

Abstract
Heart function fails when the organ is unable to pump blood at a rate proportional to the body’s need for oxygen or when this function leads to elevated cardiac chamber filling pressures (cardiogenic pulmonary edema). Despite our sophisticated knowledge of heart failure, even so-called ejection fraction-preserved heart failure has high rates of mortality and morbidity. So, novel therapies are sorely needed. This review discusses current standard therapies for heart failure and launches an exploration into emerging novel treatments on the heels of recently-approved sacubitril and ivbradine. For example, Vasoactive Intestinal Peptide (VIP) is protective of the heart, so in the absence of VIP, VIP knockout mice have dysregulation in key heart failure genes: 1) Force Generation and Propagation; 2) Energy Production and Regulation; 3) Ca+2 Cycling; 4) Transcriptional Regulators. VIP administration leads to coronary dilation in human subjects. In heart failure patients, VIP levels are elevated as a plausible endogenous protective effect. With the development of elastin polymers to stabilize VIP and prevent its degradation, VIP may therefore have a chance to satisfy the unmet need as a potential treatment for acute heart failure.
Keywords: heart failure, novel therapies, sacubitri, ivbradine, vasoactive intestinal peptide, VIP, genes, knockout mice
Introduction
Heart function fails when the organ is unable to pump blood at a rate proportional to the body’s need for oxygen or when this function leads to elevated cardiac chamber filling pressures (cardiogenic pulmonary edema). Despite our sophisticated knowledge of heart failure, even the so-called ejection fraction–preserved heart failure has high rates or mortality and morbidity. So, novel therapies are sorely needed. With over a million hospitalizations annually – up 175% over the past 25 years – and costs of nearly $15.4 billion dollars, acute heart failure is a critically important health concern. Furthermore, half of the patients discharged from the hospital are readmitted within half-a-year. In-hospital mortality remains high between 4% and 7%.1,2 Heart failure is a significant problem as the population ages. The prevalence is 2.5% of the US population or 5 million patients (from the National Health and Nutrition Education Survey).2
Common etiological mechanisms of heart failure include coronary ischemia, valvular disease, hypertension, and diastolic dysfunction. Yet, other causes include: postpartum cardiomyopathy; postinfectious, chronic tachycardia; metabolic dysregulation; adverse medication side effects (particularly adriamycin chemotherapy); orphan disease Duchenne’s muscular dystrophy; infiltrative diseases (such as amyloidosis); and inflammatory/connective tissue diseases (such as systemic lupus erythematosus). When known causes of heart failure are excluded, then heart failure is classified as idiopathic. Less often studied versus chronic heart failure, acute decompensated heart failure is associated with abrupt-onset symptoms associated with hospitalization. Nearly half of the admitted patients with heart failure have preserved ejection fraction.2,3
In order to appreciate the physiology of heart failure, a review of fundamental principles is on order. The Frank–Starling Law states that the stroke volume of the heart increases in response to an increase in the volume of blood filling the heart (the end diastolic volume) when all other factors remain constant. The increased volume of blood stretches the ventricular wall, causing cardiac muscle to contract more forcefully (the so-called Frank–Starling mechanism). The stroke volume may also increase as a result of greater contractility of the cardiac muscle during exercise, independent of the end-diastolic volume. The Frank–Starling mechanism appears to make its greatest contribution to increasing stroke volume at lower work rates, and contractility has its greatest influence at higher work rates.4
While the Frank–Starling mechanism may contribute at lower work rates, cardiac injury at lower work rates may activate additional mechanisms. Cardiac injury in heart failure leads to not only reduced left ventricular (LV) dysfunction but also activation of the renin angiotensin system and sympathetic nervous system, ultimately causing LV thickening, remodeling, vasoconstriction (with pulmonary hypertension and cell dysregulation), and, possibly, apoptosis or programmed cell death. This sequence culminates in clinical heart failure – with attendant mortality and morbidity. Severity is graded by the New York Heart Association Class, with increasing severity of classes I to IV. Another grading system is the American Heart Association Staging System with stage C, classified as the symptomatic stage. This stage C is a result of known structural heart diseases and is associated with shortness of breath, fatigue, and reduced exercise tolerance. The obvious goal is to prevent progressive LV dysfunction to end-stage D, resulting in LV assist device implantation, cardiac transplant, or palliative care.5
Current Standard Therapies
Medication management of acute heart failure often entails diuretics to reduce preload, vasodilators to reduce preload and/or afterload, and inotropes to augment contractility. Angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) and aldosterone antagonists also aim to reduce preload and afterload.6 One adverse side effect (usually nonallergic antibody, non-IgE–mediated) of ACE inhibitors is angioedema (risk up to 15.5%). The risk of angioedema from ARBs is 4.4%. For ACE inhibitors, this angioedema results from increased bradykinin production, which can occur at any time while taking these medications due to interruption of the conversion of angiotensin I to angiotensin II. Angioedema from ACE inhibitors and ARBs may lead to life-threatening swelling of the throat, requiring critical interventions, ie, intubation or tracheostomy. For ARBs, the mechanism of action of angioedema is not due to direct production of bradykinin and is unclear. Some chronic persistent angioedema may occur despite cessation of these medications. Some evidence suggests that treatment with a bradykinin B2 receptor blocker (icatibant) may attenuate angioedema attacks early.7–9 Patients with hereditary angioedema (deficiency of C1 esterase inhibitor) and spontaneous attacks of swelling should not take ACE inhibitors. Clearly, it would be prudent to design a heart failure drug that does not incur risk of angioedema.
Other medications used for heart failure include inotropes – such as dopamine and dobutamine. While inotropes increase cardiac output, they often do so at the expense of increasing myocardial oxygen demand, predisposition to arrhythmias, and neurohormonal activation. Some patients may need invasive monitoring. Furthermore, inotropes do not improve mortality. There is 50% survival at 6 months and nearly 100% mortality at 1 year for those heart failure patients who are dependent on inotropes.10
Long-term survival among heart failure patients may be improved with β-blockers,11 ACE inhibitors,12 aldosterone antagonists,13 electrophysiology devices such as automatic implantable cardiovascular defibrillators, and vasodilators. Other drugs such as digoxin and diuretics do not alter death rates – digoxin reduces hospitalizations, while diuretics (furosemide or lasix) improve symptoms.
Another aspect of heart failure is diastolic dysfunction with preserved LV ejection fraction, accounting for half of the hospitalizations. Pathophysiologically, there is concentric remodeling and increased LV end diastolic pressure from a stiff left ventricle, thereby preventing relaxation. Medications for diastolic dysfunction are similar to systolic dysfunction: ACE inhibitors, ARBs, diuretics, and β-blockers.14
Emerging Novel Therapies
LCZ696
In August of 2014, the first new drug for heart failure in many years, called LCZ696, was shown to prevent deaths and hospitalizations in heart failure patients – better than the gold standard enalipril, which is an ACE inhibitor. In the PARADIGN-HF study, LCZ696, which is comprised of two drugs – an ARB and a neprilysin inhibitor called sacubitril – reduced the risk of cardiovascular-related death to 13.3%, versus the ACE inhibitor cohort rate of 16.5%. This use of sacubitril is an example of utilizing new or different pathways to treat heart failure.15
Sacubitril is a prodrug that is activated to LBQ657 by de-ethylation via esterases.16 LBQ657 inhibits the enzyme neprilysin,17 which is responsible for the degradation of atrial and brain natriuretic peptide, two blood pressure–lowering peptides that work mainly by reducing blood volume. As a result of reduced degradation of helpful peptides, these heart failure patients on sacubitril have less circulating blood volume and have done well in terms of prevention of deaths and hospitalizations (supra vide).
Ivabradine
On April 15, 2015, the Food and Drug Administration approved Corlanor (ivabradine) to reduce hospitalizations from worsening heart failure. The indications for ivabradine are specific: symptoms of heart failure that are stable and a normal heartbeat with a resting heart rate of at least 70 beats per minute and patients must already be taking β-blockers at the highest dose they can tolerate. Ivabradine is thought to work by decreasing heart rate and represents the first approved product in this drug class. It works via the If (funny channel) and does not affect other cardiac ionic currents (not beta receptor mediated). Heart rate lowering with ivabradine reduces myocardial oxygen demand, simultaneously improving oxygen supply. There are no negative inotropic or lusitropic (myocardial relaxation) effects from ivabradine. Ventricular contractility is preserved.18 In clinical trials of 6,505 participants, hospitalizations for heart failure were reduced compared to placebo. The most notable side effects were bradycardia, hypertension, atrial fibrillation, and temporary visual disturbance entailing seeing flashes of light. The bradycardia may be severe.19
Vasoactive intestinal peptide
Other novel therapeutic agents for heart failure may consequently have plausible efficacy on the heels of sacubitril and ivabradine. For example, Vasoactive intestinal peptide (VIP), discovered by Drs Sami I. Said and Victor Mutt in the 1970s, is a 28–amino acid peptide found in nerve terminals and in mast cells.20 VIP increases cyclic adenosine monophosphate (cAMP) intracellularly to cause cardiac contraction in an analogous fashion to glucagon.21 VIP also is a smooth muscle relaxant, which may be beneficial in pulmonary hypertension associated with heart failure22 and VIP inhibits the proliferation of smooth muscle cells, attenuating vascular remodeling.23
Inasmuch as sometimes vascular remodeling is a compensatory mechanism for dysfunction in heart failure, VIP has promise to treat this deleterious component. Enhanced peripheral vascular tone is a major factor in determining deterioration of clinical heart failure. Nakamura has reviewed peripheral circulatory failure in heart failure and notes that it is caused “not only by simple arterial muscle constriction, but also by structural and functional changes, including receptor and post-receptor levels in the vasculature.” Thus, vascular remodeling potentially may be an important mechanism underlying vasodilatory failure in both limb conduit and intraskeletal muscle vessels, contributing significantly to LV dysfunction and exercise intolerance in patients with heart failure.24
What is compelling about VIP as a novel therapeutic agent for heart failure is evident in mice lacking the gene for VIP. These mice have dilated cardiomyopathy. Human heart failure gene programs are overexpressed in VIP knockout mice.25 So, the plausibility of VIP as both a treatment for heart failure in patients and a modifier of one’s biology in a pharmacogenomic fashion may have traction.
Four categories of genes triggering heart failure are increased in mice without heart-protective VIP: (1) force generation and propagation; (2) energy production and regulation; (3) Ca2+ cycling; and (4) transcriptional regulators. Force generation gene mutations encoding proteins of the muscle sarcomere have been associated with both hypertrophic and dilated cardiomyopathy. Overexpression of sarcomere protein expression in the setting of a dilated cardiomyopathy26 may suggest a compensatory mechanism. Upregulated expression of other force generation genes such as sarcoglycan and caveolin27 in VIP knockout mice supports the concept of increased force to the extracellular matrix, with ventricular hypertrophy, for apparent release of protein products of these genes in heart failure.
Regarding energy production and regulation genes, it has been increasingly recognized that nuclear-encoded metabolic gene mutations are key regulators of hypertrophic cardiac remodeling and heart failure. For example, mutations in genes encoding the lysosome-associated membrane protein are associated with heart failure in patients.28 In the absence of VIP in VIP knockout (KO) mice, upregulation of PRKAG2, for example,29 would support a compensatory response to thinning of the cardiac wall. Another category of gene program alteration – Ca2+ cycling – is critically important, since protein and RNA levels of key calcium modulators are altered in acquired and inherited forms of heart failure.
Overexpression of FKBP30 in mice supports the concept that these mice would have aberrant excessive release of calcium during the relaxation/diastolic phase of the cardiac cycle, enhancing the opportunity to generate hypertrophy. Cardiac hypertrophy is attenuated in murine models with overexpression of Caveolin-3, a potent upstream inhibitor of T-type Ca2+ current,18 and rats treated with Calhex231, an inhibitor of calcium-sensing receptor.31 Similarly, deleterious LV hypertrophy may therefore plausibly be suppressed with exogenous VIP administration. The fourth category of genetic alteration is transcriptional regulators. Mechanisms which activate or repress cardiac gene transcription may induce key molecules, which directly or indirectly lead to cardiac remodeling. Lack of VIP in VIP KO mice with the phenotype of biventricular dilated cardiomyopathy and primary pulmonary hypertension is concurrent with strong overexpression of cardiac muscle genes, supporting the concept of VIP in vivo as a maestro conductor maintaining homeostasis of the heart.26
Table 1 shows on the next page multiple heart failure gene programs which are overexpressed in VIP KO mice. An accompanying bar chart (Fig. 1) in blue demonstrates progressive right ventricular hypertrophy in these mice with age. MicroMRI (Fig. 2) additionally shows right ventricular dilation in VIP KO mice. These physiological abnormalities are associated with premature death compared to the wild type (Fig. 3). Compensatory and pathogenic gene programs are displayed in Figure 4, with overexpression of leptin as a compensatory reaction for cardiac cachexia and low body weight along with proinflammatory interleukin-6 (IL-6) and IL-1. VIP signal transduction is shown in Figure 6. VIP increases intracellular cAMP and acts via protein kinase A to activate transcriptional promoters.32,33 VIP upregulates IL-10, which is anti-inflammatory.34
Table 1.
Gene alterations related to hypertrophic/dilated cardiomyopathy in VIP KO mice compared to wild-type (WT) mice.
| GENE SYMBOL | GENE NAME | KO/WT (FOLD CHANGE) | P VALUE | DESCRIPTION | |
|---|---|---|---|---|---|
| casq1 | calsequestrin 1 | 19 | 0.004 | Calsequestrin is a major modulator of Ca2+ released from the sarcoplasmic reticulum. Overexpression of these proteins induces cellular hypertrophy in cardiac myocytes.39,40 | |
| casq2 | calsequestrin 2 | 2 | 0.050 | ||
| ttn | Titin (connectin) | 10 | 0.004 | Loss of titin homeostasis in cardiomyocytes results in myocardial stiffness observed in patients with hypertensive heart failure and a preserved ejection fraction.41 | |
| cd59a | cd59a | 3.4 | 0.002 | CD59 proteins regulate assembly of the membrane attack complex. Loss of function for both CD59a and CD59b variants result in a hemolytic phenotype.42 Elevated levels of expression may result as a molecular response to prolong erythrocyte longevity and improve oxygen distribution in the setting of heart failure. | |
| cd59b | cd59b | 1.5 | 0.007 | ||
| lum | lumican | 3.3 | 0.010 | “Lumican is increased in experimental and clinical heart failure, and its production by cardiac fibroblasts is induced by mechanical and proinflammatory stimuli.”43 | |
| TPM1 | alpha tropomyosine | 2.9 | 0.008 | Mutations that lead to dysregulation of this thin filament protein are associated with hypertrophic cardiomyopathies.44,45 | |
| DES | desmin | 2.9 | 0.024 | Upregulated in various forms of human heart failure.46 | |
| ACTC1 | Cardiac actin | 1.6 | 0.070 | Mutations of cardiac thin filament proteins eg, ACTC1 result in hypertrophic cardiomyopathy, particularly LV hypertrophy.47,48 | |
Note: Heart failure–related genes are upregulated in VIP knockout mice.26
Figure 1.
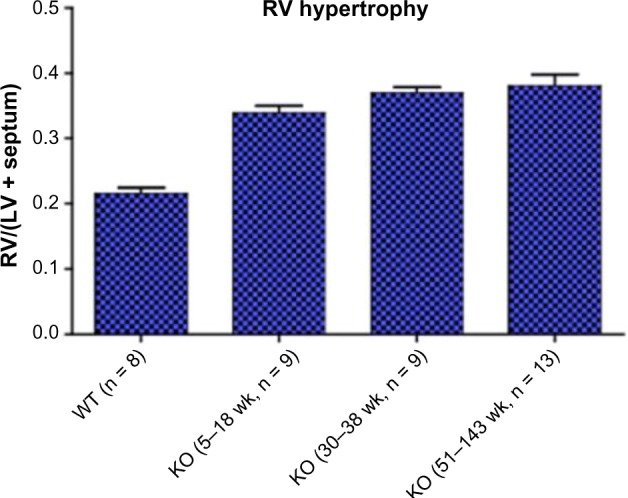
VIP knockout mice have progressive right ventricular hypertrophy with age.25 Wild-type control is the left-most bar labeled WT.
Figure 2.

Right ventricular dilation is present in VIP knockout mice, with large areas of the RV shown in red approaching the size of the left ventricle.26 These are end-diastolic multislice microMRI images acquired in the coronal plane orientation (short-axis view). A control mouse heart is shown in the top panels and a VIP−/− mouse heart is shown in the bottom panels.
Figure 3.
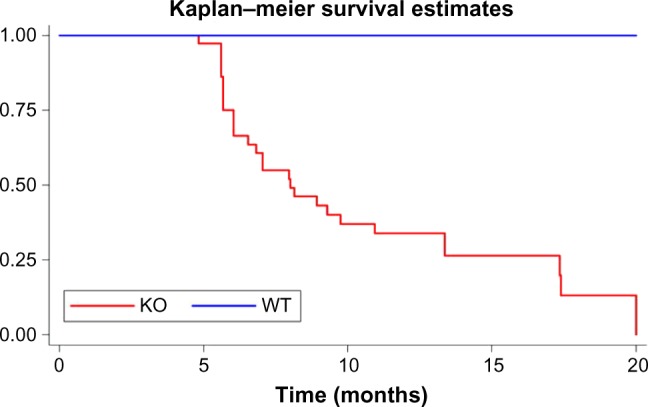
Severely increased mortality is seen in VIP KO mice compared to wild-type.25 The cause of death was not determined. However, as shown in Figure 1, there is an association of progressive right ventricular hypertrophy with age.
Figure 4.
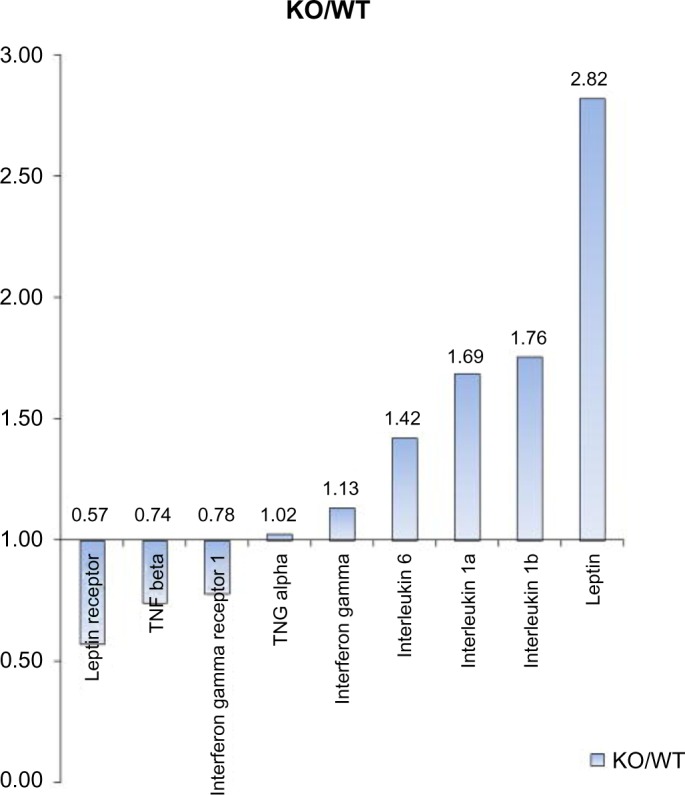
For gene microarray analyses, RNA was isolated from lung samples of five male VIP KO and five WT mice and subjected to Affymetrix gene profiling (Expression Analysis, Durham, NC, USA). Overexpression of leptin gene in VIP KO mice may be a function of compensation for the “cardiac cachexia” leading to low body weight and lack of subcutaneous adipocytes. There is also overexpression of proinflammatory genes such as IL-6 and IL-1a.26
Figure 6.

VIP is recognized by three different G-protein–coupled receptors: pituitary adenylate cyclase-activating peptide receptor (PAC1), vasoactive intestinal peptide receptor 1 (VPAC1) and vasoactive intestinal peptide receptor 2 (VPAC2).49 These receptors share a common signal transduction pathway in which activation of adenylyl cyclase (AC) upregulates cAMP production and subsequently activates protein kinase A (PKA).50–52 Downstream effects of PKA activation regulate cardiovascular function.53–60
Kupari et al found that VIP levels in serum from healthy subjects and patients with aortic stenosis and heart failure were released into the coronary sinus. VIP levels were higher in heart failure patients. These authors concluded that, although VIP was marginally elevated systemically in heart failure, it is the major neuroendocrine contributor to heart failure.35 Smitherman infused VIP into the left coronary artery of four other men at four levels. The maximum decline in coronary vascular resistance was 46% and was not associated with an increase in myocardial oxygen uptake. He concluded that “1) intravenous administration of low to intermediate doses of VIP in humans is associated with substantial coronary vasodilation, 2) the coronary bed appears to be at least as responsive as other vascular beds, 3) the coronary vasodilation is due to both direct and indirect effects, and 4) the coronary vasodilation does not appear to be mediated by prostaglandins.”36,37
So, the potential of an exogenous, sustainable VIP drug would be to help relax coronary arteries. Lucia et al also found that VIP plasma levels were higher in heart failure patients with dilated cardiomyopathy compared to healthy subjects and was higher in older subjects. Heart failure patients with higher New York Heart Association severity had lower levels of VIP. These data support a putative restorative role of VIP in treating disease. These data also suggest that heart failure patients with worse disease have an inability to produce VIP. The capacity of the elderly to produce VIP is heartening, so that low levels are not due to aging itself.38
PhaseBio Corporation recently conducted a phase 1, single ascending dose (SAD) study in patients with essential hypertension. PB1046 was administered subcutaneously and was found to be well-tolerated and demonstrated a prolonged, dose-dependent effect on blood pressure, which was used as a biomarker for activity in this study. (See http://clinicaltrials.gov/ct2/show/NCT01523067 for further details.)
With this demonstration of safety and tolerability at efficacious doses, PhaseBio conducted a second phase 1 SAD study in patients with essential hypertension, with the product administered intravenously to support the use of PB1046 in an acute setting (http://clinicaltrials.gov/show/NCT01873885). Subsequent phase 2 clinical studies will examine the efficacy of multiple ascending doses of PB1046 in patients with pulmonary arterial hypertension and heart failure.
Conclusion
Current therapies do not control progression of heart failure well and must often be used in combinations to optimize results. Novel emerging therapies show promise with improved outcomes but many still have limitations. LCZ696 utilizes a novel pathway to treat heart failure and improves mortality in comparison to the current standard of care, enalipril. It is important to remember that as a combination therapy utilizing an ARB, it carries a risk of inducing life-threatening angioedema. Ivabradine reduces rates of heart failure–related hospitalization, but the indications for use are strict and this medication carries undesirable side effects that potentiate etiologic mechanisms of heart failure. VIP, an endogenous peptide, shows widespread cardiovascular benefits as a vasodilator, an attenuator of smooth muscle proliferation, and a homeostatic regulator of heart failure gene programs thereby preventing hypertrophy of cardiac myocytes. Endogenous levels of VIP have been shown to be elevated in certain stages of heart failure. However, later stages of the disease show significantly diminished levels. Administration of exogenous VIP in early and late stages of heart failure may provide the most benefit for patients. Further research is warranted. Use of an elastin polymer to protect and stabilize VIP as a therapeutic agent shows the interface between engineering and medicine, and it bodes well for the influence of technology on society, particularly since heart failure will continue to grow as a prevalent problem as the population ages. VIP may therefore have a chance to satisfy the unmet need as a treatment for acute heart failure, thus fulfilling discoverer Dr Sami I. Said’s dream to promote human health.
Figure 5.
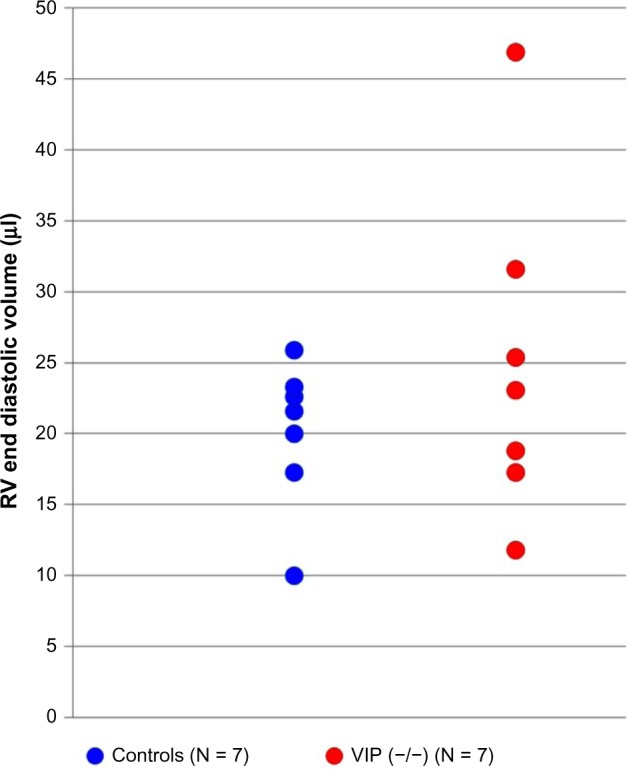
Scatter gram of RV ED volumes measured in the short-axis magnetic resonance images of individual control mice (blue) and VIP KO mice (red circles) demonstrating that the RV phenotype varied in the VIP KO mice. Right ventricular end diastolic volume is higher in some VIP KO mice, suggesting diastolic dysfunction.26
Footnotes
ACADEMIC EDITOR: Thomas E. Vanhecke, Editor in Chief.
PEER REVIEW: Three peer reviewers contributed to the peer review report. Reviewers’ reports totaled 792 words, excluding any confidential comments to the academic editor.
FUNDING: Dr Szema is a co-investigator (Principal Investigator, Adam Gonzalez, PhD) NIH R21 ES023583 Effects of Hurricane Sandy on the Respiratory and Mental Health of World Trade Center Responders; Principal Investigator Phasebio Corporation (Malvern, PA, USA) for Vasoactive Intestinal Peptide gene knockout mouse colony husbandry grant; Principal Investigator Garnett McKeen Laboratory (Bohemia, NY, USA) equipment loan for Flexivent mouse mechanical ventilator and pulmonary function with lipoic acid and RuX candidate medication; Principal Investigator NIH Lung Tissue Research Consortium (LTRC) Concept Sheet #08-99-0002, Gene Deficiency of Vasoactive Intestinal Peptide in COPD Patients with Pulmonary Arterial Hypertension. The authors confirm that the funders had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Contributed to writing of manuscript: AMS, JL. Jointly developed the structure and arguments for the paper: AMS, JL, SD. Made critical revisions: AMS, JL, SD. All authors reviewed and approved of the final manuscript.
REFERENCES
- 1.Abraham WT, Fonarow GC, Albert NM, et al. OPTIMIZE-HF Investigators and Coordinators Predictors of in-hospital mortality in patients hospitalized for heart failure: insights from the organized program to initiate lifesaving treatment in hospitalized patients with heart failure (optimize-hf) J Am Coll Cardiol. 2008;52:347–56. doi: 10.1016/j.jacc.2008.04.028. [DOI] [PubMed] [Google Scholar]
- 2.Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: executive summary: a report of the American College of Cardiology Foundation/American Heart Association task force on practice guidelines. Circulation. 2013;128:1810–52. doi: 10.1161/CIR.0b013e31829e8807. [DOI] [PubMed] [Google Scholar]
- 3.Joseph SM, Cedars AM, Ewald GA, Geltman EM, Mann DL. Acute decompensated heart failure: contemporary medical management. Tex Heart Inst J. 2009;36:510–20. [PMC free article] [PubMed] [Google Scholar]
- 4.Moss RL, Fitzsimons DP. Frank-Starling relationship: long on importance, short on mechanism. Circ Res. 2002;90:11–3. [PubMed] [Google Scholar]
- 5.Hunt SA, Baker DW, Chin MH, et al. American College of Cardiology/American Heart Association Task Force on Practice G, International Society for H, Lung T, Heart Failure Society of A ACC/AHA guidelines for the evaluation and management of chronic heart failure in the adult: executive summary a report of the American College of Cardiology/American Heart Association task force on practice guidelines (committee to revise the 1995 guidelines for the evaluation and management of heart failure): developed in collaboration with the International Society for Heart and Lung Transplantation; endorsed by the Heart Failure Society of America. Circulation. 2001;104:2996–3007. doi: 10.1161/hc4901.102568. [DOI] [PubMed] [Google Scholar]
- 6.Ferrari R, Boersma E. The impact of ace inhibition on all-cause and cardiovascular mortality in contemporary hypertension trials: a review. Expert Rev Cardiovasc Ther. 2013;11:705–17. doi: 10.1586/erc.13.42. [DOI] [PubMed] [Google Scholar]
- 7.Blaes N, Girolami JP. Targeting the ‘janus face’ of the b2-bradykinin receptor. Expert Opin Ther Targets. 2013;17:1145–66. doi: 10.1517/14728222.2013.827664. [DOI] [PubMed] [Google Scholar]
- 8.Campo P, Fernandez TD, Canto G, Mayorga C. Angioedema induced by angiotensin-converting enzyme inhibitors. Curr Opin Allergy Clin Immunol. 2013;13:337–44. doi: 10.1097/ACI.0b013e328362b835. [DOI] [PubMed] [Google Scholar]
- 9.Kyrmizakis DE, Papadakis CE, Liolios AD, et al. Angiotensin-converting enzyme inhibitors and angiotensin II receptor antagonists. Arch Otolaryngol Head Neck Surg. 2004;130:1416–9. doi: 10.1001/archotol.130.12.1416. [DOI] [PubMed] [Google Scholar]
- 10.Stevenson LW. Clinical use of inotropic therapy for heart failure: looking backward or forward? Part I: inotropic infusions during hospitalization. Circulation. 2003;108:367–72. doi: 10.1161/01.CIR.0000078348.44634.BA. [DOI] [PubMed] [Google Scholar]
- 11.Hjalmarson A, Fagerberg B. Merit-hf mortality and morbidity data. Basic Res Cardiol. 2000;95(suppl 1):I98–103. doi: 10.1007/s003950070017. [DOI] [PubMed] [Google Scholar]
- 12.Garg R, Yusuf S. Overview of randomized trials of angiotensin-converting enzyme inhibitors on mortality and morbidity in patients with heart failure. Collaborative group on ace inhibitor trials. JAMA. 1995;273:1450–6. [PubMed] [Google Scholar]
- 13.Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized aldactone evaluation study investigators. N Engl J Med. 1999;341:709–17. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]
- 14.Satpathy C, Mishra TK, Satpathy R, Satpathy HK, Barone E. Diagnosis and management of diastolic dysfunction and heart failure. Am Fam Physician. 2006;73:841–6. [PubMed] [Google Scholar]
- 15.Jessup M. Neprilysin inhibition – a novel therapy for heart failure. N Engl J Med. 2014;371:1062–4. doi: 10.1056/NEJMe1409898. [DOI] [PubMed] [Google Scholar]
- 16.Gu J, Noe A, Chandra P, et al. Pharmacokinetics and pharmacodynamics of lcz696, a novel dual-acting angiotensin receptor-neprilysin inhibitor (ARNI) J Clin Pharmacol. 2010;50:401–14. doi: 10.1177/0091270009343932. [DOI] [PubMed] [Google Scholar]
- 17.Jhund PS, Claggett B, Packer M, et al. Independence of the blood pressure lowering effect and efficacy of the angiotensin receptor neprilysin inhibitor, LCZ696, in patients with heart failure with preserved ejection fraction: an analysis of the PARAMOUNT trial. Eur J Heart Fail. 2014;16:671–7. doi: 10.1002/ejhf.76. [DOI] [PubMed] [Google Scholar]
- 18.Markandeya YS, Phelan LJ, Woon MT, et al. Caveolin-3 overexpression attenuates cardiac hypertrophy via inhibition of t-type Ca2+ current modulated by pkcalpha in cardiomyocytes. J Biol Chem. 2015 doi: 10.1074/jbc.M115.674945. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Administration USFDA . FDA Approves Corlanor to Treat Heart Failure. FDA News Release; 2015. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm453845.htm. [Google Scholar]
- 20.Harmar AJ, Fahrenkrug J, Gozes I, et al. Pharmacology and functions of receptors for vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide: IUPHAR review 1. Br J Pharmacol. 2012;166:4–17. doi: 10.1111/j.1476-5381.2012.01871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frase LL, Gaffney FA, Lane LD, et al. Cardiovascular effects of vasoactive intestinal peptide in healthy subjects. Am J Cardiol. 1987;60:1356–61. doi: 10.1016/0002-9149(87)90619-9. [DOI] [PubMed] [Google Scholar]
- 22.Said SI, Hamidi SA, Dickman KG, et al. Moderate pulmonary arterial hypertension in male mice lacking the vasoactive intestinal peptide gene. Circulation. 2007;115:1260–8. doi: 10.1161/CIRCULATIONAHA.106.681718. [DOI] [PubMed] [Google Scholar]
- 23.Maruno K, Absood A, Said SI. VIP inhibits basal and histamine-stimulated proliferation of human airway smooth muscle cells. Am J Physiol. 1995;268:L1047–51. doi: 10.1152/ajplung.1995.268.6.L1047. [DOI] [PubMed] [Google Scholar]
- 24.Nakamura M. Peripheral vascular remodeling in chronic heart failure: clinical relevance and new conceptualization of its mechanisms. J Card Fail. 1999;5:127–38. doi: 10.1016/s1071-9164(99)90035-0. [DOI] [PubMed] [Google Scholar]
- 25.Szema AM, Hamidi SA. Gene deletion of VIP leads to increased mortality associated with progressive right ventricular hypertrophy. J Cardiovasc Dis. 2014;2:131–6. [PMC free article] [PubMed] [Google Scholar]
- 26.Szema AM, Hamidi SA, Smith SD, Benveniste H. VIP gene deletion in mice causes cardiomyopathy associated with upregulation of heart failure genes. PLoS One. 2013;8:e61449. doi: 10.1371/journal.pone.0061449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He B, Tang RH, Weisleder N, et al. Enhancing muscle membrane repair by gene delivery of mg53 ameliorates muscular dystrophy and heart failure in delta-sarcoglycan-deficient hamsters. Mol Ther. 2012;20:727–35. doi: 10.1038/mt.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sabourdy F, Michelakakis H, Anastasakis A, et al. Danon disease: further clinical and molecular heterogeneity. Muscle Nerve. 2009;39:837–44. doi: 10.1002/mus.21252. [DOI] [PubMed] [Google Scholar]
- 29.Ramratnam M, Sharma RK, D’Auria S, et al. Transgenic knockdown of cardiac sodium/glucose cotransporter 1 (sglt1) attenuates prkag2 cardiomyopathy, whereas transgenic overexpression of cardiac sglt1 causes pathologic hypertrophy and dysfunction in mice. J Am Heart Assoc. 2014;3:e000899. doi: 10.1161/JAHA.114.000899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Y, Guo T, Oda T, et al. Cardiac myocyte z-line calmodulin is mainly ryr2-bound, and reduction is arrhythmogenic and occurs in heart failure. Circ Res. 2014;114:295–306. doi: 10.1161/CIRCRESAHA.114.302857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu L, Wang C, Sun D, et al. Calhex ameliorates cardiac hypertrophy by inhibiting cellular autophagy in vivo and in vivo. Cell Physiol Biochem. 2015;36:1597–612. doi: 10.1159/000430322. [DOI] [PubMed] [Google Scholar]
- 32.Cox CP, Linden J, Said SI. VIP elevates platelet cyclic amp (camp) levels and inhibits in vivo platelet activation induced by platelet-activating factor (PAF) Peptides. 1984;5:325–8. doi: 10.1016/0196-9781(84)90228-6. [DOI] [PubMed] [Google Scholar]
- 33.Lazarus SC, Basbaum CB, Barnes PJ, Gold WM. Camp immunocytochemistry provides evidence for functional VIP receptors in trachea. Am J Physiol. 1986;251:C115–9. doi: 10.1152/ajpcell.1986.251.1.C115. [DOI] [PubMed] [Google Scholar]
- 34.Szema AM, Hamidi SA, Golightly MG, Rueb TP, Chen JJ. VIP regulates the development & proliferation of Treg in vivo in spleen. Allergy Asthma Clin Immunol. 2011;7:19. doi: 10.1186/1710-1492-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kupari M, Mikkola TS, Turto H, Lommi J, Ylikorkala O. Vasoactive intestinal peptide – release from the heart and response in heart failure due to left ventricular pressure overload. Eur J Heart Fail. 2006;8:361–5. doi: 10.1016/j.ejheart.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 36.Popma JJ, Smitherman TC, Bedotto JB, Eichhorn EJ, Said SI, Dehmer GJ. Direct coronary vasodilation induced by intracoronary vasoactive intestinal peptide. J Cardiovasc Pharmacol. 1990;16:1000–6. doi: 10.1097/00005344-199012000-00021. [DOI] [PubMed] [Google Scholar]
- 37.Smitherman TC, Popma JJ, Said SI, Krejs GJ, Dehmer GJ. Coronary hemodynamic effects of intravenous vasoactive intestinal peptide in humans. Am J Physiol. 1989;257:H1254–62. doi: 10.1152/ajpheart.1989.257.4.H1254. [DOI] [PubMed] [Google Scholar]
- 38.Lucia P, Caiola S, Coppola A, et al. Vasoactive intestinal peptide in heart failure. Ital Heart J Suppl. 2000;1:679–85. [PubMed] [Google Scholar]
- 39.Guo A, Cala SE, Song LS. Calsequestrin accumulation in rough endoplasmic reticulum promotes perinuclear Ca2+ release. J Biol Chem. 2012;287:16670–80. doi: 10.1074/jbc.M112.340927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zarain-Herzberg A, Estrada-Aviles R, Fragoso-Medina J. Regulation of sarco(endo) plasmic reticulum Ca2+-atpase and calsequestrin gene expression in the heart. Can J Physiol Pharmacol. 2012;90:1017–28. doi: 10.1139/y2012-057. [DOI] [PubMed] [Google Scholar]
- 41.Zile MR, Baicu CF, Ikonomidis JS, et al. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation. 2015;131:1247–59. doi: 10.1161/CIRCULATIONAHA.114.013215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baalasubramanian S, Harris CL, Donev RM, et al. Cd59a is the primary regulator of membrane attack complex assembly in the mouse. J Immunol. 2004;173:3684–92. doi: 10.4049/jimmunol.173.6.3684. [DOI] [PubMed] [Google Scholar]
- 43.Engebretsen KV, Lunde IG, Strand ME, et al. Lumican is increased in experimental and clinical heart failure, and its production by cardiac fibroblasts is induced by mechanical and proinflammatory stimuli. FEBS J. 2013;280:2382–98. doi: 10.1111/febs.12235. [DOI] [PubMed] [Google Scholar]
- 44.Selvi Rani D, Nallari P, Dhandapany PS, et al. Coexistence of digenic mutations in both thin (tpm1) and thick (myh7) filaments of sarcomeric genes leads to severe hypertrophic cardiomyopathy in a South Indian FHCM. DNA Cell Biol. 2015;34:350–9. doi: 10.1089/dna.2014.2650. [DOI] [PubMed] [Google Scholar]
- 45.Gupte TM, Haque F, Gangadharan B, et al. Mechanistic heterogeneity in contractile properties of alpha-tropomyosin (tpm1) mutants associated with inherited cardiomyopathies. J Biol Chem. 2015;290:7003–15. doi: 10.1074/jbc.M114.596676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chugh S, Ouzounian M, Lu Z, et al. Pilot study identifying myosin heavy chain 7, desmin, insulin-like growth factor 7, and annexin a2 as circulating bio-markers of human heart failure. Proteomics. 2013;13:2324–34. doi: 10.1002/pmic.201200455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coppini R, Ho CY, Ashley E, et al. Clinical phenotype and outcome of hypertrophic cardiomyopathy associated with thin-filament gene mutations. J Am Coll Cardiol. 2014;64:2589–600. doi: 10.1016/j.jacc.2014.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Debold EP, Saber W, Cheema Y, et al. Human actin mutations associated with hypertrophic and dilated cardiomyopathies demonstrate distinct thin filament regulatory properties in vivo. J Mol Cell Cardiol. 2010;48:286–92. doi: 10.1016/j.yjmcc.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang K, Trepanier CH, Li H, et al. Vasoactive intestinal peptide acts via multiple signal pathways to regulate hippocampal NMDA receptors and synaptic transmission. Hippocampus. 2009;19:779–89. doi: 10.1002/hipo.20559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Picketts DJ. Neuropeptide signaling and hydrocephalus: SCO with the flow. J Clin Invest. 2006;116:1828–32. doi: 10.1172/JCI29148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Delgado M, Pozo D, Ganea D. The significance of vasoactive intestinal peptide in immunomodulation. Pharmacol Rev. 2004;56:249–90. doi: 10.1124/pr.56.2.7. [DOI] [PubMed] [Google Scholar]
- 52.Murthy KS, Mahavadi S, Huang J, Zhou H, Sriwai W. Phosphorylation of grk2 by PKA augments grk2-mediated phosphorylation, internalization, and desensitization of vpac2 receptors in smooth muscle. Am J Physiol Cell Physiol. 2008;294:C477–87. doi: 10.1152/ajpcell.00229.2007. [DOI] [PubMed] [Google Scholar]
- 53.Taggart P, Boyett MR, Logantha S, Lambiase PD. Anger, emotion, and arrhythmias: from brain to heart. Front Physiol. 2011;2:67. doi: 10.3389/fphys.2011.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang JH, Saucerman JJ. Computational models reduce complexity and accelerate insight into cardiac signaling networks. Circ Res. 2011;108:85–97. doi: 10.1161/CIRCRESAHA.110.223602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saucerman JJ, McCulloch AD. Mechanistic systems models of cell signaling networks: a case study of myocyte adrenergic regulation. Prog Biophys Mol Biol. 2004;85:261–78. doi: 10.1016/j.pbiomolbio.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 56.Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol. 2009;54:1747–62. doi: 10.1016/j.jacc.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 57.Schaub MC, Hefti MA, Zaugg M. Integration of calcium with the signaling network in cardiac myocytes. J Mol Cell Cardiol. 2006;41:183–214. doi: 10.1016/j.yjmcc.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 58.Roberts OL, Dart C. Camp signalling in the vasculature: the role of EPAC (exchange protein directly activated by camp) Biochem Soc Trans. 2014;42:89–97. doi: 10.1042/BST20130253. [DOI] [PubMed] [Google Scholar]
- 59.Fimia GM, Sassone-Corsi P. Cyclic amp signalling. J Cell Sci. 2001;114:1971–2. doi: 10.1242/jcs.114.11.1971. [DOI] [PubMed] [Google Scholar]
- 60.Lauder JM. Neurotransmitters as growth regulatory signals: role of receptors and second messengers. Trends Neurosci. 1993;16:233–40. doi: 10.1016/0166-2236(93)90162-f. [DOI] [PubMed] [Google Scholar]