3 (2.0) | 3.3 (3.1) | 3.8 (3.8) |
| Refinement | |||
| Resolution, Å | 20–1.50 | 15–1.25 | 33–1.50 |
| No. reflections | 56,100 | 105,797 | 31,711 |
| Rwork/Rfree | 0.192/0.223 | 0.141/0.168 | 0.174/0.179 |
| No. atoms | |||
| Protein | 2,943 | 3,042 | 1,473 |
| Iodide | 6 | 5 | 5 |
| Ligand | 8 (Tris) | N/A | 12 (Mes) |
| Water | 459 | 597 | 234 |
| B-factors | |||
| Protein | 17.4 | 12.7 | 18.1 |
| Iodide | 23.9 | 16.5 | 25.2 |
| Ligand | 26.8 (Tris) | N/A | 18.8 (Mes) |
| Water | 27.4 | 26.2 | 28.0 |
| rms deviations | |||
| Bond lengths, Å | 0.006 | 0.008 | 0.006 |
| Bond angles, ° | 1.122 | 1.318 | 1.191 |
Values in parentheses are for the highest-resolution shell. Data were collected from 1 crystal for each structure.
Neutron Crystallography.
Crystals were mounted in quartz capillaries and subjected to H/D exchange using buffers made with 100% D2O for several weeks before data collection. All neutron diffraction data were collected at room temperature. Time-of-flight wavelength-resolved Laue images were collected and processed at the Protein Crystallography Station (PCS) at LANSCE (Los Alamos National Laboratory, Los Alamos, NM) for pD8.9, WT-Mes, and N44D as described previously (48). Monochromatic neutron diffraction data were collected using the BIODIFF beamline at the FRM II research reactor at the Heinz Maier-Leibnitz Zentrum (MLZ) for pD6.2. At a wavelength of 2.66 Å, 303 frames of 0.3° oscillation and 50 min exposure time were recorded. To increase the data completeness and redundancy, the crystal was reoriented, and a second series of 243 frames were collected. The diffraction data were indexed and integrated using DENZO and then scaled with SCALEPACK (49). For pD4.8, neutron diffraction data were collected on the macromolecular neutron diffractometer MaNDi (50) located at the Spallation Neutron Source (Oak Ridge National Laboratory, Oak Ridge, TN) taking advantage of a time-of-flight Laue technique similar to that used at PCS. A total of 20 crystal settings were collected, with the exposure time for each setting of 16 h and using neutrons with wavelengths in the range of 2–4 Å. Between each exposure a φ step of 10° was carried out. As is usual for a Laue experiment, the crystal was held stationary at a different φ setting for each exposure. The diffraction images were processed using Mantid software and scaled for detector response using the Argonne National Laboratory variable wavelength data reduction program Andrev (50). The resulting reflection file was examined and merged with XPREP (a program from the SHELXTL suite).
Joint XN Structure Determination and Refinement.
The joint XN structures of pD4.8, pD6.2, pD8.9, and WT-Mes were determined and refined using nCNS (51) whereas PHENIX (47) was used for N44D. After rigid body refinement, several cycles of positional, atomic displacement parameter and occupancy refinement followed. D atoms were placed on the conformationally flexible -OD, -ND2, and -ND3+ groups, and their orientations were based on the FO − FC difference neutron scattering length density maps. After further refinement, the structures were checked for the correctness of -OD, -ND2, and -ND3+ conformations. The protonation states of His and Lys residues were also examined. The protonation states of some mobile (disordered) side chains could not be directly determined. In the following rounds of refinement, the water building was performed. All water molecules were refined as D2O. Water oxygen atoms were initially placed according to their electron density peaks. The D2O orientations were inspected and altered based on the FO − FC difference neutron scattering length density maps and the potential hydrogen-bond donors and acceptors. Some water molecules were eliminated from, or added to, the joint XN refinement based on the lack or observation of peaks in the 2FO − FC or FO − FC neutron scattering length density maps, respectively, relative to the starting X-ray structure. The level of H/D exchange at labile positions on the enzyme was refined by modeling all labile H positions as D and refining the occupancies of D within the range of −0.56 to 1.00 in nCNS, or by placing H and D at every exchangeable position and refining their occupancies, restraining the sum to 1.0 in PHENIX. The coherent scattering length of H is −3.739 fm, and that of D is 6.671 fm; thus, the scattering length of H is −0.56 times the scattering length of D. Before depositing the final structures refined with nCNS in the PDB, a script was run that converts a record for the coordinate of a labile D atom into two records corresponding to an H and a D atom partially occupying the same coordinate, both with positive partial occupancies that add up to unity. This step was not necessary for the structure refined with PHENIX. The refinement statistics are represented in Table S1.
pKa Calculations and MD Simulations.
The H++ web server version 3.1 (52), which uses a Poisson–Boltzmann continuum electrostatics approach to approximate the electrostatic environment of biological macromolecules, was used to compute the pKas of ionizable residues in the pD 4.8 and pD 6.2 neutron structures of xylanase, pD4.8 and pD6.2, respectively. The pKa calculations were carried out assuming a salinity of 0.15 M, an internal dielectric constant of either 10 or 4 (Results), and an external dielectric constant of 80. Umbrella sampling, or stratification, simulations (53) were used to compute the potential of mean force for the rotation of the χ2 dihedral of Glu177 in its protonated state from the downward to the upward conformation in the presence of a xylohexaose substrate. In the enzyme-substrate cocrystal structure (PDB ID code 4HK8), the catalytic Glu177 was mutated to Gln. Thus, we back-mutated Gln177 to Glu, protonated it, and positioned it in a downward conformation with χ2 ∼ −90°. The CHARMM-GUI web server was used to set up the model for the simulations, which were then carried out with NAMD version 2.9 (54). The CHARMM 36 Additive Force Field (55) was used to describe the protein, and the CHARMM Carbohydrate Force Field (56) and TIP3P water model were used to describe the xylohexaose substrate and water, respectively. The system was solvated in a rectangular box of TIP3P water molecules with a minimum distance of 10 Å from the protein to the nearest face of the box, which corresponded to a box size of ∼64 Å3. Five Cl− anions were added to neutralize the charge of the system. Periodic boundary conditions were used, and the Particle Mesh Ewald method was used to describe long-range electrostatics interactions. A time step of 2.0 fs was used for all simulations. The temperature and pressure were maintained at 300 K and 1 atm, respectively, with the Langevin temperature and Langevin Piston methods. After equilibration, we ran 50 ns unrestrained MD and used a snapshot near the end of the simulation as a starting configuration for the umbrella sampling simulations. In these simulations, Glu177 was restrained to 15 discrete values ranging from −200 to −60 degrees in 10-degree increments using a harmonic biasing potential with a force constant of 0.06 kcal·mol−1·degree−2. Each window was equilibrated for 2 ns, followed by 8 ns of production sampling. The program WHAM (membrane.urmc.rochester.edu/content/wham), which implements the weighted histogram analysis method (54), was used to compute the potential of mean force. A total of 150 bins were used, and the temperature was set at 300 K for WHAM. Five hundred steps of bootstrapping were used to estimate the statistical error in the simulations.
Results
Apo-XynII at Different pD Values.
Structures of the native XynII and N44D variant at pD values 6.2 and 6.4 (Table S1), respectively, which are close to the optimum pD for XynII activity, were expected to confirm protonation of Glu177 and the lack of proton on Glu86. Surprisingly, in the neutron structure of native XynII at pD 6.2, named pD6.2 henceforth, the side chains of both Glu177 and Glu86 are deprotonated and therefore are negatively charged. Glu177 is in the upward conformation facing Glu86, with the closest distance between the side chains being 5.9 Å. The carboxylates of Glu177 and Glu86 accept D atoms from the phenolic oxygens of Tyr88 and Tyr77 in two hydrogen bonds with D…O distances of 1.9 Å and 1.7 Å, respectively (Fig. 1A). Tyr88 is also hydrogen bonded to a water molecule whereas Tyr77 makes a weak interaction with Trp79. Glu177 accepts two more hydrogen bonds with nearby water molecules but is 4.4 Å away from the side-chain amide of Asn44. The Glu86 carboxylate in pD6.2 forms a 1.9 Å hydrogen bond with the adjacent Gln136 side chain and is also exposed to the bulk solvent.
Fig. 1.

Neutron scattering length density maps for the active sites in XynII structures. (A) pD6.2, (B) N44D, (C) pD8.9, and (D) pD4.8. 2FO − FC maps are shown as green meshes contoured at 1.2 σ. FO − FC omit maps are shown as red meshes contoured at 3.0 σ in A, B, and C, and at 2.5 σ in D. Hydrogen bonds are shown as blue dotted lines. D atoms are dark cyan whereas H atoms are light gray. Distances are in Å.
Previous 13C-NMR experiments using the homologous Bacillus circulans xylanase suggested that replacing an Asn residue with Asp near the acidic Glu residue increases the pKa of Glu due to the possibility of formation of a short hydrogen bond (33). Therefore, we expected that an equivalent N44D variant of the T. reesei enzyme would have a Glu177 pKa above 8. However, we still did not observe a D atom on the carboxylate of Glu177. The active site of the N44D variant at pD 6.4 (N44D) is very similar to that in pD6.2 (Fig. 1B). In both structures, Asp44 remains over 4 Å away from the deprotonated Glu177 positioned in the upward conformation. The omit FO − FC neutron scattering length density maps clearly demonstrate that the D atoms of the Tyr88 and Tyr77 phenolic groups are covalently bound to the oxygen atoms, indicating normal hydrogen bonds with Glu177 and Glu86 in both pD6.2 and N44D (Fig. 1 A and B).
In the neutron structure at the basic pD of 8.9, pD8.9 (Table S1), the omit FO − FC nuclear density map showed that the D atom taking part in the hydrogen bond between Glu177 and Tyr88 is located halfway between the two oxygens, with D…O distances of 1.5 and 1.6 Å, respectively (Fig. 1C). This observation indicates the possible presence of a low-barrier hydrogen bond induced by the increased pD. These changes in the hydrogen bonding at the active site might explain low catalytic activity of XynII in basic conditions.
Under acidic conditions, a significant change occurs in the XynII active site of the neutron structure at pD 4.8, pD4.8 (Fig. 1D). The Glu177 side chain rotates ∼90° about the χ2 dihedral angle so that the carboxylate moves away from Glu86 to assume a downward conformation. The distance between the side chains of Glu177 and Glu86 increases to 9.3 Å. In this conformation, the Glu177 side chain is protonated and positioned in a pocket lined by the backbone of Ser72 and Gly178, and by the side chain groups of Trp18, Asn44, Val46, Asn71, Tyr73, Tyr96, Ala175, and Tyr179, but makes no hydrogen bonding contacts with any of them. The COOH is hydrogen bonded at a distance of 1.5 Å to a D2O molecule, which is quite mobile as indicated by its high B-factor of ∼60 Å2. In pD4.8, Asn44 is located closer to Glu177 than in pD6.2, with the side-chain amide N being 3.7 Å away from the carboxylate O, but this distance is still too long for a hydrogen bond. There is a concerted movement of the aromatic rings of Tyr73, Tyr77, and Tyr88 in the opposite direction of Glu177 rotation upon decreasing the pD (Fig. 2). Surprisingly, the hydroxyl group of Tyr88 is oriented similarly in both the pD4.8 and pD6.2 even though its phenol group moves closer to, and is positioned within, hydrogen bonding distance of the Glu86 carboxylate in pD4.8 (O…O distance = 3.2 Å). The atomic positions are very similar in the low-temperature X-ray structures at low pH of the recombinant WT XynII at pH 4.0 and N44D variant at pH 4.6 (Table S2) (the rmsd of the main-chain atoms for the two structures is 0.1 Å). Interestingly, in the pH 4.0 X-ray structure, an Na+ cation was found instead of a water molecule observed in pD4.8 between Asn71 and Glu177 in the downward conformation. This observation can be explained by the large amount of NaCl (∼1 M) used in the crystallization conditions for the recombinant enzyme. Na+ is coordinated to the oxygen of the amide side chain of Asn71 and a water molecule and is 3.75 Å away from Glu177.
Fig. 2.

Superposition of pD4.8 (atoms colored by atom type: carbon is cyan) and pD6.2 (atoms colored by atom type: carbon is magenta).
Complexes with Mes.
We were not able to grow large crystals of an XynII complex with a bound substrate (30); however, we obtained a neutron structure with Mes to mimic proton donation by Glu177 to the glycosidic oxygen of the substrate in the first step of glycoside hydrolysis. A Mes molecule was found in the active site of the native XynII neutron structure at pD 6.4, denoted WT-Mes, and in the X-ray structure of N44E variant at high concentrations of Mes buffer (>0.5 M). The active sites of WT-Mes and N44E-Mes superimpose almost perfectly, with the ligand occupying almost identical positions. The only difference is in the orientation of the side chain of Glu44 relative to Asn44 in the two structures. Asn44 is oriented toward Glu177 in WT-Mes. The longer side chain of Glu44 cannot be accommodated in the same orientation. Thus, it rotates about 90° around the χ2 dihedral angle to face the bulk solvent, making a short C–H…O contact of 3.1 Å with the morpholinic ring of Mes (Fig. S3). In WT-Mes, Mes forms three hydrogen bonds with the active-site residues. The morpholinic nitrogen is protonated and donates its D atom in a strong hydrogen bond with Oε2 of Glu177 (D…O distance = 1.7 Å) (Fig. S3). The sulfonate makes two hydrogen bonds with the side-chain groups of Tyr73 and Gln136 (D…O distances = 1.9 Å). Additionally, the CH2 group vicinal to the nitrogen of the morpholinic ring makes a C–H…O contact with the Glu86 carboxylate. These interactions are similar to those made by xylohexaose substrate in E177Q-X6 (30). Oε1 of Glu177 is hydrogen bonded to Tyr88 whereas Glu86 forms two hydrogen bonds with Tyr77 and Gln136 in a similar fashion as in pD6.2. These two complexes are good proxies for the substrate-bound XynII protein, illustrating the upward conformation of the deprotonated Glu177 residue.
Fig. S3.

(A) Superposition of the active sites in N44E-Mes (green carbon atoms, dark magenta; 2FO − FC electron density map contoured at 2.0σ level) and WT-Mes (cyan carbon atoms). (B) Neutron scattering length density maps for the active site in WT-Mes. 2FO − FC map is shown as green mesh (cyan for the ligand) contoured at 1.5 σ. FO − FC omit map is shown as red mesh contoured at 3.0 σ. Hydrogen bonds are depicted as blue dotted lines. D atoms are dark teal whereas H atoms are light gray. Distances are in Å.
H/D Exchange Analysis.
Fig. S4 depicts the hydrogen/deuterium (H/D) exchange pattern for the main-chain amides in pD6.2 (see Fig. S5 for the analysis of the other neutron structures). Overall, xylanase exhibits moderate levels of H/D exchange, with about half of the residues showing full exchange and the rest being fully nonexchanged. Only a few residues are partially exchanged. Notably, β strands B3–B6 close to the catalytic glutamates are almost fully nonexchanged. β-sheets are rigid structures, stabilized by extensive hydrogen bonding between the strands, which typically undergo very little H/D exchange (34). The sole α-helix in the XynII structure contains fully exchanged amides on the outer side whereas those on the inner side facing β strands B7 and B8 are completely nonexchanged. The H/D exchange patterns in all other neutron structures are similar to that in pD6.2, except for pD8.9 with slightly higher exchange in the A2–A5 β-strands and the α-helix. This observation suggests that pD8.9 is more dynamic than the other structures, which may be due to the increased pD. The H/D exchange patterns for the studied structures are indicative of an enzyme whose backbone is well ordered (Figs. S4 and S5), in agreement with the previous NMR measurements (35).
Fig. S4.

Backbone H/D exchange patterns for pD6.2 depicted with the enzyme in diagram representation (A) and as topology (B). Magenta corresponds to fully exchanged main-chain amides (D atom occupancy of 0.60–1.00), yellow is for partially exchanged (D occupancy of 0.00–0.60), and blue indicates nonexchanged NH (D occupancy of −0.56 to 0.00). Proline residues lack the main-chain amide's hydrogen and are depicted in green.
Fig. S5.

Backbone H/D exchange patterns for N44D, pD8.9, pD4.8, and WT-Mes molecules depicted as topology. Magenta corresponds to fully exchanged main-chain amides (D atom occupancy of 0.60–1.00), yellow is for partially exchanged (D occupancy of 0.00–0.60), and blue indicates nonexchanged NH (D occupancy of −0.56 to 0.00). Proline residues lack the main-chain amide's hydrogen and are depicted in green.
pKa Calculations and Molecular Dynamics Simulations.
Using Poisson–Boltzmann continuum electrostatics calculations (see SI Methods for details), we observed a conformational dependence in the pKa of Glu177, with the downward state bearing a higher pKa than the upward state. We used pD4.8 and pD6.2 to estimate the pKa values of selected ionizable residues (Tables S3–S5). To investigate the dependence of the computed pKas on the internal dielectric constant (εint), we computed their values with εint = 10 and εint = 4. The pKa of Glu86 with εint = 10 is similar for both pD4.8 and pD6.2, with values of 3.5 and 3.9, respectively. Reducing εint to 4 had a minor effect for both structures, lowering the pKa by only 0.6 and 0.2 units, respectively, indicating that Glu86 is relatively insensitive to the choice of εint. The computed pKa values are consistent with the assumption that Glu86 is deprotonated throughout the reaction.
Table S3.
Computed pKa values of ionizable residues in apo-XynII and its Michaelis complex containing xylohexaose substrate
| Glu177 in downward conformation | Glu177 in upward conformation | Glu177 in downward conformation | Glu177 in upward conformation | |||||||||
| Residue | pD4.8 | pD4.8 | pD6.2 | pD6.2 | ΔpKaI | ΔpKaI | Michaelis complex | Michaelis complex | Michaelis complex | Michaelis complex | ΔpKaII | ΔpKaII |
| εint = 10 | εint = 4 | εint = 10 | εint = 4 | εint = 10 | εint = 4 | εint = 10 | εint = 4 | εint = 10 | εint = 4 | εint = 10 | εint = 4 | |
| Glu86 | 3.5 | 2.9 | 3.9 | 3.7 | −0.4 | −0.8 | 2.2 | 1.0 | 2.6 | 2.0 | −0.4 | −1.0 |
| Glu177 | 6.0 | 8.3 | 5.3 | 5.9 | +0.7 | +2.4 | 8.5 | >12.0 | 7.8 | >12.0 | +0.7 | N/A |
ΔpKaI is the difference in pKa values between the structures with Glu177 in the upward (pD6.2) and downward (pD4.8) conformations. ΔpKaII is the difference in pKa values in the Michaelis complexes with Glu177 in the downward or upward conformations.
Table S5.
Computed pKa values of selected ionizable residues in the Michaelis complex of XynII constructed from the E177Q-xylohexaose co-crystal structure (PDB ID code 4hk8)
| Residue | Glu177 downward | Glu177 downward | Glu177 upward | Glu177 upward |
| εint = 10 | εint = 4 | εint = 10 | εint = 4 | |
| Asp20 | 3.3 | 3.7 | 3.3 | 3.8 |
| Asp111 | 0.7 | 0.8 | <0.0 | <0.0 |
| Asp116 | 0.5 | <0.0 | 0.2 | <0.0 |
| Asp170 | 3.0 | 2.6 | 3.1 | 2.8 |
| Glu86 | 2.2 | 1.0 | 2.6 | 2.0 |
| Glu91 | <0.0 | <0.0 | 1.5 | 2.9 |
| Glu107 | 3.0 | 2.6 | 3.0 | 2.5 |
| Glu177 | 8.5 | >12.0 | 7.8 | >12.0 |
| His22 | 7.6 | 8.4 | 7.7 | 8.5 |
| His144 | 6.2 | 5.5 | 6.3 | 5.6 |
| His155 | 5.2 | <0.0 | 5.1 | <0.0 |
Q177 was mutated back to the native E177 and was oriented in either a downward or upward conformation.
Table S4.
Computed pKa values of selected ionizable residues in apo-Xyn II
| Residue | pD4.8 | pD4.8 | pD6.2 | pD6.2 |
| εint = 10 | εint = 4 | εint = 10 | εint = 4 | |
| Asp20 | 2.7 | 2.1 | 2.7 | 1.9 |
| Asp111 | 0.9 | 0.8 | 2.7 | 5.7 |
| Asp116 | 0.2 | <0.0 | 0.4 | <0.0 |
| Asp170 | 2.8 | 2.6 | 2.5 | 1.9 |
| Glu86 | 3.5 | 2.9 | 3.9 | 3.7 |
| Glu91 | 1.5 | 4.1 | 0.0 | <0.0 |
| Glu107 | 2.9 | 2.4 | 2.9 | 2.6 |
| Glu177 | 6.0 | 8.3 | 5.3 | 5.9 |
| His22 | 7.1 | 7.1 | 7.1 | 7.0 |
| His144 | 6.4 | 5.5 | 6.3 | 5.5 |
| His155 | 5.5 | <0.0 | 5.5 | <0.0 |
The pKa values of Glu177 in pD4.8 and pD6.2 were computed to be 6.0 and 5.3, respectively, when εint was set equal to 10. For pD4.8, the Glu177 carboxylate occupies a relatively buried, hydrophobic site when oriented downward, as evidenced by the computed desolvation penalty of ∼2.0 pK units. The desolvation penalty for the same residue in pD6.2 is 1.35 pK units, indicating that the carboxylate occupies a more hydrophilic site when in the upward conformation. To account for the apparent hydrophobicity increase of the Glu177 side chain in the downward state, we reduced the internal dielectric constant from 10 to 4 and recomputed the pKas. The result is a relatively large shift of +2.4 pK units, increasing the pKa of Glu177 in the downward conformation to 8.3. However, when the same procedure was carried out for pD6.2, the pKa was perturbed upward by only 0.6 pK units, indicating a noticeable difference in hydrophobicity between the two conformations. Thus, on the basis of the upper and lower limiting values considered here for the internal dielectric constant, we estimate that the pKa of Glu177 is 0.7–2.4 pK units higher in the downward than in the upward orientation.
To investigate whether a bound substrate alters the pKa of Glu177, we computed pKas using the 1.15-Å resolution X-ray crystal structure E177Q-X6 (Tables S3–S5). We replaced Gln with Glu and positioned its side chain in an energy-minimized downward conformation to mimic the Michaelis complex. The pKa of Glu177 calculated with εint = 4 increased by 3.7 pK units relative to the apo-enzyme whereas the pKa of Glu86 shifted down by −1.9 pK units. Although we expect errors in the absolute pKas, the direction of the pKa shifts should be meaningful. Thus, in the Michaelis complex, the substrate seems to perturb the pKas of Glu177 up and of Glu86 down relative to the apo-enzyme.
We used the same Michaelis complex model to estimate the free energy profile for converting the side chain of Glu177 from a downward to an upward conformation by performing classical umbrella sampling molecular dynamics (MD) simulations (see SI Methods for details). The χ2 dihedral of the Glu177 side chain was used as the reaction coordinate in the simulations, and Glu177 was modeled in the neutral, protonated state. Consistent with pD4.8, these simulations revealed a free energy minimum at χ2 = −78° (Fig. S6), which corresponds to the downward conformation of Glu177. For the rotation of χ2 toward the upward conformation, we obtained a free energy barrier of 4.3 kcal/mol with a maximum at χ2 = −135°. A second minimum at χ2 of −177°, corresponding to the upward state, was located at 1.4 kcal/mol above the first minimum.
Fig. S6.

Computed free energy profile for side chain rotation of protonated Glu177 from the downward (χ2 = −78) to the upward (χ2 = −177) conformation in the presence of the xylohexaose substrate. Error bars obtained from Bootstrapping analysis are shown.
Discussion
Enzymes function much more efficiently than hydronium (H3O+) or hydroxide (OH−) acid/base catalysts (36). GHs achieve their rate enhancements by converting the specific acid-catalyzed bimolecular hydrolysis of glycosides in solution into a unimolecular general acid catalysis in the active site of the enzyme (10). In GH11 enzymes, the general acid catalyst is a glutamic acid (Glu177 in XynII), which is initially protonated; a second Glu residue (Glu86 in XynII) serves the role of the nucleophile and is supposed to be negatively charged. To date, the protonation of the general acid catalyst has not been directly observed in any GH enzyme.
The interplay of the two catalytic Glu residues is crucial for initiating the hydrolysis reaction. As a consequence, the general acid must be a significantly weaker acid than the nucleophile, with a microscopic pKa above 6. The apparent pKa values for Glu177 and Glu86 in XynII have been estimated to be around 7 and 4 on the basis of the pH dependence of activity (37). The pKa values of corresponding Glu residues in B. circulans xylanase (BCX) have been determined using infrared spectroscopy and 13C NMR spectroscopy to be very similar to those in XynII, at 6.7 and 4.6, respectively (33, 38, 39). The active site residues are conserved in both enzymes; therefore, similar pKa values are expected. These pKa values indicate that, in slightly acidic conditions (pH ≤ 6), the protonation of Glu177 can be anticipated. Surprisingly, we did not observe a D atom on Oε2 of Glu177 in pD6.2 when the side chain adopts the upward conformation (Fig. 1A). The other oxygen of the carboxylate, Oε1, accepts D atoms in two hydrogen bonds with Tyr88 and a D2O and therefore cannot be protonated. We note that Gln177 occupies the same position in the E177Q-X6 Michaelis complex mimic, in which Nε2 of the side-chain amide makes a hydrogen bond with the glycosidic oxygen (30). Therefore, Oε2 of Glu177 must be the oxygen atom that accepts a proton from the bulk solvent and delivers it to the glycosidic oxygen. Furthermore, no protonation of Glu177 was found in N44D at pD 6.4 (Fig. 1B), which is even more surprising because NMR measurements indicated that the pKa of the general acid increases by about 1.5 pK units in the equivalent N35D variant of BCX relative to the WT enzyme (33, 40).
To reconcile the apparent inconsistencies between the experimental pKa values of the catalytic Glu residues requiring Glu177 protonation in XynII at pD ∼6, and the lack of D atoms on this glutamate in the pD6.2 and N44D neutron structures, we analyzed in more detail how the surroundings of both Glu177 and Glu86 may affect their pKa values. Both carboxylates are exposed to solvent and are similarly hydrated. Glu86 makes weak interactions with nearby mobile water molecules and is tightly hydrogen bonded to Tyr77 and Gln136. The hydrogen bonding lowers the pKa of Glu86, as does the 5 Å proximity to the positively charged Arg122, as was previously proposed (41). In the upward conformation, Glu177 is hydrogen bonded to two D2O molecules, makes weak contacts with several mobile waters, and is hydrogen bonded with Tyr88. The hydrogen bond between Tyr88 and Glu177 is an important feature of the catalytic site. Site-directed mutagenesis experiments demonstrated that substituting the equivalent Tyr80 in BCX with Phe crippled enzyme activity (33). In addition, the strength of the Glu177…Tyr88 hydrogen bond seems to be pH-dependent, with Tyr88 possibly having a pKa lower than the standard value of ∼10. When the pD was increased to 8.9, this interaction transformed into a possible low-barrier hydrogen bond, as observed in pD8.9, with the D atom of the phenolic oxygen situated halfway between the latter and Oε1 of Glu177 (Fig. 1C). Glu86 and Glu177 have comparable hydrophobic surroundings. Glu86 has hydrophobic interactions with Trp79 and Phe134 whereas Glu177 contacts Val46 and Tyr73. Consequently, the reason for the very different experimental pKa values of Glu86 and Glu177 (and the equivalent residues in other GH11 enzymes) seems unclear. The only features that set the carboxylates apart are one versus two hydrogen bonds made by Glu177 and Glu86, respectively, with enzyme residues, and the proximity of Arg122 to Glu86. Our pKa calculations revealed that, when Glu177 is in the upward conformation, its pKa is below 6, which would explain why protonation was not observed in our neutron structures, whereas Glu86 is expectedly more acidic than Glu177 (Table S3). The same trend in computed pKa values was found for the catalytic residues in BCX and Bacillus agaradhaerens xylanases (41).
Törrönen and Rouvinen previously proposed that Glu177 changes its conformation downward at pH ≤ 6 by rotating about χ2, placing it in a different environment that can alter its pKa (28, 29). We have directly observed that Glu177 is indeed protonated when it occupies the downward conformation in the pD4.8 neutron structure, and we argue that the downward conformation is functionally relevant. In this orientation the side-chain carboxylic group is positioned in a hydrophobic pocket, where it donates a D atom in a short hydrogen bond with a mobile D2O but makes no other hydrogen-bonding interactions (Figs. 1D and 2). Its computed pKa increases by 0.7–2.4 pK units relative to the value in the upward conformation, demonstrating that the carboxyl is more basic when positioned downward. The pKa of Glu86 is decreased by 0.4–0.8 pK units due to the negative charge on Glu177 moving away from it, which weakens their electrostatic interaction. Thus, the apparent pKa value of the general acid catalyst measured previously by NMR can be considered as an average of the intrinsic pKas of the two glutamate conformations. Sequence analysis of GH11 enzymes reveals that residues are highly conserved in the pocket surrounding the downward conformation of Glu177 (42). In acidophilic xylanases active well below pH 5.0, the Asn71, Gly178, and Tyr179 residues from this pocket are frequently replaced by Ser, Ala, and Trp, respectively, whereas Asn44 is universally substituted with Asp.
Based on the neutron structures and pKa calculations reported here, we propose that Glu177 obtains a proton from the solvent when in the downward conformation. In our X-ray structure of the recombinant WT XynII obtained using a high concentration of NaCl, an Na+ cation is located in the site occupied by the D2O hydrogen bonded to the protonated Glu177 in pD4.8. It is possible that this site specifically binds cations and could be occupied by an H3O+ ion that protonates Glu177 when it rotates downward. The side chain then rotates up to assume the upward conformation, in which Glu177 can act as a general acid catalyst to transfer its proton to the glycosidic oxygen of the substrate (Fig. 3). This conformational change requires ∼4 kcal/mol of free energy according to umbrella sampling simulations of the Michaelis complex, with the protonated Glu177 being slightly more stable in the downward than the upward conformation (Fig. S6). The energy barrier is low enough, however, that the downward and upward conformations of the side chain of Glu177 are in dynamic equilibrium before the hydrolysis reaction begins. Interestingly, upon downward rotation of Glu177, there is a significant movement of nearby tyrosine residues Tyr73, Tyr77, and Tyr88, whose side chains shift in the opposite direction to that of the glutamate motion (Fig. 2). This movement brings the phenolic oxygen of Tyr88 within hydrogen bonding distance of the carboxylate of Glu86 (O…O distance = 3.2 Å). However, the O-D group of Tyr88 does not rotate toward Glu86 but remains oriented toward Glu177. This behavior of Tyr88 would ensure that, when Glu177 returns back to the upward conformation, there is no extra energy cost associated with breaking a hydrogen bond with Glu86 and that Tyr88 always stays correctly positioned to quickly capture the moving Glu177 in the correct orientation for the reaction to be initiated.
Fig. 3.
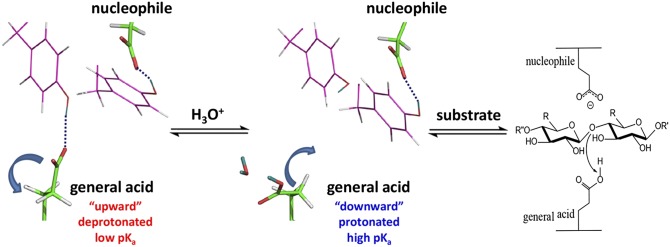
Proposed cycling of the general acid between two conformations controlling pKa values in GH11 enzymes that ensure Glu protonation and delivery of the proton to the substrate’s glycosidic oxygen. A Tyr residue that is hydrogen bonded to the general acid moves toward the nucleophile when the general acid changes its conformation downward, but the phenolic group of the Tyr makes no hydrogen bond with the nucleophile.
Changes in the computed pKa values of the catalytic glutamates due to the rotation of Glu177 in the Michaelis complex are similar to those calculated for apo-XynII (Table S3). Glu177 becomes a stronger acid and Glu86 a weaker acid when the side-chain orientation of Glu177 changes from the downward to the upward conformation. In the WT-Mes neutron structure, Glu177 forms a short hydrogen bond with the protonated amino nitrogen of Mes whereas Glu86 is 3.3 Å from the carbon atom vicinal to the nitrogen (Fig. S3). These two contacts represent interactions that the catalytic glutamates would make in the real reactant complex although the hydrogen would be donated by Glu177 to the glycosidic oxygen. In WT-Mes, Oε1 of Glu177 makes an identical hydrogen bond with Tyr88 to that in pD6.2. Asn44 moves 0.7 Å closer to Oε2 relative to its position in pD6.2 but does not form a hydrogen bond with Glu177 as observed in E177Q-X6 (30). Repulsive interactions of Asn44 with the morpholinic ring, whose orientation is different from the xylose unit at position −1, may be responsible for its inability to slide closer to Glu177 in WT-Mes.
It has been previously demonstrated that a single amino acid substitution of Asn35 to Asp in BCX reduces the pH optimum of activity from 5.7 to 4.6 (40). It was proposed that the N35D variant follows a reverse protonation mechanism (43). In that scenario, the catalytically active state of the enzyme would contain the minor protonated species of Asp35 with a pKa of 3.7, acting as the general acid instead of Glu172, and the minor deprotonated species of the nucleophilic Glu78 with a pKa of 5.7. Because the pKa of the general acid Glu172 was measured to be 8.4 in the N35D variant, both residues Asp35 and Glu172 were considered to be protonated in the catalytic site, serving the role of the general acid in unison because Glu172 would be too weak an acid to function alone. We did not observe protonation of the corresponding general acid Glu177 in our N44D neutron structure, indicating that the upward conformation has a pKa lower than 6.0, as it does in the native XynII. We suggest that, in N44D (corresponding to N35D of BCX and other similarly substituted xylanases), Glu177 can still act as the general acid catalyst, while maintaining dynamic equilibrium between the downward conformation with the higher pKa and the upward conformation with the lower pKa. The downward conformation may have a higher pKa than that in the native enzyme due to the presence of a nearby negative charge on the Asp, explaining the value of 8.4 for the general acid determined by NMR in the B. circulans N35D variant. With the general acid being able to cycle its pKa by adopting two different conformations, and its intrinsic pKa in the upward conformation being close to that in the native enzyme according to our neutron structure, a reverse protonation mechanism is unnecessary to explain the activity of the Asn-to-Asp variant at pH values above 5. However, protonation of this Asp may become important at pH values below 5 when the population of the neutral species can be sufficient to affect the rate of catalysis.
Conclusions
To our knowledge, this study is the first to report direct determination of proton positions in a GH enzyme using neutron crystallography. We have obtained five neutron structures of a GH11 xylanase at different pD values in the apo- and ligand-bound forms and performed pKa calculations and free energy simulations. Our findings have led us to propose that, in GH11 enzymes, the Glu general acid side chain dynamically moves between the downward conformation, with its high pKa facilitating its protonation, and the upward conformation, with its low pKa facilitating the delivery of a proton to the glycosidic oxygen (Fig. 3). We believe that our findings resolve decades of confusion surrounding the question of why the general acid and nucleophilic glutamates in GH11 enzymes, while having similar hydrogen-bonding interactions and solvent accessibilities, exhibit very different pKa values and assume their corresponding catalytic roles. On the basis of the H/D exchange analysis of XynII and the dynamics of BCX measured with NMR, we hypothesize that GH11 enzymes do not require palm dynamics for function, but the conformational flexibility of the thumb and fingers may be crucial for substrate binding and product release.
Methods
For neutron diffraction, single crystals of XynII and recombinant N44D variant of 2–4 mm3 in volume were obtained using vapor diffusion methods in sitting drops. Neutron crystallographic data were collected at the Protein Crystallography Station (PCS), the Los Alamos Neutron Science Center (LANSCE), the Los Alamos National Laboratory (LANL), BioDiff [Forschungs-Neutronenquelle Heinz Maier-Leibnitz (FRM II)], and the Macromolecular Neutron Diffractometer (MaNDi) [Spallation Neutron Source (SNS), Oak Ridge National Laboratory (ORNL)]. Corresponding room-temperature X-ray diffraction data for joint X-ray/neutron refinement were collected in-house. Low-temperature X-ray data were obtained on beamline ID19 at 100 K at the Advanced Photon Source (Argonne, IL). The neutron structures were refined in nCNS and PHENIX using joint X-ray/neutron refinement. The H++ web server version 3.1, which uses a Poisson–Boltzmann continuum electrostatics approach to approximate the electrostatic environment of biological macromolecules, was used to compute the pKas of ionizable residues in the pD 4.8 and pD 6.2 neutron structures of xylanase, pD4.8 and pD6.2, respectively. Methods are described in detail in SI Methods.
Acknowledgments
We thank Dr. Demian M. Riccardi for his contributions to pKa calculations. The research was partly supported by the Laboratory Directed Research and Development Program at Oak Ridge National Laboratory (ORNL), which is managed by UT-Battelle LLC for the US Department of Energy (DOE) under DOE Contract DE-AC05-00OR22725. We thank the Center for Structural Molecular Biology at ORNL, supported by the DOE Office of Biological and Environmental Research (BER) for using their facilities. The Macromolecular Neutron Diffractometer beamline at the Spallation Neutron Source was supported by the Scientific User Facilities Division, DOE Basic Energy Sciences. The Protein Crystallography Station is funded by the DOE BER. We thank ID19 beamline at the Advanced Photon Source at Argonne National Laboratory for assistance with data collection. Argonne is operated by UChicago Argonne LLC for the DOE Office of Science under Contract DE-AC02-06CH11357. L.C., P.L., and A.K. were partly supported by the DOE BES. Q.W. is partly supported by the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry, and the Innovation Fund of Yangzhou University (Grant 2014CXJ058). This manuscript has been authored by UT-Battelle, LLC under Contract DE-AC05-00OR22725 with the US Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (energy.gov/downloads/doe-public-access-plan).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. D.R. is a guest editor invited by the Editorial Board.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4S2H, 4S2G, 4S2F, 4S2D, 4XPV, 4XQD, 4XQ4, and 4XQW).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1504986112/-/DCSupplemental.
References
- 1.Wolfenden R, Lu X, Young G. Spontaneous hydrolysis of glycosides. J Am Chem Soc. 1998;120(27):6814–6815. [Google Scholar]
- 2.Wilson DB. Cellulases and biofuels. Curr Opin Biotechnol. 2009;20(3):295–299. doi: 10.1016/j.copbio.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 3.Dodd D, Cann IKO. Enzymatic deconstruction of xylan for biofuel production. Glob Change Biol Bioenergy. 2009;1(1):2–17. doi: 10.1111/j.1757-1707.2009.01004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharma V, Nagpal M, Jain UK, Mangotia A, Kumar R. Antidiabetic drug and combination therapy. Adv Res Pharm Biol. 2013;3:389–394. [Google Scholar]
- 5.Gloster TM, Davies GJ. Glycosidase inhibition: Assessing mimicry of the transition state. Org Biomol Chem. 2010;8(2):305–320. doi: 10.1039/b915870g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Speciale G, Thompson AJ, Davies GJ, Williams SJ. Dissecting conformational contributions to glycosidase catalysis and inhibition. Curr Opin Struct Biol. 2014;28:1–13. doi: 10.1016/j.sbi.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith MB, March J. March’s Advanced Organic Chemistry: Reactions, Mehcanisms, and Structure. Wiley Interscience; Hoboken, NJ: 2007. [Google Scholar]
- 8.Selwood T, Sinnott ML. A solvent-isotope-effect study of proton transfer during catalysis by Escherichia coli (lacZ) beta-galactosidase. Biochem J. 1990;268(2):317–323. doi: 10.1042/bj2680317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenberg S, Kirsch JF. Oxygen-18 leaving group kinetic isotope effects on the hydrolysis of nitrophenyl glycosides. 1. Beta-galactosidease-catalyzed hydrolysis. Biochemistry. 1981;20(11):3189–3196. doi: 10.1021/bi00514a031. [DOI] [PubMed] [Google Scholar]
- 10.Legler G. Glycoside hydrolases: Mechanistic information from studies with reversible and irreversible inhibitors. Adv Carbohydr Chem Biochem. 1990;48:319–384. doi: 10.1016/s0065-2318(08)60034-7. [DOI] [PubMed] [Google Scholar]
- 11.Koshland DE., Jr Stereochemistry and the mechanism of enzymatic reactions. Biol Rev Camb Soc. 1953;28:416–435. [Google Scholar]
- 12.Zechel DL, Withers SG. Glycosidase mechanisms: Anatomy of a finely tuned catalyst. Acc Chem Res. 2000;33(1):11–18. doi: 10.1021/ar970172+. [DOI] [PubMed] [Google Scholar]
- 13.Phillips DC. The Henn egg-white lysozyme molecule. Proc Natl Acad Sci USA. 1967;57(3):483–495. [Google Scholar]
- 14.Rupley JA, Gates V. Studies of the enzymatic activity of lysozyme, II. The hydrolysis and transfer reactions of N-acetylglucosamine oligosaccharides. Proc Natl Acad Sci USA. 1967;57(3):496–510. [Google Scholar]
- 15.Chiba S. A historical perspective for the catalytic reaction mechanism of glycosidase; so as to bring about breakthrough in confusing situation. Biosci Biotechnol Biochem. 2012;76(2):215–231. doi: 10.1271/bbb.110713. [DOI] [PubMed] [Google Scholar]
- 16.Banait NS, Jencks WP. Reaction of anionic nucleophiles with α-D-glucopyranosyl fluoride in aqueous solution through a concerted, ANDN (SN2) mechanism. J Am Chem Soc. 1991;113(21):7951–7958. [Google Scholar]
- 17.Notenboom V, et al. Insights into transition state stabilization of the β-1,4-glycosidase Cex by covalent intermediate accumulation in active site mutants. Nat Struct Biol. 1998;5(9):812–818. doi: 10.1038/1852. [DOI] [PubMed] [Google Scholar]
- 18.Davies GJ, et al. Snapshots along an enzymatic reaction coordinate: Analysis of a retaining β-glycoside hydrolase. Biochemistry. 1998;37(34):11707–11713. doi: 10.1021/bi981315i. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki R, et al. Crystallographic snapshots of an entire reaction cycle for a retaining xylanase from Streptomyces olivaceoviridis E-86. J Biochem. 2009;146(1):61–70. doi: 10.1093/jb/mvp047. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka Y, Tao W, Blanchard JS, Hehre EJ. Transition state structures for the hydrolysis of alpha-D-glucopyranosyl fluoride by retaining and inverting reactions of glycosylases. J Biol Chem. 1994;269(51):32306–32312. [PubMed] [Google Scholar]
- 21.Huang X, Tanaka KSE, Bennet AJ. Glucosidase-catalyzed hydrolysis of α-d-glucopyranosyl pyridinium salts: Kinetic evidence for nucleophilic involvement at the glucosidation transition state. J Am Chem Soc. 1997;119(46):11147–11154. [Google Scholar]
- 22.Mori H, et al. Catalytic reaction mechanism based on α-secondary deuterium isotope effects in hydrolysis of trehalose by European honeybee trehalase. Biosci Biotechnol Biochem. 2009;73(11):2466–2473. doi: 10.1271/bbb.90447. [DOI] [PubMed] [Google Scholar]
- 23.Vuong TV, Wilson DB. Glycoside hydrolases: Catalytic base/nucleophile diversity. Biotechnol Bioeng. 2010;107(2):195–205. doi: 10.1002/bit.22838. [DOI] [PubMed] [Google Scholar]
- 24.White A, Rose DR. Mechanism of catalysis by retaining β-glycosyl hydrolases. Curr Opin Struct Biol. 1997;7(5):645–651. doi: 10.1016/s0959-440x(97)80073-5. [DOI] [PubMed] [Google Scholar]
- 25.Niimura N, Podjarny A. Neutron Protein Crystallography. Hydrogen, Protons, and Hydration in Bio-Macromolecules. Oxford Univ Press; Oxford: 2011. p. 232. [Google Scholar]
- 26.Paës G, Berrin J-G, Beaugrand J. GH11 xylanases: Structure/function/properties relationships and applications. Biotechnol Adv. 2012;30(3):564–592. doi: 10.1016/j.biotechadv.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 27.Törrönen A, et al. The two major xylanases from Trichoderma reesei: Characterization of both enzymes and genes. Biotechnology (N Y) 1992;10(11):1461–1465. doi: 10.1038/nbt1192-1461. [DOI] [PubMed] [Google Scholar]
- 28.Törrönen A, Harkki A, Rouvinen J. Three-dimensional structure of endo-1,4-β-xylanase II from Trichoderma reesei: Two conformational states in the active site. EMBO J. 1994;13(11):2493–2501. doi: 10.1002/j.1460-2075.1994.tb06536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Törrönen A, Rouvinen J. Structural comparison of two major endo-1,4-xylanases from Trichoderma reesei. Biochemistry. 1995;34(3):847–856. doi: 10.1021/bi00003a019. [DOI] [PubMed] [Google Scholar]
- 30.Wan Q, et al. X-ray crystallographic studies of family 11 xylanase Michaelis and product complexes: Implications for the catalytic mechanism. Acta Crystallogr D Biol Crystallogr. 2014;70(Pt 1):11–23. doi: 10.1107/S1399004713023626. [DOI] [PubMed] [Google Scholar]
- 31.Davies GJ, Wilson KS, Henrissat B. Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem J. 1997;321(Pt 2):557–559. doi: 10.1042/bj3210557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mhlongo NN, Skelton AA, Kruger G, Soliman MES, Williams IHA. A critical survey of average distances between catalytic carboxyl groups in glycoside hydrolases. Proteins. 2014;82(9):1747–1755. doi: 10.1002/prot.24528. [DOI] [PubMed] [Google Scholar]
- 33.Joshi MD, et al. Dissecting the electrostatic interactions and pH-dependent activity of a family 11 glycosidase. Biochemistry. 2001;40(34):10115–10139. [PubMed] [Google Scholar]
- 34.Blum M-M, et al. Rapid determination of hydrogen positions and protonation states of diisopropyl fluorophosphatase by joint neutron and X-ray diffraction refinement. Proc Natl Acad Sci USA. 2009;106(3):713–718. doi: 10.1073/pnas.0807842106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Connelly GP, Withers SG, McIntosh LP. Analysis of the dynamic properties of Bacillus circulans xylanase upon formation of a covalent glycosyl-enzyme intermediate. Protein Sci. 2000;9(3):512–524. doi: 10.1110/ps.9.3.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ghanem E, Raushel FM (2007) Enzymes: The active site. Handbook of Proteins: Structure, Function and Methods, eds Cox MM, Phillips GN (Wiley, Chichester, UK), Vol 1, pp 396–403.
- 37.Tenkanen M, Puls J, Poutanen K. Two major xylanases from Trichoderam reesei. Enzyme Microb Technol. 1992;14:566–574. [Google Scholar]
- 38.Davoodi J, Wakarchuk WW, Campbell RL, Carey PR, Surewicz WK. Abnormally high pKa of an active-site glutamic acid residue in Bacillus circulans xylanase: The role of electrostatic interactions. Eur J Biochem. 1995;232(3):839–843. [PubMed] [Google Scholar]
- 39.McIntosh LP, et al. The pKa of the general acid/base carboxyl group of a glycosidase cycles during catalysis: A 13C-NMR study of bacillus circulans xylanase. Biochemistry. 1996;35(31):9958–9966. doi: 10.1021/bi9613234. [DOI] [PubMed] [Google Scholar]
- 40.Joshi MD, et al. Hydrogen bonding and catalysis: A novel explanation for how a single amino acid substitution can change the pH optimum of a glycosidase. J Mol Biol. 2000;299(1):255–279. doi: 10.1006/jmbi.2000.3722. [DOI] [PubMed] [Google Scholar]
- 41.Kongsted J, Ryde U, Wydra J, Jensen JH. Prediction and rationalization of the pH dependence of the activity and stability of family 11 xylanases. Biochemistry. 2007;46(47):13581–13592. doi: 10.1021/bi7016365. [DOI] [PubMed] [Google Scholar]
- 42.Sapag A, et al. The endoxylanases from family 11: Computer analysis of protein sequences reveals important structural and phylogenetic relationships. J Biotechnol. 2002;95(2):109–131. doi: 10.1016/s0168-1656(02)00002-0. [DOI] [PubMed] [Google Scholar]
- 43.Mock WL. Theory of enzymatic reverse protonation catalysis. Bioorg Chem. 1992;20(4):377–381. [Google Scholar]
- 44.Wan Q, et al. Heterologous expression, purification, crystallization and preliminary X-ray analysis of Trichoderma reesei xylanase II and four variants. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2013;69(Pt 3):320–323. doi: 10.1107/S1744309113001164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: The integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 8):859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 46.Sheldrick GM, Schneider TR. SHELXL: High-resolution refinement. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 47.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kovalevsky AY, et al. Preliminary joint X-ray and neutron protein crystallographic studies of endoxylanase II from the fungus Trichoderma longibrachiatum. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2011;67(Pt 2):283–286. doi: 10.1107/S174430911005075X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Otwinoski Z, Minor W. 1997. Macromolecular Crystallography, Part A. Methods in Enzymology, eds Carter CW, Jr, Sweet RM (Academic, New York), Vol 276, pp 307–326.
- 50.Coates L, et al. The macromolecular neutron diffractometer MaNDi at the Spallation Neutron Source. J Appl Cryst. 2015;48:1302–1306. [Google Scholar]
- 51.Adams PD, Mustyakimov M, Afonine PV, Langan P. Generalized X-ray and neutron crystallographic analysis: More accurate and complete structures for biological macromolecules. Acta Crystallogr D Biol Crystallogr. 2009;65(Pt 6):567–573. doi: 10.1107/S0907444909011548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anandakrishnan R, Aguilar B, Onufriev AVH. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012;40(Web Server issue):W537–W541. doi: 10.1093/nar/gks375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J Comput Chem. 1992;13(8):1011–1021. [Google Scholar]
- 54.Phillips JC, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brooks BR, et al. CHARMM: The biomolecular simulation program. J Comput Chem. 2009;30(10):1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guvench O, Hatcher ER, Venable RM, Pastor RW, MacKerell AD., Jr CHARMM additive all-atom force field for glycosidic linkages between hexopyranoses. J Chem Theory Comput. 2009;5(9):2353–2370. doi: 10.1021/ct900242e. [DOI] [PMC free article] [PubMed] [Google Scholar]