

Abstract
Many viruses cause disease within an infected host after spread from an initial portal of entry to secondary sites of replication. Viruses can disseminate via the bloodstream or through nerves. Mammalian orthoreoviruses (reoviruses) are neurotropic viruses that use both bloodborne and neural pathways to spread systemically within their hosts to cause disease. Using a robust mouse model and a dynamic reverse genetics system, we have identified a viral receptor and a viral nonstructural protein that are essential for hematogenous reovirus dissemination. Junctional adhesion molecule-A (JAM-A) is a member of the immunoglobulin superfamily expressed in tight junctions and on hematopoietic cells that serves as a receptor for all reovirus serotypes. Expression of JAM-A is required for infection of endothelial cells and development of viremia in mice, suggesting that release of virus into the bloodstream from infected endothelial cells requires JAM-A. Nonstructural protein σ1s is implicated in cell cycle arrest and apoptosis in reovirus-infected cells but is completely dispensable for reovirus replication in cultured cells. Surprisingly, a recombinant σ1s-null reovirus strain fails to spread hematogenously in infected mice, suggesting that σ1s facilitates apoptosis of reovirus-infected intestinal epithelial cells. It is possible that apoptotic bodies formed as a consequence of σ1s expression lead to reovirus uptake by dendritic cells for subsequent delivery to the mesenteric lymph node and the blood. Thus, both host and viral factors are required for efficient hematogenous dissemination of reovirus. Understanding mechanisms of reovirus bloodborne spread may shed light on how microbial pathogens invade the bloodstream to disseminate and cause disease in infected hosts.
I. Introduction
Many pathogenic human and animal viruses disseminate from mucosal sites to peripheral tissues where they cause organ-specific disease (Nathanson and Tyler, 1997). The capacity of a virus to spread systemically can correlate with increased virulence (de Jong et al., 2006; Gu et al., 2007; Kuiken et al., 2003; Pallansch and Roos, 2001). Systemic dissemination requires that the virus effectively navigate diverse intracellular and extracellular environments to infect, replicate, and evade immune detection in multiple cell types and tissues (Adair et al., 2012; Antar et al., 2009; Boehme et al., 2011; Boehme, Guglielmi and Dermody, 2009). Although some general principles of virus dissemination are understood, little is known about the precise viral and cellular determinants that govern virus spread. Defining mechanisms by which viruses disseminate within their hosts is of fundamental importance to an understanding of viral pathogenesis.
Mammalian orthoreoviruses (reoviruses) are highly tractable models for studies of viral pathogenesis. Studies of reovirus neural spread have provided important information about mechanisms by which neurotropic viruses cause disease in the central nervous system (CNS). The recent identification of new viral and host determinants that govern reovirus spread by the blood provides new insights into how hematogenous dissemination contributes to viral disease.
A. Reoviruses
Viruses of the Reoviridae family infect a wide range of host organisms, including mammals, birds, insects, and plants (Dermody, In Press). The Reoviridae includes rotaviruses, the most common diarrheal pathogen among children (Parashar et al., 1998), orbiviruses, which are economically important pathogens of sheep, cattle, and horses (Coetzee et al., 2012), and reoviruses. Three reovirus serotypes (T1, T2, and T3) currently circulate in humans and other mammals. The serotypes are distinguished on the basis of antibody-mediated neutralization of infectivity and inhibition of hemagglutination. Each serotype is represented by a prototype strain isolated from a human host: type 1 Lang (T1L), type 2 Jones (T2J), and type 3 Dearing (T3D). These strains differ dramatically in host cell tropism, mechanisms of cell killing, modes of dissemination, and CNS disease. In particular, studies of T1 and T3 reoviruses have generated foundational knowledge about strategies used by viruses to replicate and cause neural injury. Development of a plasmid-based reverse genetics system allows introduction of mutations into the viral genome to test specific hypotheses about the structure and function of viral proteins and RNAs (Kobayashi et al., 2007; Kobayashi et al., 2010). In concert with an experimentally facile mouse model of infection (Fields, 1992; Parashar, Tarlow and McCrae, 1992), reovirus is an ideal experimental platform for studies of virus-host interactions.
Reoviruses are nonenveloped, icosahedral viruses that contain a genome consisting of 10 segments of double-stranded (ds) RNA (Fig. 1) (Dermody, In Press). There are three large (L1, L2, L3), three medium (M1, M2, M3), and four small (S1, S2, S3, S4) dsRNA segments that are packaged in an equimolar stoichiometric relationship with one copy of each per virion. With the exception of the M3 and S1 gene segments, each of the reovirus gene segments is monocistronic. Reovirus virions are composed of two concentric protein shells, the outer capsid and core (Fig. 1) (Dryden et al., 1993). The outer capsid consists of heterohexameric complexes of the μ1 (encoded by M2) and σ3 (encoded by S4) proteins. At each of the icosahedral five-fold symmetry axes, the attachment protein σ1 (encoded by S1) extends from turret-like structures formed by pentamers of λ2 (encoded by L2) protein. The inner core shell is formed by parallel asymmetric dimers of λ1 (encoded by L3) protein that are stabilized by σ2 (encoded by S2) protein. The λ3 (encoded by L1) and μ2 (encoded by M1) proteins are anchored to the inner surface of the core via interactions with λ1. Lastly, the M3 gene segment encodes nonstructural proteins μNS and μNSC, the S3 gene segment encodes nonstructural protein σNS, and the S1 gene segment encodes nonstructural protein σ1s.
Figure 1.
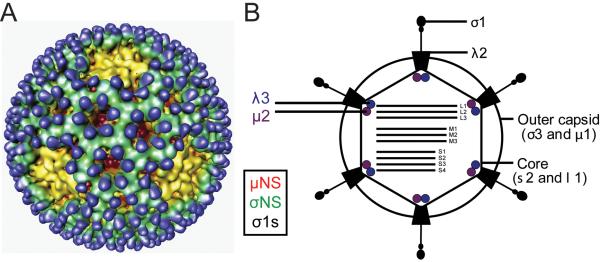
The reovirus virion. (A) Cryo-electron micrograph image reconstruction of a reovirus virion. Outer-capsid protein σ3 (blue), is the initial target for virion disassembly in infected cells. Pentameric λ2 protein (yellow) forms an insertion pedestal for σ1, which is the viral attachment protein. Copyright © American Society for Microbiology, Journal of Virology, vol. 75, 2001, pages 146625–6634, doi: 10.1128/JVI.75.14.6625-6634.2001 (Nason et al., 2001). (B) Schematic of a reovirus virion. Reovirus particles are formed from two concentric protein shells, the outer capsid and core. The core contains the viral genome, which consists of ten dsRNA segments. Reovirus also encodes nonstructural proteins, σNS, μNS, μNSC, and σ1s.
Viral attachment protein σ1 is a long filamentous molecule with head-and-tail morphology (Fig. 2A) (Chappell et al., 2002; Fraser et al., 1990; Mercier et al., 2004; Reiter et al., 2011). The σ1 protein is comprised of three distinct structural domains: an N-terminal α-helical coiled-coil tail, a central β-spiral body, and a C-terminal globular head (Chappell et al., 2002; Reiter et al., 2011). Short regions of undefined structure separate each domain and are hypothesized to permit molecular flexibility required to engage cellular receptors during viral entry (Bokiej et al., 2012; (Chappell et al., 2002; Fraser et al., 1990; Reiter et al., 2011). Attachment of the σ1 protein to cell-surface receptors initiates reovirus infection of susceptible host cells (Lee, Hayes and Joklik, 1981; Weiner, Ault and Fields, 1980). The σ1 protein of T3 reovirus targets two different receptors, α-linked sialic acid (SA) (Armstrong, Paul and Lee, 1984; Dermody et al., 1990a; Pacitti and Gentsch, 1987; Paul, Choi and Lee, 1989; Paul and Lee, 1987) and junctional adhesion molecule-A (JAM-A) (Barton et al., 2001b; Campbell et al., 2005; Prota et al., 2003). Residues in the T3 σ1 β-spiral body domain bind SA (Chappell et al., 2000; Reiter et al., 2011), whereas sequences in the σ1 globular head domain engage JAM-A (Barton et al., 2001b; Kirchner et al., 2008).
Figure 2. Structure of σ1 and JAM-A.

(A) Full-length model of attachment protein σ1 bound to JAM-A. A model of full-length σ1 extending from the virion is shown as a ribbon drawing, with the known structure of the C-terminus (Chappell et al., 2002) in tricolor and the predicted structure of the N-terminus in grey. Arrows indicate predicted regions of flexibility. A model of full-length JAM-A is shown in green as a ribbon drawing of the known structure of the extracellular domain (Prota et al., 2003) and a schematic representation of the transmembrane and intracellular domains. For clarity, only two JAM-A monomers are shown bound to σ1. Adapted from Kirchner et al. (2008, Fig. 1). (B) Structure of human JAM-A D1 and D2 domains. Ribbon drawings of a JAM-A homodimer, with one monomer shown in yellow and the other in green. Two orthogonal views are displayed. Adapted from Prota et al. (2003). Copyright (2003) National Academy of Sciences, USA.
After receptor binding, virions are internalized into endosomes via a process dependent on β1 integrin (Maginnis et al., 2006) and distributed to organelles marked by Rab7 and Rab9 where viral disassembly takes place (Mainou and Dermody, 2012). During viral disassembly, outer-capsid protein σ3 is degraded by cathepsin proteases, attachment protein σ1 undergoes a conformational change, and outer-capsid protein μ1 is cleaved to form infectious subvirion particles (ISVPs) (Danthi et al., 2010). The μ1 cleavage fragments undergo conformational rearrangement to facilitate endosome penetration and delivery of transcriptionally active core particles into the cytoplasm (Nibert et al., 2005; Odegard et al., 2004). Primary transcription occurs within the viral core, and nascent RNAs are translated or encapsidated into new viral cores, where they serve as templates for negative-strand synthesis. Within new viral cores, secondary rounds of transcription occur. Outer-capsid proteins are added to nascent cores, which silences viral transcription and yields progeny viral particles. Reovirus release from host cells is hypothesized to occur via a lytic mechanism, but the egress pathway is not understood (Dermody, In Press).
B. Junctional adhesion molecule-A
JAM-A is the only known proteinaceous receptor for reovirus. It mediates entry of prototype and field-isolate strains of all three reovirus serotypes (Barton et al., 2001b; Campbell et al., 2005). JAM-A is a member of the immunoglobulin (Ig) superfamily of proteins that functions in cell-cell adhesion (Bazzoni, 2003). It is expressed on the surface of endothelial and epithelial cells as a component of tight junctions that maintain the integrity of barriers formed between polarized cells (Martin-Padura et al., 1998; Woodfin et al., 2007). JAM-A also is expressed on hematopoietic cells, where it mediates leukocyte extravasation (Corada et al., 2005; Ghislin et al., 2011), and on platelets, where it functions in platelet activation during blood clot formation (Bazzoni, 2003; Sobocka et al., 2004). JAM-A contains three distinct structural domains: an N-terminal ectodomain, a single-span transmembrane anchor, and a C-terminal cytoplasmic tail (Fig. 2B) (Prota et al., 2003). The ectodomain consists of two Ig-like domains, a membrane-distal D1 domain and a membrane-proximal D2 domain. The cytoplasmic tail terminates in a PDZ-binding domain that interacts with intracellular tight junction components (Bazzoni et al., 2005; Nomme et al., 2011). JAM-A participates in homotypic interactions between D1 domains on opposing monomers (Prota et al., 2003). An interaction between two JAM-A monomers on adjacent cells promotes cell adhesion (Iden et al., 2012; Mandell et al., 2005; Ostermann et al., 2005).
The σ1 protein interacts with the JAM-A D1 domain to adhere reovirus virions to the surface of target cells (Kirchner et al., 2008). Interestingly, the σ1-JAM-A interaction is substantially stronger (approximately 1000-fold) than the interaction between JAM-A monomers (Kirchner et al., 2008). Consequently, σ1 binding to JAM-A likely disrupts JAM-A homodimers. Studies using JAM-A-deficient mice indicate that JAM-A is required for the establishment of viremia, which is essential for dissemination and disease in newborn mice following peroral inoculation of reovirus (Antar et al., 2009). JAM-A is not required for reovirus replication in the murine CNS or development of encephalitis (Antar et al., 2009). These findings suggest that reovirus utilizes other cell-surface receptors to mediate entry into specific cell types.
C. Reovirus pathogenesis
Reoviruses are highly virulent in newborn mice and cause injury to a variety of host organs, including the CNS, heart, and liver (Dermody, In Press). T1 and T3 reovirus strains invade the CNS but use different routes and produce distinct pathologic consequences following peroral or intramuscular inoculation. T1 reoviruses spread by hematogenous routes and infect ependymal cells, causing nonlethal hydrocephalus (Tyler, McPhee and Fields, 1986; Weiner et al., 1977; Weiner, Powers and Fields, 1980). T3 reoviruses spread to the CNS by both hematogenous and neural routes and infect neurons (Antar et al., 2009; Boehme et al., 2011; Tyler, McPhee and Fields, 1986). In the brain, T3 reoviruses induce neuronal apoptosis, which results in fatal encephalitis (Morrison, Sidman and Fields, 1991a; Tyler, McPhee and Fields, 1986; Weiner et al., 1977; Weiner, Powers and Fields, 1980). Studies using T1L × T3D reassortant viruses mapped the major determinant of CNS pathology to the viral S1 gene (Dichter and Weiner, 1984; Tardieu and Weiner, 1982), which encodes attachment protein σ1 and nonstructural protein σ1s (Sarkar et al., 1985; Weiner, Ault and Fields, 1980). Because of its role in viral attachment and entry, these serotype-specific differences in dissemination and disease have largely been ascribed to the σ1 protein. However, σ1s plays a critical role in promoting reovirus spread by the bloodstream (Boehme et al., 2011; Boehme, Guglielmi and Dermody, 2009).
D. Nonstructural protein σ1s
Protein σ1s is a 14 kDa nonstructural protein encoded by the viral S1 gene segment (Cashdollar et al., 1985; Ernst and Shatkin, 1985; Sarkar et al., 1985). The σ1s open-reading frame (ORF) completely overlaps the σ1 coding sequence; however, σ1s lies in a different reading frame (Cashdollar et al., 1985; Cenatiempo et al., 1984; Dermody et al., 1990b; Ernst and Shatkin, 1985; Sarkar et al., 1985). Although every reovirus strain sequenced to date contains a σ1s ORF, little amino acid sequence identity exists between the σ1s proteins from the different reovirus serotypes (Cashdollar et al., 1985; Dermody et al., 1990b). The only conserved sequence among σ1s proteins is a cluster of basic amino acids near the amino terminus (Cashdollar et al., 1985; Dermody et al., 1990b). The basic cluster from T3 σ1s functions as an autonomous nuclear localization signal that can redirect an appended heterologous protein to the nucleus (Hoyt, Bouchard and Tyler, 2004). While the majority of native σ1s localizes to the nucleus (Rodgers et al., 1998), it is not known whether the basic cluster mediates nuclear translocation in the context of reovirus infection. Functionally, the σ1s protein is implicated in reovirus-induced cell cycle arrest at the G2/M boundary (Poggioli, Dermody and Tyler, 2001; Poggioli et al., 2000) and may influence reovirus neurovirulence by promoting reovirus-induced apoptosis in the murine CNS (Hoyt et al., 2005). Initial studies to define the function of σ1s in reovirus pathogenesis were complicated by the use of non-isogenic σ1s-null mutant and parental virus strains (Rodgers et al., 1998).
Development of a plasmid-based reverse genetics system for mammalian reovirus (Kobayashi et al., 2007; Kobayashi et al., 2010) made it possible to elucidate the function of σ1s in reovirus replication and pathogenesis. Recombinant reoviruses deficient in σ1s expression were engineered by incorporating a single nucleotide change (AUG to ACG) to disrupt the σ1s translational start site into the plasmid containing the cDNA encoding the S1 gene segment. Importantly, the mutation does not affect the coding sequence of the overlapping σ1 ORF. Thus, except for σ1s expression, the resultant viruses are isogenic with the parental strain. Viruses deficient in σ1s expression have been generated in the T1 and T3 S1 gene backgrounds. In both cases, the σ1s-null viruses are viable and replicate with equivalent kinetics and produce yields of progeny virus comparable to the corresponding wild-type viruses, indicating that the σ1s protein is dispensable for reovirus replication in cultured cells (Boehme et al., 2011; Boehme, Guglielmi and Dermody, 2009). These viruses were used to uncover a role for σ1s in promoting hematogenous reovirus dissemination.
II. Dynamics of reovirus infection in the intestine and lung
Reoviruses infect their hosts by the fecal-oral and respiratory routes. Virus enters the host by ingestion of contaminated food or inhalation of virus-containing aerosols. At both portals of entry, reoviruses infect epithelial cells and disseminate to peripheral sites where they cause disease.
A. Infection via the gastrointestinal tract
Reoviruses have been isolated from the stools of healthy (Ramos-Alvarez, 1956; Ramos-Alvarez and Sabin, 1954) and ill (Ramos-Alvarez and Sabin, 1958) children as well as a variety of animals (Ramos-Alvarez and Sabin, 1958). These findings suggest that reovirus is ingested into and shed from the gastrointestinal tract. The dynamics of reovirus infection in vivo have largely been elucidated using experimental mouse and rat model systems. Following entry into the gastrointestinal tract, intestinal proteases rapidly convert reovirus virions to ISVPs, suggesting that the form of the reovirus particle that initiates infection in the intestine is the ISVP (Bass et al., 1990; Bodkin, Nibert and Fields, 1989; Chappell et al., 1998). In newborn mice, cells at the tips of microvilli are readily infected, whereas cells in the intestinal crypts are spared (Antar et al., 2009; Boehme, Guglielmi and Dermody, 2009). In contrast, intestinal crypt cells are infected in adult mice, and cells at the villus tips are uninfected (Rubin, Kornstein and Anderson, 1985). Infectious reovirus can be recovered following peroral inoculation from the duodenum, jejunum, ileum, and colon (Rubin, Eaton and Anderson, 1986; Rubin, Kornstein and Anderson, 1985). However, the vast majority of virus is produced in the ileum. This differential production of virus may be due to the capacity of reovirus to infect Peyer patches. Reoviruses are thought to penetrate the intestinal barrier via transport across microfold (M) cells, which are specialized cells of the follicle-associated epithelium (FAE) that overlay the Peyer patches (Amerongen et al., 1994; Wolf et al., 1987; Wolf et al., 1983; Wolf et al., 1981). M cells transfer antigens from the intestinal lumen to lymphoid cells of the gut-associated lymphoid tissue (GALT) (van de Pavert and Mebius, 2010) and serve to monitor luminal contents by exposing Peyer patch lymphoid cells to food antigens, the intestinal microbiota, and intestinal microbial pathogens. This process is essential for induction of oral tolerance and activation of immune responses to pathogenic microorganisms (van de Pavert and Mebius, 2010). The preferential targeting of crypt cells observed in adult mice is hypothesized to result from transcytosis of virus across M cells and subsequent infection of crypt cells via the basolateral surface (Rubin, 1987). However, M cells also take up reovirus in neonatal mice (Antar et al., 2009; Boehme, Guglielmi and Dermody, 2009; Wolf et al., 1981), suggesting that viral transcytosis across M cells is unlikely to explain the difference in intestinal cell tropism observed in adult and newborn mice. It is possible that the proliferative status of stem cells in the crypts of adult mice may recapitulate the cellular environment of neonatal intestinal cells, thereby facilitating reovirus infection of intestinal crypt cells.
B. Infection via the respiratory tract
Reovirus also infects the respiratory tract (Sabin, 1959). In rats, both T1 and T3 reovirus strains cause a pneumonia that is characterized by destruction of type I alveolar epithelial cells and infiltration of leukocytes into the alveolar spaces (Morin, Warner and Fields, 1996). The pathology associated with reovirus infection closely mimics disease progression in bronchiolitis obliterans organizing pneumonia, which is notable for fibrous extensions into alveolar spaces in the context of an organizing pneumonia (Bellum et al., 1997). Following inoculation into the respiratory tract, lung proteases convert reovirus virions to ISVPs (Golden and Schiff, 2005; Nygaard, Golden and Schiff, 2012). Similar to infection in the intestine, reovirus infects the lung by transcytosis through M cells that overlie the bronchus-associated lymphoid tissue (BALT) (Morin, Warner and Fields, 1994; Morin, Warner and Fields, 1996).
III. Hematogenous spread of reovirus
A. Transport of reovirus from the intestine to the bloodstream
Systemic reovirus infection is thought to originate from infected lymphoid cells in the Peyer patch. From the Peyer patch, reovirus transits intestinal lymphatics to the mesenteric lymph node (MLN) and ultimately enters the bloodstream via the thoracic duct (Antar et al., 2009; Boehme, Guglielmi and Dermody, 2009; Wolf et al., 1981). Many pathogens that cause systemic disease, including poliovirus (Bodian, 1955; Sabin, 1956) and Salmonella (Carter and Collins, 1974; Galan and Curtiss, 1989; Jones, Ghori and Falkow, 1994), initiate extraintestinal infection and access the bloodstream via this route.
Reovirus reaches the Peyer patches early after infection; viral antigen is detected in Peyer patches within 24 hours after peroral inoculation (Antar et al., 2009; Bass et al., 1988; Boehme, Guglielmi and Dermody, 2009; Wolf et al., 1987; Wolf et al., 1983; Wolf et al., 1981). However, the mechanism by which reovirus infects Peyer patch cells is not known. It is possible that dendritic cells in the Peyer patches take up reovirus virions immediately following viral transcytosis across M cells. This is the most direct route from the intestinal lumen to the Peyer patch and the primary pathway used for processing of intestinal antigens for immune surveillance. A second possibility is that progeny virions released from the basolateral surface of infected FAE cells are taken up by lymphoid cells in Peyer patches. Both viral structural and nonstructural proteins are detected in FAE cells (Fleeton et al., 2004), indicating that active viral replication occurs in these cells. However, it is not known whether FAE cells produce virus. A third possibility is that dendritic cells in Peyer patches take up apoptotic fragments from infected FAE cells, which undergo apoptosis following reovirus infection (Fleeton et al., 2004). Dendritic cells in the underlying Peyer patches immediately adjacent to apoptotic FAE cells contain both active caspase-3 and reovirus structural proteins (Fleeton et al., 2004). These observations suggest that Peyer patch dendritic cells take up apoptotic bodies from infected FAE cells. Additionally, apoptosis induction in the FAE may signal Peyer patch cells to phagocytose the apoptotic remnants, along with reovirus particles.
Regardless of the mechanism by which reovirus accesses Peyer patches, reovirus antigen is detected in the MLN 24 hours after peroral inoculation. Little is known about the cell types that support reovirus growth within the intestine and dissemination to the MLN. In adult mice, CD11c+ dendritic cells harbor reovirus antigen, but these cells are not thought to be actively infected (Fleeton et al., 2004). Viral nonstructural proteins are not present in these cells (Fleeton et al., 2004), suggesting that active replication does not occur. CD11c+ dendritic cells are present in neonatal animals (Muthukkumar, Goldstein and Stein, 2000), but it is not known whether these cells internalize reovirus following peroral inoculation of newborn mice. Reovirus productively infects bulk splenocytes isolated from newborn mice (Tardieu, Powers and Weiner, 1983), suggesting that reovirus can replicate in primary lymphoid cells. Reovirus cannot productively infect splenocytes explanted after the mouse reaches 7 days of age (Tardieu, Powers and Weiner, 1983). Thus, the lack of viral replication in Peyer patch cells in older animals may contribute to the age restriction to reovirus infection.
From Peyer patches, reovirus is hypothesized to traffic via afferent lymphatics to the MLN, then through efferent lymphatics to the blood. It is possible that infected lymphoid cells or lymphoid cells harboring virus mediate transport from the Peyer patches to the bloodstream. However, migrating dendritic cells rarely exit lymph nodes once they enter and present antigen to B and T cells (Iwasaki, 2007). Thus, the cells responsible for transport of reovirus from the Peyer patch are likely retained in the MLN. Reovirus titers in the MLN increase rapidly after peroral inoculation (Antar et al., 2009; Boehme, Guglielmi and Dermody, 2009), suggesting that active viral replication occurs in the MLN. However, it also is possible that the increase in viral load in the MLN represents migration of infected lymphoid cells from the Peyer patches. Dissemination from the MLN to the bloodstream may occur as free virus or within another lymphoid cell subset.
An alternative mechanism for accessing the blood is direct uptake of viral particles from the gut. CD18+ phagocytes extend cellular processes between enterocytes to directly sample luminal contents. Dendritic cells also extend processes through the epithelial monolayer while maintaining barrier integrity to sample gut pathogens (Rescigno et al., 2001). A number of pathogens, including Salmonella (Vazquez-Torres et al., 1999) and Yersinia (Isberg and Barnes, 2001), use macrophages or dendritic cells to invade the bloodstream and cause extraintestinal infection. Following uptake of luminal pathogens, CD18+ phagocytes traffic across the lamina propria and directly into the blood allowing for rapid entry of the pathogen into the bloodstream.
B. Reovirus viremia
Although virus is detected in the blood of infected animals, it is not known whether reovirus virions within the blood are free in the plasma or associated with hematopoietic cells. Other Reoviridae family members, including bluetongue virus (BTV) and Colorado tick fever virus, produce cell-associated viremia during infection. BTV infects and replicates in mononuclear cells, lymphocytes, and endothelial cells (Barratt-Boyes and MacLachlan, 1994; Ellis et al., 1993; MacLachlan et al., 1990; Mahrt and Osburn, 1986; Veronesi et al., 2009). Colorado tick fever virus is detected in mature erythrocytes (Oshiro et al., 1978). However, arthropod vectors transmit BTV and Colorado tick fever virus, making viremia a necessary part of the viral infectious cycle in nature. Mammalian reoviruses are not transmitted by arthropod vectors and may produce a distinctly different type of viremia. Studies in which oncolytic reovirus was delivered intravenously to persons with cancer revealed that virus is largely found in hematopoietic cells, specifically mononuclear cells, granulocytes, and platelets (Adair et al., 2012). Each of these cell types express JAM-A (Martin-Padura et al., 1998; Naik et al., 2001; Sobocka et al., 2000), suggesting that reovirus associates with or infects blood cells to disseminate through the blood to target organs. However, in these studies, virus was delivered directly into the bloodstream by intravenous inoculation. It is not known how reovirus spreads systemically following infection from a natural portal, such as the intestine or lung.
C. Role of receptors in reovirus dissemination
Interactions between viral attachment proteins and host cell receptors play a pivotal role in viral pathogenesis. Receptor engagement is a primary mechanism to define cells targeted by viruses. Therefore, patterns of receptor expression are a key determinant of viral disease. Reoviruses engage two types of cellular receptors: cell-surface carbohydrate (Paul, Choi and Lee, 1989) and JAM-A (Barton et al., 2001b; Paul, Choi and Lee, 1989). Both T1 and T3 (Dermody et al., 1990a; Pacitti and Gentsch, 1987; Paul, Choi and Lee, 1989) reoviruses bind cell-surface SA (Armstrong, Paul and Lee, 1984; Dermody et al., 1990a; Pacitti and Gentsch, 1987; Paul, Choi and Lee, 1989; Paul and Lee, 1987). However, the domains of σ1 that engage glycans differ between the serotypes (Dermody et al., 1990a), as do the specific glycans bound.
SA engagement enhances reovirus infection through an adhesion-strengthening mechanism in which viral particles are tethered to the cell surface via a low-affinity interaction with the carbohydrate (Barton et al., 2001a). This interaction maintains the virus on the cell surface and increases the opportunity to engage JAM-A. SA-binding reovirus strains have an increased capacity to infect cells compared with non-SA-binding viruses; pre-treatment of cells with neuraminidase to remove cell-surface SA eliminates this advantage (Barton et al., 2001a). SA engagement also enhances reovirus tropism for bile duct epithelial cells in mice following peroral inoculation (Barton et al., 2003). The resulting disease closely mimics biliary atresia in human infants (Barton et al., 2003), an illness epidemiologically associated with reovirus (Richardson, Bishop and Smith, 1994; Tyler et al., 1998).
Reovirus strains circulating in nature vary in the capacity to bind SA (Dermody et al., 1990a; Dermody et al., 1990b). This finding suggests that SA binding comes with a fitness cost. Accordingly, SA binding appears to inhibit the capacity of reovirus to establish infection at mucosal portals of entry. Non-SA-binding viruses infect primary human airway epithelial cells substantially more efficiently than SA-binding strains (Excoffon et al., 2008). Moreover, infection of primary human airway epithelial cells by SA-binding viruses is enhanced by removal of cell-surface SA with neuraminidase. Mucosal surfaces are covered with a glycocalyx consisting of polysaccharides and glycoproteins that are rich in SA (Excoffon et al., 2008). SA-binding viruses may be trapped by SA within the glycocalyx and incapable of reaching the underlying epithelium (Excoffon et al., 2008). However, once infection is established, SA binding may enhance the capacity of reovirus to cause disease. In addition to the capacity to target bile duct epithelium, SA-binding strains are more neurovirulent than non-SA-binding viruses following intracranial inoculation (Barton et al., 2003). This increase in virulence is likely due to more efficient infection of neurons, which results in neuronal apoptosis and encephalitis. The function of SA binding in reovirus hematogenous spread remains to be determined.
Although all reoviruses bind JAM-A, T1 and T3 reoviruses infect distinct cells and cause serotype-specific patterns of pathologic injury within the CNS. These observations suggest that JAM-A binding does not influence serotype-specific differences in reovirus neural tropism and CNS disease. Following peroral inoculation, reovirus produces similar viral titers in the intestine of wild-type and JAM-A-deficient mice, suggesting that JAM-A is not required for reovirus replication in the mouse gastrointestinal tract (Antar et al., 2009). In sharp contrast, viral titers at all sites of secondary replication are significantly lower in JAM-A-deficient animals compared with wild-type controls (Fig. 3). Viral loads are comparable within the brains of wild-type and JAM-A-deficient animals after intracranial inoculation, suggesting that JAM-A is not required for viral replication at this site of secondary replication (Antar et al., 2009). These results suggest that JAM-A is required for dissemination of the virus from the intestine to replication sites in target organs.
Figure 3. JAM-A is required for hematogenous reovirus dissemination.
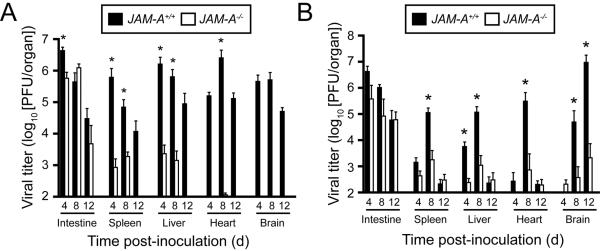
(A) Newborn JAM-A+/+ and JAM-A−/− mice were inoculated perorally with 106 PFU of strain T1L. At days 4, 8, and 12 after inoculation, mice were euthanized, organs were resected, and viral titers were determined by plaque assay. Results are expressed as mean viral titers for 6 animals for each time point. Error bars indicate SD. *, P < 0.005 by Student's t test. When all values are less than the limit of detection (spleen, liver, heart, and brain in JAM-A−/− mice), a Student's t test P value cannot be calculated. (B) Newborn JAM-A+/+ and JAM-A−/− mice were inoculated perorally with 104 PFU of strain T3SA-. At days 4, 8, and 12 after inoculation, mice were euthanized, organs were resected, and viral titers were determined by plaque assay. Results are expressed as mean viral titers for 6–13 animals for each time point. Error bars indicate SD. *, P < 0.05 by Student's t test in comparison to JAM-A−/− mice.1
How might JAM-A promote hematogenous dissemination? Substantially lower reovirus titers are detected in the blood of JAM-A-deficient mice compared with wild-type mice (Fig. 4), suggesting that JAM-A is involved in the establishment of viremia (Antar et al., 2009). Diminished viremia is detected in mice inoculated with either T1 or T3 reovirus, indicating that JAM-A functions in promoting bloodborne spread of T1 viruses that disseminate by strictly hematogenous mechanisms as well as neurotropic T3 reoviruses. Primary pulmonary endothelial cells isolated from JAM-A-deficient mice are refractory to reovirus infection compared with those harvested from wild-type mice (Fig. 5). These data suggest that reovirus engages JAM-A to infect endothelial cells following infection, likely in the lymphatics or vasculature of the gastrointestinal tract. It is possible that virus released from endothelial cells invades the bloodstream to disseminate to peripheral target organs either free in the plasma or associated with hematopoietic cells.
Figure 4. JAM-A is required for reovirus viremia.
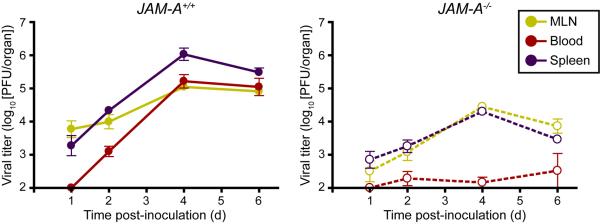
Newborn JAM-A+/+ and JAM-A−/− mice were inoculated perorally with 108 PFU of T1L. At days 1, 2, 4, and 6 after inoculation, mice were euthanized, mesenteric lymph node (MLN), blood, and spleen were collected, and viral titers were determined by plaque assay. Results are expressed as mean viral titers for 3–8 animals for each time point. Error bars indicate SD.1
Figure 5. JAM-A is required efficient reovirus infection of endothelial cells.
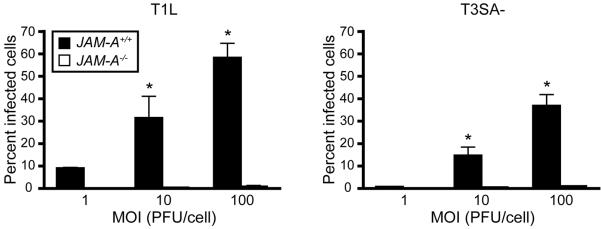
JAMA+/+ and JAM-A−/− primary endothelial cells were adsorbed with T1L or T3SA- at MOIs of 1, 10, and 100 PFU/cell and incubated for 20 h. The percentage of infected cells was quantified by dividing the number of cells exhibiting reovirus staining by the total number of cell nuclei exhibiting DAPI staining in whole 96 wells for triplicate experiments. Wells contained between 200 and 1600 nuclei. Error bars indicate SD. *, P < 0.05 as determined by Student's t test in comparison to JAM-A−/− endothelial cells inoculated at the same MOI.1
D. Function of nonstructural protein σ1s in reovirus dissemination
Studies using T1 σ1s-null virus uncovered a role for σ1s in promoting bloodborne reovirus spread (Boehme, Guglielmi and Dermody, 2009). The σ1s protein is not required for the initial establishment of reovirus infection in the gut. Wild-type and σ1s-null viruses replicate to comparable levels in the gastrointestinal tract following peroral inoculation (Fig. 6) (Boehme, Guglielmi and Dermody, 2009). Reovirus antigen is evident in the intestinal epithelium and Peyer patches of mice inoculated with wild-type or σ1s-null virus, indicating that σ1s does not influence reovirus tropism in the intestine. In contrast to wild-type virus, the σ1s-null mutant fails to produce substantial titers in the MLN. The σ1s-null virus is detected at low titer in the MLN, but viral titers do not increase over the course of infection. These findings indicate that σ1s either is essential for transit through lymphatic channels to the MLN or serves to promote replication in MLN cells. Wild-type virus is detected in the blood and sites of secondary viral replication, including the brain, heart, liver, and spleen (Fig. 6). The σ1s-null virus is not detected in the blood or any of the target organs examined. This difference is probably not due to a requirement for σ1s in mediating replication at these sites, as wild-type and σ1s-null viruses produce comparable titers in the brain following intracranial inoculation. Together, these findings suggest that the σ1s protein performs a function that is essential for reovirus to spread from the gut through intestinal lymphatics and ultimately to the blood where it gains access peripheral organs.
Figure 6. The σ1s protein is required for systemic reovirus dissemination following peroral inoculation.
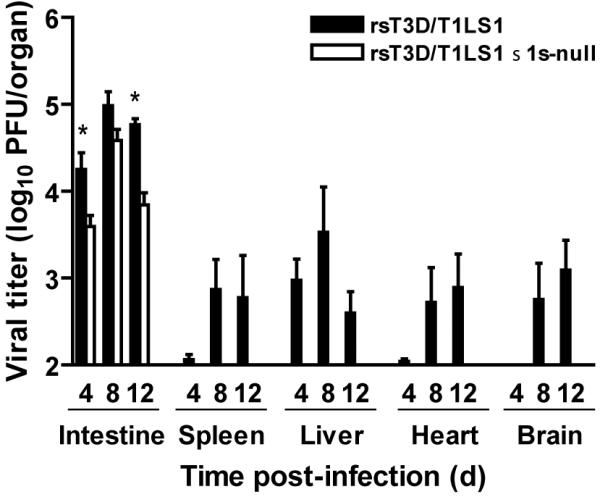
Newborn C57/BL6 mice were inoculated perorally with 104 PFU of wild-type or σ1s-null reovirus. At days 4, 8, and 12 post-inoculation, viral titers in the organs shown were determined by plaque assay. Error bars indicate SEM. *, P < 0.05 as determined by Mann-Whitney test in comparison to wild-type virus. When all values are less than the limit of detection, a Mann-Whitney test P value cannot be calculated. Adapted from Boehme et al. (2009). Copyright (2009) National Academy of Sciences, USA.
In contrast to T1 reoviruses that spread by strictly hematogenous mechanisms, T3 reoviruses disseminate by both hematogenous and neural pathways. The amino acid sequences of the σ1s proteins from the different reovirus serotypes differ substantially (Dermody et al., 1990b). Therefore, it is possible that the σ1s proteins perform serotype-specific functions. In nature, reovirus infects by the peroral route and spreads to the CNS in infant animals resulting in neuropathology. However, infectivity of T3 prototype strain T3D is diminished significantly within the gastrointestinal tract (Bodkin and Fields, 1989; Bodkin, Nibert and Fields, 1989) due to cleavage of its σ1 protein by intestinal proteases (Bodkin and Fields, 1989; Bodkin, Nibert and Fields, 1989; Nibert, Chappell and Dermody, 1995). Consequently, intramuscular inoculation into the hindlimb is used to assess mechanisms of T3D dissemination. Inoculation of type 3 reoviruses intramuscularly leads to invasion of the brain by neural routes (Tyler, McPhee and Fields, 1986). Following intramuscular inoculation, wild-type T3D produces substantially higher titers than the σ1s-null virus in peripheral organs including the heart, liver, intestine, and spleen, similar to results obtained using wild-type and σ1s-null T1 viruses (Boehme et al., 2011). Moreover, wild-type T3D but not the T3D σ1s-null mutant virus is detected in the blood. Together, these data suggest that σ1s functions to promote the establishment of reovirus viremia in a serotype-independent manner, which ultimately leads to infection of peripheral target tissues.
In contrast to its function in hematogenous spread, the σ1s protein is dispensable for reovirus spread to the CNS by neural routes. Both wild-type and σ1s-null viruses produce comparable titers in the spinal cord following inoculation into the hindlimb muscle (Boehme, 2011). Both viruses also produce comparable titers in the brain following direct intracranial inoculation and in cultured primary neurons. Together, these findings indicate that σ1s is not required for reovirus neural spread or replication in the murine CNS. Thus, although T3 viruses spread via neural and hematogenous mechanisms, the T3 σ1s protein only influences the efficiency of hematogenous dissemination.
IV. Neural dissemination of reovirus
In addition to bloodborne spread, T3 reoviruses use neural circuits to disseminate to the CNS (Boehme et al., 2011; Tyler, McPhee and Fields, 1986). Spread via neural routes is a fundamental mechanism of reovirus pathogenesis that is essential for development of reovirus-induced encephalitis (Boehme et al., 2011; Tyler, McPhee and Fields, 1986). Direct infection of neurons at peripheral sites provides the virus with access to the CNS and serves as a conduit to the brain. Although the importance of neural spread to reovirus pathogenesis is well-appreciated, the cellular and molecular mechanisms that underlie neuronal reovirus trafficking are not well understood.
In contrast to hematogenous spread, JAM-A is dispensable for neural dissemination. Although JAM-A is expressed in the brain, the cell types on which it is present have not been defined. JAM-A is found on NG2-glia cells, which are a subset of stem cells that give rise to oligodendrocytes (Nomme et al., 2011). It is unclear whether JAM-A is expressed on peripheral or CNS neurons. Viral titers in the brains of wild-type and JAM-A-deficient mice are comparable after intracranial inoculation (Antar et al., 2009). Viral tropism in the brain for hippocampal, thalamic, and cortical regions also does not differ between wild-type and JAM-A-deficient mice. Concordantly, primary cortical neurons isolated from wild-type and JAM-A-deficient mice are equally susceptible to reovirus infection and produce equivalent yields of viral progeny (Antar et al., 2009). Together, these data indicate JAM-A is not required for reovirus infection of neural tissue and suggest that JAM-A is dispensable for reovirus spread by neural routes. These findings further suggest that a cellular receptor distinct from JAM-A mediates reovirus infection of neurons.
Some evidence exists about the means by which reovirus traverses neural circuits. Treatment of animals with colchicine to inhibit fast axonal transport impairs reovirus spread to the spinal cord following hindlimb inoculation (Sjostrand and Karlsson, 1969; Tyler, McPhee and Fields, 1986). However, treatment with β-β′-iminodipropronitrile to inhibit slow axonal transport does not affect reovirus dissemination to the spinal cord (Hansson et al., 1971; Tyler, McPhee and Fields, 1986). These findings suggest that reovirus traffics in neurons along fast axonal transport pathways. However, these inhibitors may act non-specifically to impair other aspects of viral replication. It also is not known whether reovirus uses afferent or efferent neurons to traffic to the CNS or whether virions can travel using both retrograde and anterograde pathways within neurons. Finally, it is not known where or how progeny virions exit neurons. Much work is required to fully elucidate how reoviruses replicate and traffic in neurons.
V. Function of hematogenous and neural spread in reovirus pathogenesis
T1 reoviruses disseminate to sites of secondary viral replication solely by hematogenous pathways. Following peroral inoculation of JAM-A-deficient mice, T1 reovirus does not reach the blood or peripheral organs (Antar et al., 2009). Similarly, T1 σ1s-null virus fails to disseminate from the intestine to sites of secondary replication (Boehme, Guglielmi and Dermody, 2009). Because T1 reoviruses utilize a single mechanism to spread within its host, inhibiting that mode of dissemination prevents virus-induced systemic disease.
T3 reoviruses, in contrast, disseminate to peripheral organs using a combination of hematogenous and neural mechanisms. Neural spread is essential for maximal neural injury induced by T3 reovirus (Tyler, McPhee and Fields, 1986). Following peroral inoculation or inoculation into the hindlimb muscle, T3 reovirus infects peripheral neurons and travels along nerve fibers to infect the CNS and cause disease (Morrison, Sidman and Fields, 1991b; Tyler, McPhee and Fields, 1986). Inhibiting neural spread by sectioning the sciatic nerve prior to hindlimb inoculation prevents virus spread to the spinal cord (Tyler, McPhee and Fields, 1986). This finding indicates that T3 reovirus spreads along neural routes to the CNS and suggests that neural dissemination is essential for reovirus neuropathogenesis.
The importance of hematogenous spread in reovirus neuropathogenesis is evident from studies that identified host and viral factors that mediate reovirus transport through the blood. JAM-A-deficient mice are completely resistant to reovirus-induced disease following peroral inoculation with T3 reovirus, whereas wild-type mice succumb to infection (Antar et al., 2009). Viral titers in the brains of JAM-A-deficient mice are substantially reduced in comparison to those in wild-type controls. Concordantly, viral loads in the blood of JAM-A-deficient mice are lower than those detected in wild-type mice (Antar et al., 2009). However, following intracranial inoculation, wild-type and JAMA-deficient mice are equally susceptible to reovirus disease, and equivalent viral yields are produced in the brains of wild-type and JAM-A-deficient mice (Antar et al., 2009). These results indicate that reduced reovirus virulence in JAM-A-deficient mice following peroral inoculation is not the result of differences in reovirus replication in the brain.
Studies of T3 σ1s-null viruses also highlight the requirement of hematogenous dissemination for reovirus neuropathogenesis. Wild-type T3 reovirus is substantially more virulent than the T3 σ1s-null virus following hindlimb inoculation (Fig. 7) (Boehme et al., 2011). Approximately 75% of animals inoculated with wild-type virus succumb to infection compared with 25% of mice inoculated with the σ1s-null virus (Boehme et al., 2011). Wild-type and σ1s-null T3 reoviruses induced 100% mortality following intracranial inoculation, although animals inoculated with wild-type virus succumbed to CNS disease with slightly faster kinetics than those inoculated with the σ1s-null virus. Wild-type and σ1s-null viruses also produce equivalent titers in the brain following intracranial inoculation, indicating that σ1s is dispensable for viral replication in the murine CNS. Thus, the disparity in virulence between wild-type and σ1s-null viruses following intramuscular inoculation does not result from differences in replication in the CNS between the two viral strains.
Figure 7.

The σ1s protein enhances reovirus virulence following intramuscular inoculation. (A) Newborn C57/BL6 mice were inoculated in the left hindlimb with 106 PFU of wild-type or σ1s-null T3 reovirus. Mice (n = 19 for each virus strain) were monitored for survival for 25 days. *, P < 0.001 as determined by log-rank test in comparison to wild-type T3 reovirus. (B) The σ1s protein is not required for reovirus spread by neural routes. Newborn C57/BL6 mice were inoculated in the left hindlimb with 106 PFU of wild-type or σ1s-null T3 reovirus. At days 1, 2, 4, 8, and 12 post-inoculation, mice were euthanized, hindlimb muscle, spinal cord, and brain were resected, and viral titers were determined by plaque assay. Results are expressed as mean viral titers for 6–9 animals for each time point. Error bars indicate SEM. *, P < 0.05 as determined by Mann-Whitney test in comparison to wild-type T3 reovirus. Copyright © American Society for Microbiology, Journal of Virology, vol. 85, 2011, pages 11781 1790, doi: 10.1128/JVI.02289-10.
Following hindlimb inoculation, wild-type virus is detected in the brain 1 day after infection (Figure 7). In contrast, the σ1s-null virus is not found in the brain until 2 days after inoculation. At days 2 and 4 post-inoculation, viral titers in the brains of animals inoculated with wild-type virus are markedly higher than those observed in mice inoculated with the σ1s-null mutant. This finding correlates with significantly higher loads of wild-type virus in the blood of infected animals at early times post-inoculation compared with the σ1s-null virus (Boehme et al., 2011). Comparable titers of wild-type and σ1s-null viruses are found in the spinal cord at days 1, 2, and 4 post-inoculation. This observation suggests that transport of the σ1s-null virus to the CNS by neural pathways is not impaired. At day 8 post-inoculation, peak titers of wild-type and σ1s-null viruses in the brain are equivalent, possibly reflecting delivery of virus to the brain via neural routes. Collectively, these findings suggest that hematogenous spread is required for reovirus transport to the brain at early times after infection. These results also suggest that the timing of reovirus delivery to the brain is critical for neuropathogenesis. Viral transport by neural routes does not differ between wild-type and σ1s-null viruses, and both virus strains produce equivalent peak titers in the brain. However, peak titers of the σ1s-null virus appear to be achieved after the mice reach the age-imposed limit to reovirus infection, and these animals are no longer susceptible to reovirus-induced CNS disease (Mann et al., 2002). Thus, reovirus transport to the brain by the blood at early times after infection is critical for neuropathogenesis.
Reovirus spreads to the spinal cord via the sciatic nerve following intramuscular inoculation into the hindlimb (Tyler, McPhee and Fields, 1986). Transection of the sciatic nerve prior to inoculation inhibits neural transmission of the virus to the spinal cord; however, viral dissemination by the blood is unaffected (Tyler, McPhee and Fields, 1986). T3 reovirus retains the capacity to spread to the brain after sciatic nerve section (Boehme et al., 2011), suggesting that reovirus can access the brain even in the absence of neural spread, likely via the bloodstream (Fig. 8). In addition, almost no virus is detected in the brain following hindlimb inoculation with the σ1s-null virus when the sciatic nerve is sectioned. Thus, virus cannot access the brain when both hematogenous and neural pathways of spread are inhibited.
Figure 8. Reovirus disseminates to the CNS by hematogenous and neural routes.

The left sciatic nerve of newborn C57/BL6 mice was sectioned prior to inoculation in the left hindlimb with 106 PFU of wild-type or σ1s-null T3 reovirus. In parallel, mice in which the left sciatic nerve was not sectioned were inoculated in the left hindlimb with 106 PFU of wild-type or σ1s-null T3 reovirus. At days 2 and 4 post-inoculation, mice were euthanized, (A) hindlimb muscle, spinal cord, and brain and (B) heart, intestine, liver, and spleen were resected, and viral titers were determined by plaque assay. Results are expressed as mean viral titers for 6 animals for each time point. Error bars indicate SEM. *, P < 0.05 as determined by Mann-Whitney test in comparison to animals in which the sciatic nerve was not sectioned. Copyright © American Society for Microbiology, Journal of Virology, vol. 85, 2011, pages 11781 1790, doi: 10.1128/JVI.02289-10.
Together, these findings suggest that (i) spread by neural routes alone is not sufficient to cause reovirus CNS disease, (ii) bloodborne spread is required for delivery of reovirus to the brain at early times post-infection, (iii) hematogenous viral dissemination to the brain is an essential mechanism of reovirus neuropathogenesis, and (iv) virus must be delivered to the brain by the blood early after inoculation for full reovirus neurovirulence.
VI. Unanswered questions and future directions
We have identified host and viral factors essential for the hematogenous dissemination of reovirus. However, many unanswered questions remain. Because viruses capable of bloodstream spread may share similar mechanisms of dissemination, understanding how reovirus spreads in the infected host may aid in the development of therapeutics that target this critical step in viral pathogenesis.
A. How does reovirus enter and exit the bloodstream?
To spread to peripheral organs by hematogenous pathways, reovirus must first enter the bloodstream. Studies of reovirus pathogenesis suggest that following peroral inoculation, reovirus infects Peyer patch lymphoid cells that transport virus to the bloodstream (Figure 9). However, reovirus also disseminates hematogenously following intracranial inoculation (Boehme et al., 2011; Boehme, Guglielmi and Dermody, 2009). This observation suggests that reovirus has the capacity to cross endothelial barriers to enter the blood. Little is known about how reovirus infects polarized cells, such as those that constitute the endothelium. JAM-A localizes to tight junctions linking endothelial cells and functions in maintaining the barrier between the tissue and blood compartments. JAM-A is required for hematogenous spread of reovirus and infection of primary cultures of pulmonary vascular endothelial cells (Antar et al., 2009). We envision several possible mechanisms to explain how reovirus uses JAM-A to facilitate entry into the blood. First, JAM-A may function as a gatekeeper for reovirus entry into the bloodstream (Figure 9). Although reovirus infection does not change the barrier function of primary human airway epithelial cells (Excoffon et al., 2008), it is unclear whether the same phenomenon occurs during reovirus infection of the endothelium. Free reovirus virions may interact with JAM-A in endothelial cell tight junctions, transiently disrupt these structures, and cause focal breaches of the endothelial barrier to allow viral invasion of the bloodstream. Other viruses are known to disrupt polarized cell barriers during infection. Mouse adenovirus-1 infection of endothelial cells reduces tight junction protein expression and decreases barrier function in polarized endothelial cell monolayers (Gralinski et al., 2009). Coxsackieviruses engage decay-accelerating factor, an apically distributed protein of polarized epithelial cells, to disrupt tight junctions (Coyne et al., 2007). In doing so, coxsackieviruses gain access to the basolaterally located coxsackievirus and adenovirus receptor (CAR) (Coyne et al., 2007). HIV-1 gp120 diminishes expression of tight junction proteins and increases vascular permeability (Kanmogne et al., 2009).
Figure 9. Model of reovirus hematogenous spread from the intestine.

(1) Following peroral inoculation, reovirus infects intestinal epithelial cells (2) and is taken up by lymphoid cells in the Peyer patch. (3) Infected dendritic cells or lymphocytes carry reovirus from the Peyer patch through the lymphatics and finally to the blood. (4) Phagocytic cells that extend processes into the lumen of the intestine also might be infected for subsequent transport of virus through the lymphatics. (5) Reovirus may enter directly into the blood by passing between endothelial cells or via release into the bloodstream from infected cells.
Second, it is possible that reovirus infects endothelial cells to allow progeny virus to be released directly into the blood (Figure 9). Endothelial cells function as sites of amplification for many viruses that spread via the bloodstream. Murine cytomegalovirus dissemination occurs after an episode of secondary viremia that requires viral replication in endothelial cells (Sacher et al., 2008). It is possible that reovirus productively infects endothelium from the basolateral surface on the abluminal side of the endothelium and is released from the apical surface into the blood. Many viruses that infect polarized cells egress apically (Roberts 1995). This mechanism is common for respiratory viruses, in which release from the apical surface of infected respiratory epithelial cells ensures that the virus will be shed into the respiratory tract to facilitate transmission to susceptible hosts (Gerl et al., 2012; Brock et al., 2003; Rodriguez and Sabatini, 1978). Studies of reovirus infection of polarized endothelial cells will shed light on mechanisms used by the virus to traverse endothelial monolayers.
Third, reovirus spread may involve infection or association with hematopoietic cells (Figure 9). Hematopoietic cells express JAM-A as an adhesin to allow monocyte extravasation across endothelial barriers (Martin-Padura et al., 1998; Williams et al., 1999). It is not known whether hematopoietic cells are infected or whether infected blood cells transport reovirus systemically following infection by a natural route of inoculation. However, in cancer patients treated with an intravenous infusions of reovirus, virions associate with mononuclear cells, granulocytes, and platelets to allow dissemination to tumors localized in the viscera (Adair et al., 2012). If hematopoietic cells are responsible for hematogenous reovirus dissemination, age-dependent restriction of reovirus replication in these cells may be one mechanism to explain the limitation of reovirus disease to newborn animals (Tardieu, Powers and Weiner, 1983).
Reovirus exit from the bloodstream is required for infection and replication in target tissues and development of organ-specific disease. After peroral inoculation of reovirus, high viral titers are found in virtually all organs (Antar et al., 2009; Boehme et al., 2011; Boehme, Guglielmi and Dermody, 2009). Mechanisms similar to those that facilitate reovirus entry into the vasculature may mediate reovirus escape from the blood. Reovirus interactions with JAM-A may induce localized perturbations of tight junction integrity that permit virus escape into tissues. Reovirus virions in the blood may infect endothelial cells from the apical surface and progeny virions may be released basolaterally. Finally, infected hematopoietic cells may transport virus from the blood into target organs. None of these possibilities is mutually exclusive; reovirus may use multiple strategies to enter and exit the bloodstream. Studies using mice with tissue-specific expression of JAM-A may help to elucidate mechanisms by which JAM-A facilitates reovirus spread through the bloodstream.
B. How does σ1s promote hematogenous spread?
Mechanisms by which σ1s promotes dissemination have not been determined. The σ1s protein is required for reovirus-induced cell cycle arrest at the G2/M boundary (Poggioli et al., 2002; Poggioli, Dermody and Tyler, 2001) and has been implicated in apoptosis in vivo (Hoyt et al., 2005). It not known whether inhibition of cell cycle progression is related to the induction of apoptosis following reovirus infection. Cells respond to replication stress or DNA damage by activating checkpoints that arrest the cell cycle. For cells in which genomic damage cannot be repaired, apoptosis is induced to ensure that only faithfully replicated DNA is passed to daughter cells. The relationship between cell cycle arrest and apoptosis in the context of reovirus infection has not been examined. It is possible that σ1s-mediated cell cycle arrest contributes to reovirus-induced apoptosis. Interaction of σ1s with components of the host cell cycle machinery that inhibit normal cell cycle progression could cause the cell to undergo apoptosis.
It is not known whether σ1s-dependent cell cycle arrest and apoptosis are responsible for σ1s-mediated reovirus dissemination. It is possible that σ1s-dependent apoptosis in intestinal epithelial cells promotes reovirus uptake by phagocytic cells at the site of inoculation, and these cells in turn traffic virus to the bloodstream where the virus has access to JAM-A. Although σ1s is not required for reovirus growth in cultured cells (Boehme et al., 2011; Boehme, Guglielmi and Dermody, 2009; Rodgers et al., 1998), it is possible that σ1s is necessary for efficient reovirus replication in specific cell types that are required for viral dissemination. Defining the cell types used by reovirus to spread through the blood may help uncover how σ1s promotes hematogenous spread. Finally, σ1s may mediate evasion of the host immune response, thereby allowing viral spread. Differences in viral dissemination between wild-type and σ1s are evident at early times post-inoculation. This suggests that σ1s would impact host innate immune mechanisms, as opposed to adaptive responses that develop at later times after infection. Determining how σ1s promotes hematogenous reovirus spread is essential to understand how an enteric, neurotropic virus circuits from the intestine to the CNS.
C. Clinical implications
Defining factors that govern reovirus dissemination in the blood is essential for optimum use of reovirus in clinical applications. Reovirus efficiently replicates in and kills cancer cells (Adair et al., 2012; Karapanagiotou et al., 2012). Phase II and III clinical trials are underway to test the efficacy of reovirus as an adjunct to conventional cancer therapies (Adair et al., 2012; Karapanagiotou et al., 2012; Kottke et al., 2011). Following intravenous administration, reovirus must navigate and exit the bloodstream to infect solid organ tumors. Intratumoral injection of reovirus may allow for enhanced replication in tumor cells and subsequent spread through the blood to target metastatic tumor foci. Thus, determining viral and cellular determinants underlying how reoviruses gain access to the blood compartment, spread within the bloodstream, and exit from the circulation may aid in oncolytic design. Use of the reverse genetics system may allow engineering of reovirus therapeutics with mutations that increase vector potency or safety by manipulating dissemination determinants (Kobayashi et al., 2007).
We have uncovered a central role for hematogenous dissemination in reovirus neuropathogenesis and elucidated molecular mechanisms that govern reovirus spread by the blood. However, we have much more to learn. Understanding mechanisms of reovirus dissemination will provide broader insight into events at the pathogen-host interface that lead to systemic disease and may aid in the development of therapeutics that target this critical step in viral pathogenesis.
Acknowledgements
We thank members of our laboratories and Dr. J. Craig Forrest for many useful discussions. This research was supported by Public Health Service awards K22 AI094079 (K.W.B.), F31 NS074596 (C.M.L.), R37 AI38296 (T.S.D.), and the Elizabeth B. Lamb Center for Pediatric Research.
Footnotes
Reprinted from Cell Host & Microbe, 5, Antar, A.A.R., Konopka, J.L., Campbell, J.A., Henry, R.A., Perdigoto, A.L., Carter, B.D., Pozzi, A., Abel, T.W., Dermody, T.S. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus, 59–71, Copyright (2009), with permission from Elsevier.
References
- Adair RA, Roulstone V, Scott KJ, Morgan R, Nuovo GJ, Fuller M, Beirne D, West EJ, Jennings VA, Rose A, Kyula J, Fraser S, Dave R, Anthoney DA, Merrick A, Prestwich R, Aldouri A, Donnelly O, Pandha H, Coffey M, Selby P, Vile R, Toogood G, Harrington K, Melcher AA. Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Science translational medicine. 2012;4:138ra177. doi: 10.1126/scitranslmed.3003578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amerongen HM, Wilson GAR, Fields BN, Neutra MR. Proteolytic processing of reovirus is required for adherence to intestinal M cells. J. Virol. 1994;68:8428–8432. doi: 10.1128/jvi.68.12.8428-8432.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antar AAR, Konopka JL, Campbell JA, Henry RA, Perdigoto AL, Carter BD, Pozzi A, Abel TW, Dermody TS. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe. 2009;5:59–71. doi: 10.1016/j.chom.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong GD, Paul RW, Lee PW. Studies on reovirus receptors of L cells: virus binding characteristics and comparison with reovirus receptors of erythrocytes. Virology. 1984;138:37–48. doi: 10.1016/0042-6822(84)90145-4. [DOI] [PubMed] [Google Scholar]
- Barratt-Boyes SM, MacLachlan NJ. Dynamics of viral spread in bluetongue virus infected calves. Vet. Microbiol. 1994;40:361–371. doi: 10.1016/0378-1135(94)90123-6. [DOI] [PubMed] [Google Scholar]
- Barton ES, Connolly JL, Forrest JC, Chappell JD, Dermody TS. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J. Biol. Chem. 2001a;276:2200–2211. doi: 10.1074/jbc.M004680200. [DOI] [PubMed] [Google Scholar]
- Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y, Schnell F, Nusrat A, Parkos CA, Dermody TS. Junction adhesion molecule is a receptor for reovirus. Cell. 2001b;104:441–451. doi: 10.1016/s0092-8674(01)00231-8. [DOI] [PubMed] [Google Scholar]
- Barton ES, Youree BE, Ebert DH, Forrest JC, Connolly JL, Valyi-Nagy T, Washington K, Wetzel JD, Dermody TS. Utilization of sialic acid as a coreceptor is required for reovirus-induced biliary disease. J. Clin. Invest. 2003;111:1823–1833. doi: 10.1172/JCI16303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass DM, Bodkin D, Dambrauskas R, Trier JS, Fields BN, Wolf JL. Intraluminal proteolytic activation plays an important role in replication of type 1 reovirus in the intestines of neonatal mice. J. Virol. 1990;64:1830–1833. doi: 10.1128/jvi.64.4.1830-1833.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass DM, Trier JS, Dambrauskas R, Wolf JL. Reovirus type 1 infection of small intestinal epithelium in suckling mice and its effect on M cells. Laboratory Investigations. 1988;58:226–235. [PubMed] [Google Scholar]
- Bazzoni G. The JAM family of junctional adhesion molecules. Curr. Opin. Cell Biol. 2003;15:525–530. doi: 10.1016/s0955-0674(03)00104-2. [DOI] [PubMed] [Google Scholar]
- Bazzoni G, Tonetti P, Manzi L, Cera MR, Balconi G, Dejana E. Expression of junctional adhesion molecule-A prevents spontaneous and random motility. J. Cell Sci. 2005;118:623–632. doi: 10.1242/jcs.01661. [DOI] [PubMed] [Google Scholar]
- Bellum SC, Dove D, Harley RA, Greene WB, Judson MA, London L, London SD. Respiratory reovirus 1/L induction of intraluminal fibrosis. A model for the study of bronchiolitis obliterans organizing pneumonia. Am. J. Pathol. 1997;150:2243–2254. [PMC free article] [PubMed] [Google Scholar]
- Bodian D. Emerging concept of poliomyelitis infection. Science. 1955;122:105–108. doi: 10.1126/science.122.3159.105. [DOI] [PubMed] [Google Scholar]
- Bodkin DK, Fields BN. Growth and survival of reovirus in intestinal tissue: role of the L2 and S1 genes. J. Virol. 1989;63:1188–1193. doi: 10.1128/jvi.63.3.1188-1193.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodkin DK, Nibert ML, Fields BN. Proteolytic digestion of reovirus in the intestinal lumens of neonatal mice. J. Virol. 1989;63:4676–4681. doi: 10.1128/jvi.63.11.4676-4681.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehme KW, Frierson JM, Konopka JL, Kobayashi T, Dermody TS. The reovirus sigma1s protein is a determinant of hematogenous but not neural virus dissemination in mice. J. Virol. 2011;85:11781–11790. doi: 10.1128/JVI.02289-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehme KW, Guglielmi KM, Dermody TS. Reovirus nonstructural protein σ1s is required for establishment of viremia and systemic dissemination. Proc. Natl. Acad. Sci. U. S. A. 2009;106:19986–19991. doi: 10.1073/pnas.0907412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JA, Shelling P, Wetzel JD, Johnson EM, Wilson GAR, Forrest JC, Aurrand-Lions M, Imhof B, Stehle T, Dermody TS. Junctional adhesion molecule-A serves as a receptor for prototype and field-isolate strains of mammalian reovirus. J. Virol. 2005;79:7967–7978. doi: 10.1128/JVI.79.13.7967-7978.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter PB, Collins FM. The route of enteric infection in normal mice. J. Exp. Med. 1974;139:1189–1203. doi: 10.1084/jem.139.5.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashdollar LW, Chmelo RA, Wiener JR, Joklik WK. Sequences of the S1 genes of the three serotypes of reovirus. Proc. Natl. Acad. Sci. U. S. A. 1985;82:24–28. doi: 10.1073/pnas.82.1.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenatiempo Y, Twardowski T, Shoeman R, Ernst H, Brot N, Weissbach H, Shatkin AJ. Two initiation sites detected in the small s1 species of reovirus mRNA by dipeptide synthesis in vitro. Proc. Natl. Acad. Sci. U. S. A. 1984;81:1084–1088. doi: 10.1073/pnas.81.4.1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell JD, Barton ES, Smith TH, Baer GS, Duong DT, Nibert ML, Dermody TS. Cleavage susceptibility of reovirus attachment protein σ1 during proteolytic disassembly of virions is determined by a sequence polymorphism in the σ1 neck. J. Virol. 1998;72:8205–8213. doi: 10.1128/jvi.72.10.8205-8213.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell JD, Duong JL, Wright BW, Dermody TS. Identification of carbohydrate-binding domains in the attachment proteins of type 1 and type 3 reoviruses. J. Virol. 2000;74:8472–8479. doi: 10.1128/jvi.74.18.8472-8479.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell JD, Prota A, Dermody TS, Stehle T. Crystal structure of reovirus attachment protein σ1 reveals evolutionary relationship to adenovirus fiber. EMBO J. 2002;21:1–11. doi: 10.1093/emboj/21.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corada M, Chimenti S, Cera MR, Vinci M, Salio M, Fiordaliso F, De Angelis N, Villa A, Bossi M, Staszewsky LI, Vecchi A, Parazzoli D, Motoike T, Latini R, Dejana E. Junctional adhesion molecule-A-deficient polymorphonuclear cells show reduced diapedesis in peritonitis and heart ischemia-reperfusion injury. Proc. Natl. Acad. Sci. U. S. A. 2005;102:10634–10639. doi: 10.1073/pnas.0500147102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne CB, Shen L, Turner JR, Bergelson JM. Coxsackievirus entry across epithelial tight junctions requires occludin and the small GTPases Rab34 and Rab5. Cell Host Microbe. 2007;2:181–192. doi: 10.1016/j.chom.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danthi P, Guglielmi KM, Kirchner E, Mainou B, Stehle T, Dermody TS. From touchdown to transcription: the reovirus cell entry pathway. Curr. Top. Microbiol. Immunol. 2010;343:91–119. doi: 10.1007/82_2010_32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, Hoang DM, Chau NV, Khanh TH, Dong VC, Qui PT, Cam BV, Ha do Q, Guan Y, Peiris JS, Chinh NT, Hien TT, Farrar J. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 2006;12:1203–1207. doi: 10.1038/nm1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermody TS, Parker J, Sherry B. Orthoreovirus. In: Howley D. M. K. a. P. M., editor. Fields Virology. Lippincott Williams & Wilkins; Philadelphia: In Press. [Google Scholar]
- Dermody TS, Nibert ML, Bassel-Duby R, Fields BN. A σ1 region important for hemagglutination by serotype 3 reovirus strains. J. Virol. 1990a;64:5173–5176. doi: 10.1128/jvi.64.10.5173-5176.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermody TS, Nibert ML, Bassel-Duby R, Fields BN. Sequence diversity in S1 genes and S1 translation products of 11 serotype 3 reovirus strains. J. Virol. 1990b;64:4842–4850. doi: 10.1128/jvi.64.10.4842-4850.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichter MA, Weiner HL. Infection of neuronal cell cultures with reovirus mimics in vitro patterns of neurotropism. Ann. Neurol. 1984;16:603–610. doi: 10.1002/ana.410160512. [DOI] [PubMed] [Google Scholar]
- Dryden KA, Wang G, Yeager M, Nibert ML, Coombs KM, Furlong DB, Fields BN, Baker TS. Early steps in reovirus infection are associated with dramatic changes in supramolecular structure and protein conformation: analysis of virions and subviral particles by cryoelectron microscopy and image reconstruction. J. Cell Biol. 1993;122:1023–1041. doi: 10.1083/jcb.122.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis JA, Coen ML, MacLachlan NJ, Wilson WC, Williams ES, Leudke AJ. Prevalence of bluetongue virus expression in leukocytes from experimentally infected ruminants. Am. J. Vet. Res. 1993;54:1452–1456. [PubMed] [Google Scholar]
- Ernst H, Shatkin AJ. Reovirus hemagglutinin mRNA codes for two polypeptides in overlapping reading frames. Proc. Natl. Acad. Sci. U. S. A. 1985;82:48–52. doi: 10.1073/pnas.82.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffon KJDA, Guglielmi KM, Wetzel JD, Gansemer ND, Campbell JA, Dermody TS, Zabner J. Reovirus preferentially infects the basolateral surface and is released from the apical surface of polarized human respiratory epithelial cells. J. Infect. Dis. 2008;197:1189–1197. doi: 10.1086/529515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields BN. Studies of reovirus pathogenesis reveal potential sites for antiviral intervention. Adv. Exp. Med. Biol. 1992;312:1–14. doi: 10.1007/978-1-4615-3462-4_1. [DOI] [PubMed] [Google Scholar]
- Fleeton M, Contractor N, Leon F, Wetzel JD, Dermody TS, Kelsall B. Peyer's patch dendritic cells process viral antigen from apoptotic epithelial cells in the intestine of reovirus-infected mice. J. Exp. Med. 2004;200:235–245. doi: 10.1084/jem.20041132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser RDB, Furlong DB, Trus BL, Nibert ML, Fields BN, Steven AC. Molecular structure of the cell-attachment protein of reovirus: correlation of computer-processed electron micrographs with sequence-based predictions. J. Virol. 1990;64:2990–3000. doi: 10.1128/jvi.64.6.2990-3000.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan JE, Curtiss R., 3rd Cloning and molecular characterization of genes whose products allow Salmonella typhimurium to penetrate tissue culture cells. Proc. Natl. Acad. Sci. U.S.A. 1989;86:6383–6387. doi: 10.1073/pnas.86.16.6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghislin S, Obino D, Middendorp S, Boggetto N, Alcaide-Loridan C, Deshayes F. Junctional adhesion molecules are required for melanoma cell lines transendothelial migration in vitro. Pigment cell & melanoma research. 2011;24:504–511. doi: 10.1111/j.1755-148X.2011.00856.x. [DOI] [PubMed] [Google Scholar]
- Golden JW, Schiff LA. Neutrophil elastase, an acid-independent serine protease, facilitates reovirus uncoating and infection in U937 promonocyte cells. Virol. J. 2005;2:48. doi: 10.1186/1743-422X-2-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Xie Z, Gao Z, Liu J, Korteweg C, Ye J, Lau LT, Lu J, Zhang B, McNutt MA, Lu M, Anderson VM, Gong E, Yu AC, Lipkin WI. H5N1 infection of the respiratory tract and beyond: a molecular pathology study. Lancet. 2007;370:1137–1145. doi: 10.1016/S0140-6736(07)61515-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson G, Kristensson K, Olsson Y, Sjostrand J. Embryonal and postnatal development of mast cells in rat peripheral nerve. Acta Neuropathol. 1971;17:139–149. doi: 10.1007/BF00687489. [DOI] [PubMed] [Google Scholar]
- Hoyt CC, Bouchard RJ, Tyler KL. Novel nuclear herniations induced by nuclear localization of a viral protein. J. Virol. 2004;78:6360–6369. doi: 10.1128/JVI.78.12.6360-6369.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt CC, Richardson-Burns SM, Goody RJ, Robinson BA, Debiasi RL, Tyler KL. Nonstructural protein σ1s is a determinant of reovirus virulence and influences the kinetics and severity of apoptosis induction in the heart and central nervous system. J. Virol. 2005;79:2743–2753. doi: 10.1128/JVI.79.5.2743-2753.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iden S, Misselwitz S, Peddibhotla SSD, Tuncay H, Rehder D, Gerke V, Robenek H, Suzuki A, Ebnet K. aPKC phosphorylates JAM-A at Ser285 to promote cell contact maturation and tight junction formation. J. Cell Biol. 2012;196:623–639. doi: 10.1083/jcb.201104143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isberg RR, Barnes P. Subversion of integrins by enteropathogenic Yersinia. J. Cell Sci. 2001;114:21–28. doi: 10.1242/jcs.114.1.21. [DOI] [PubMed] [Google Scholar]
- Iwasaki A. Mucosal dendritic cells. Annu Rev Immunol. 2007;25:381–418. doi: 10.1146/annurev.immunol.25.022106.141634. [DOI] [PubMed] [Google Scholar]
- Jones BD, Ghori N, Falkow S. Salmonella typhimurium initiates murine infection by penetrating and destroying the specialized epithelial M cells of the Peyer's patches. J. Exp. Med. 1994;180:15–23. doi: 10.1084/jem.180.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchner E, Guglielmi KM, Strauss HM, Dermody TS, Stehle T. Structure of reovirus σ1 in complex with its receptor junctional adhesion molecule-A. PLoS Path. 2008;4:e1000235. doi: 10.1371/journal.ppat.1000235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Antar AAR, Boehme KW, Danthi P, Eby EA, Guglielmi KM, Holm GH, Johnson EM, Maginnis MS, Naik S, Skelton WB, Wetzel JD, Wilson GJ, Chappell JD, Dermody TS. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe. 2007;1:147–157. doi: 10.1016/j.chom.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Ooms LS, Ikizler M, Chappell JD, Dermody TS. An improved reverse genetics system for mammalian orthoreoviruses. Virology. 2010;2:194–200. doi: 10.1016/j.virol.2009.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiken T, Fouchier RA, Schutten M, Rimmelzwaan GF, van Amerongen G, van Riel D, Laman JD, de Jong T, van Doornum G, Lim W, Ling AE, Chan PK, Tam JS, Zambon MC, Gopal R, Drosten C, van der Werf S, Escriou N, Manuguerra JC, Stohr K, Peiris JS, Osterhaus AD. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet. 2003;362:263–270. doi: 10.1016/S0140-6736(03)13967-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PW, Hayes EC, Joklik WK. Protein sigma 1 is the reovirus cell attachment protein. Virology. 1981;108:156–163. doi: 10.1016/0042-6822(81)90535-3. [DOI] [PubMed] [Google Scholar]
- MacLachlan NJ, Jagels G, Rossitto PV, Moore PF, Heidner HW. The pathogenesis of experimental bluetongue virus infection of calves. Vet. Pathol. 1990;27:223–229. doi: 10.1177/030098589002700402. [DOI] [PubMed] [Google Scholar]
- Maginnis MS, Forrest JC, Kopecky-Bromberg SA, Dickeson SK, Santoro SA, Zutter MM, Nemerow GR, Bergelson JM, Dermody TS. Beta1 integrin mediates internalization of mammalian reovirus. J Virol. 2006;80:2760–2770. doi: 10.1128/JVI.80.6.2760-2770.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahrt CR, Osburn BI. Experimental bluetongue virus infection of sheep; effect of vaccination: pathologic, immunofluorescent, and ultrastructural studies. Am. J. Vet. Res. 1986;47:1198–1203. [PubMed] [Google Scholar]
- Mainou BA, Dermody TS. Transport to late endosomes is required for efficient reovirus infection. J. Virol. 2012 doi: 10.1128/JVI.00100-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell KJ, Babbin BA, Nusrat A, Parkos CA. Junctional adhesion molecule-1 (JAM1) regulates epithelial cell morphology through effects on β1 integrins and Rap1 activity. J. Biol. Chem. 2005;280:11665–11674. doi: 10.1074/jbc.M412650200. [DOI] [PubMed] [Google Scholar]
- Mann MA, Knipe DM, Fischbach GD, Fields BN. Type 3 reovirus neuroinvasion after intramuscular inoculation: direct invasion of nerve terminals and age-dependent pathogenesis. Virology. 2002;303:222–231. doi: 10.1006/viro.2002.1699. [DOI] [PubMed] [Google Scholar]
- Martin-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A, Ruco L, Villa A, Simmons D, Dejana E. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J. Cell Biol. 1998;142:117–127. doi: 10.1083/jcb.142.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier GT, Campbell JA, Chappell JD, Stehle T, Dermody TS, Barry MA. A chimeric adenovirus vector encoding reovirus attachment protein σ1 targets cells expressing junctional adhesion molecule 1. Proc. Natl. Acad. Sci. U. S. A. 2004;101:6188–6193. doi: 10.1073/pnas.0400542101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin MJ, Warner A, Fields BN. A pathway for entry of reoviruses into the host through M cells of the respiratory tract. J. Exp. Med. 1994;180:1523–1527. doi: 10.1084/jem.180.4.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin MJ, Warner A, Fields BN. Reovirus infection in rat lungs as a model to study the pathogenesis of viral pneumonia. J. Virol. 1996;70:541–548. doi: 10.1128/jvi.70.1.541-548.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison LA, Sidman RL, Fields BN. Direct spread of reovirus from the intestinal lumen to the central nervous system through vagal autonomic nerve fibers. Proc Natl Acad Sci U S A. 1991a;88:3852–3856. doi: 10.1073/pnas.88.9.3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison LA, Sidman RL, Fields BN. Direct spread of reovirus from the intestinal lumen to the central nervous system through vagal autonomic nerve fibers. Proc. Natl. Acad. Sci. U. S. A. 1991b;88:3852–3856. doi: 10.1073/pnas.88.9.3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthukkumar S, Goldstein J, Stein KE. The ability of B cells and dendritic cells to present antigen increases during ontogeny. J. Immunol. 2000;165:4803–4813. doi: 10.4049/jimmunol.165.9.4803. [DOI] [PubMed] [Google Scholar]
- Naik UP, Naik MU, Eckfeld K, Martin-DeLeon P, Spychala J. Characterization and chromosomal localization of JAM-1, a platelet receptor for a stimulatory monoclonal antibody. J. Cell Sci. 2001;114:539–547. doi: 10.1242/jcs.114.3.539. [DOI] [PubMed] [Google Scholar]
- Nason EL, Wetzel JD, Mukherjee SK, Barton ES, Prasad BVV, Dermody TS. A monoclonal antibody specific for reovirus outer-capsid protein σ3 inhibits σ1-mediated hemagglutination by steric hindrance. J. Virol. 2001;75:6625–6634. doi: 10.1128/JVI.75.14.6625-6634.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanson N, Tyler KL. Entry, dissemination, shedding, and transmission of viruses. In: Nathanson N, editor. Viral Pathogenesis. Lippincott-Raven; Philadelphia: 1997. pp. 13–33. [Google Scholar]
- Nibert ML, Chappell JD, Dermody TS. Infectious subvirion particles of reovirus type 3 Dearing exhibit a loss in infectivity and contain a cleaved σ1 protein. J. Virol. 1995;69:5057–5067. doi: 10.1128/jvi.69.8.5057-5067.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibert ML, Odegard AL, Agosto MA, Chandran K, Schiff LA. Putative autocleavage of reovirus mu1 protein in concert with outer-capsid disassembly and activation for membrane permeabilization. J Mol Biol. 2005;345:461–474. doi: 10.1016/j.jmb.2004.10.026. [DOI] [PubMed] [Google Scholar]
- Nomme J, Fanning AS, Caffrey M, Lye MF, Anderson JM, Lavie A. The Src homology 3 domain is required for junctional adhesion molecule binding to the third PDZ domain of the scaffolding protein ZO-1. J. Biol. Chem. 2011;286:43352–43360. doi: 10.1074/jbc.M111.304089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard R, Golden JW, Schiff LA. Impact of host proteases on reovirus infection in the respiratory tract. J. Virol. 2012 doi: 10.1128/JVI.06429-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegard AL, Chandran K, Zhang X, Parker JS, Baker TS, Nibert ML. Putative autocleavage of outer capsid protein micro1, allowing release of myristoylated peptide micro1N during particle uncoating, is critical for cell entry by reovirus. J Virol. 2004;78:8732–8745. doi: 10.1128/JVI.78.16.8732-8745.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshiro LS, Dondero DV, Emmons RW, Lennette EH. The development of Colorado tick fever virus within cells of the haemopoietic system. J Gen Virol. 1978;39:73–79. doi: 10.1099/0022-1317-39-1-73. [DOI] [PubMed] [Google Scholar]
- Ostermann G, Fraemohs L, Baltus T, Schober A, Lietz M, Zernecke A, Liehn EA, Weber C. Involvement of JAM-A in mononuclear cell recruitment on inflamed or atherosclerotic endothelium: inhibition by soluble JAM-A. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25:729–735. doi: 10.1161/01.ATV.0000157154.14474.3b. [DOI] [PubMed] [Google Scholar]
- Pacitti A, Gentsch JR. Inhibition of reovirus type 3 binding to host cells by sialylated glycoproteins is mediated through the viral attachment protein. J. Virol. 1987;61:1407–1415. doi: 10.1128/jvi.61.5.1407-1415.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallansch MA, Roos RP. Enteroviruses: Polioviruses, Coxsacieviruses, Echoviruses. In: Knipe DM, Howley PM, editors. Fields Virology. Lippincott-Raven Press; Philadelphia: 2001. pp. 723–775. [Google Scholar]
- Parashar K, Tarlow MJ, McCrae MA. Experimental reovirus type 3-induced murine biliary tract disease. J. Pediatr. Surg. 1992;27:843–847. doi: 10.1016/0022-3468(92)90380-p. [DOI] [PubMed] [Google Scholar]
- Parashar UD, Bresee JS, Gentsch JR, Glass RI. Rotavirus. Emerg Infect Dis. 1998;4:561–570. doi: 10.3201/eid0404.980406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul RW, Choi AH, Lee PWK. The α-anomeric form of sialic acid is the minimal receptor determinant recognized by reovirus. Virology. 1989;172:382–385. doi: 10.1016/0042-6822(89)90146-3. [DOI] [PubMed] [Google Scholar]
- Paul RW, Lee PWK. Glycophorin is the reovirus receptor on human erythrocytes. Virology. 1987;159:94–101. doi: 10.1016/0042-6822(87)90351-5. [DOI] [PubMed] [Google Scholar]
- Poggioli GJ, DeBiasi RL, Bickel R, Jotte R, Spalding A, Johnson GL, Tyler KL. Reovirus-induced alterations in gene expression related to cell cycle regulation. J. Virol. 2002;76:2585–2594. doi: 10.1128/JVI.76.6.2585-2594.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poggioli GJ, Dermody TS, Tyler KL. Reovirus-induced σ1s-dependent G2/M cell cycle arrest results from inhibition of p34cdc2. J. Virol. 2001;75:7429–7434. doi: 10.1128/JVI.75.16.7429-7434.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poggioli GJ, Keefer CJ, Connolly JL, Dermody TS, Tyler KL. Reovirus-induced G2/M cell cycle arrest requires σ1s and occurs in the absence of apoptosis. J. Virol. 2000;74:9562–9570. doi: 10.1128/jvi.74.20.9562-9570.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prota AE, Campbell JA, Schelling P, Forrest JC, Peters TR, Watson MJ, Aurrand-Lions M, Imhof B, Dermody TS, Stehle T. Crystal structure of human junctional adhesion molecule 1: implications for reovirus binding. Proc. Natl. Acad. Sci. U. S. A. 2003;100:5366–5371. doi: 10.1073/pnas.0937718100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Alvarez M, Sabin AB. Intestinal flora of healthy children demonstrable by monkey kidney tissue culture. Amer. J. Publ. Hlth. 1956;46:295–299. doi: 10.2105/ajph.46.3.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Alvarez M, Sabin AB. Characteristics of poliomyelitis and other enteric viruses recovered in tissue culture from healthy American children. Proc. Soc. Exp. Biol. Med. 1954;87:655–661. doi: 10.3181/00379727-87-21474. [DOI] [PubMed] [Google Scholar]
- Ramos-Alvarez M, Sabin AB. Enteropathogenic viruses and bacteria. Role in summer diarrheal diseases of infancy and early childhood. JAMA. 1958;167:147–158. doi: 10.1001/jama.1958.02990190001001. [DOI] [PubMed] [Google Scholar]
- Reiter DM, Frierson JM, Halvorson EE, Kobayashi T, Dermody TS, Stehle T. Crystal structure of reovirus attachment protein σ1 in complex with sialylated oligosaccharides. PLoS Path. 2011;7:e1002166. doi: 10.1371/journal.ppat.1002166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson SC, Bishop RF, Smith AL. Reovirus serotype 3 infection in infants with extrahepatic biliary atresia or neonatal hepatitis. Journal of Gastroenterology & Hepatology. 1994;9:264–268. doi: 10.1111/j.1440-1746.1994.tb01721.x. [DOI] [PubMed] [Google Scholar]
- Rodgers SE, Connolly JL, Chappell JD, Dermody TS. Reovirus growth in cell culture does not require the full complement of viral proteins: identification of a σ1s-null mutant. J. Virol. 1998;72:8597–8604. doi: 10.1128/jvi.72.11.8597-8604.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin DH. Reovirus serotype 1 binds to the basolateral membrane of intestinal epithelial cells. Microb. Pathog. 1987;3:215–220. doi: 10.1016/0882-4010(87)90098-2. [DOI] [PubMed] [Google Scholar]
- Rubin DH, Eaton MA, Anderson AO. Reovirus infection in adult mice: the virus hemagglutinin determines the site of intestinal disease. Microb. Pathog. 1986;1:79–87. doi: 10.1016/0882-4010(86)90034-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin DH, Kornstein MJ, Anderson AO. Reovirus serotype 1 intestinal infection: a novel replicative cycle with ileal disease. J. Virol. 1985;53:391–398. doi: 10.1128/jvi.53.2.391-398.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabin AB. Pathogenesis of poliomyelitis; reappraisal in the light of new data. Science. 1956;123:1151–1157. doi: 10.1126/science.123.3209.1151. [DOI] [PubMed] [Google Scholar]
- Sabin AB. Reoviruses: a new group of respiratory and enteric viruses formerly classified as ECHO type 10 is described. Science. 1959;130:1387–1389. doi: 10.1126/science.130.3386.1387. [DOI] [PubMed] [Google Scholar]
- Sacher T, Podlech J, Mohr CA, Jordan S, Ruzsics Z, Reddehase MJ, Koszinowski UH. The major virus-producing cell type during murine cytomegalovirus infection, the hepatocyte, is not the source of virus dissemination in the host. Cell Host Microbe. 2008;3:263–272. doi: 10.1016/j.chom.2008.02.014. [DOI] [PubMed] [Google Scholar]
- Sarkar G, Pelletier J, Bassel-Duby R, Jayasuriya A, Fields BN, Sonenberg N. Identification of a new polypeptide coded by reovirus gene S1. J. Virol. 1985;54:720–725. doi: 10.1128/jvi.54.3.720-725.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjostrand J, Karlsson JO. Axoplasmic transport in the optic nerve and tract of the rabbit: a biochemical and radioautographic study. J. Neurochem. 1969;16:833–844. doi: 10.1111/j.1471-4159.1969.tb08971.x. [DOI] [PubMed] [Google Scholar]
- Sobocka MB, Sobocki T, Babinska A, Hartwig J, Li M, Ehrlich YH, Kornecki E. Signaling pathways of the F11 receptor (F11R; a.k.a. JAM-1, JAM-A) in human platelets: F11R dimerization, phosphorylation and complex formation with the integrin GPIIIa. J. Recept. Signal Transduct. Res. 2004;24:85–105. doi: 10.1081/rrs-120034252. [DOI] [PubMed] [Google Scholar]
- Sobocka MB, Sobocki T, Banerjee P, Weiss C, Rushbrook JI, Norin AJ, Hartwig J, Salifu MO, Markell MS, Babinska A, Ehrlich YH, Kornecki E. Cloning of the human platelet F11 receptor: a cell adhesion molecule member of the immunoglobulin superfamily involved in platelet aggregation. Blood. 2000;95:2600–2609. [PubMed] [Google Scholar]
- Tardieu M, Powers ML, Weiner HL. Age-dependent susceptibility to reovirus type 3 encephalitis: role of viral and host factors. Ann. Neurol. 1983;13:602–607. doi: 10.1002/ana.410130604. [DOI] [PubMed] [Google Scholar]
- Tardieu M, Weiner HL. Viral receptors on isolated murine and human ependymal cells. Science. 1982;215:419–421. doi: 10.1126/science.6276976. [DOI] [PubMed] [Google Scholar]
- Tyler KL, McPhee DA, Fields BN. Distinct pathways of viral spread in the host determined by reovirus S1 gene segment. Science. 1986;233:770–774. doi: 10.1126/science.3016895. [DOI] [PubMed] [Google Scholar]
- Tyler KL, Sokol RJ, Oberhaus SM, Le M, Karrer FM, Narkewicz MR, Tyson RW, Murphy JR, Low R, Brown WR. Detection of reovirus RNA in hepatobiliary tissues from patients with extrahepatic biliary atresia and choledochal cysts. Hepatology. 1998;27:1475–1482. doi: 10.1002/hep.510270603. [DOI] [PubMed] [Google Scholar]
- van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat. Rev. Immunol. 2010;10:664–674. doi: 10.1038/nri2832. [DOI] [PubMed] [Google Scholar]
- Vazquez-Torres A, Jones-Carson J, Baumler AJ, Falkow S, Valdivia R, Brown W, Le M, Berggren R, Parks WT, Fang FC. Extraintestinal dissemination of Salmonella by CD18-expressing phagocytes. Nature. 1999;401:804–808. doi: 10.1038/44593. [DOI] [PubMed] [Google Scholar]
- Veronesi E, Darpel KE, Hamblin C, Carpenter S, Takamatsu HH, Anthony SJ, Elliott H, Mertens PP, Mellor PS. Viraemia and clinical disease in Dorset Poll sheep following vaccination with live attenuated bluetongue virus vaccines serotypes 16 and 4. Vaccine. 2009;28:1397–1403. doi: 10.1016/j.vaccine.2009.10.107. [DOI] [PubMed] [Google Scholar]
- Weiner HL, Ault KA, Fields BN. Interaction of reovirus with cell surface receptors. I. Murine and human lymphocytes have a receptor for the hemagglutinin of reovirus type 3. J Immunol. 1980;124:2143–2148. [PubMed] [Google Scholar]
- Weiner HL, Drayna D, Averill DR, Jr., Fields BN. Molecular basis of reovirus virulence: role of the S1 gene. Proc Natl Acad Sci U S A. 1977;74:5744–5748. doi: 10.1073/pnas.74.12.5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner HL, Powers ML, Fields BN. Absolute linkage of virulence and central nervous system cell tropism of reoviruses to viral hemagglutinin. J Infect Dis. 1980;141:609–616. doi: 10.1093/infdis/141.5.609. [DOI] [PubMed] [Google Scholar]
- Williams LA, Martin-Padura I, Dejana E, Hogg N, Simmons DL. Identification and characterisation of human junctional adhesion molecule (JAM) Mol. Immunol. 1999;36:1175–1188. doi: 10.1016/s0161-5890(99)00122-4. [DOI] [PubMed] [Google Scholar]
- Wolf JL, Dambrauskas R, Sharpe AH, Trier JS. Adherence to and penetration of the intestinal epithelium by reovirus type 1 in neonatal mice. Gastroenterology. 1987;92:82–91. doi: 10.1016/0016-5085(87)90842-0. [DOI] [PubMed] [Google Scholar]
- Wolf JL, Kauffman RS, Finberg R, Dambrauskas R, Fields BN, Trier JS. Determinants of reovirus interaction with the intestinal M cells and absorptive cells of murine intestine. Gastroenterology. 1983;85:291–300. [PubMed] [Google Scholar]
- Wolf JL, Rubin DH, Finberg R, Kaufman RS, Sharpe AH, Trier JS, Fields BN. Intestinal M cells: a pathway of entry of reovirus into the host. Science. 1981;212:471–472. doi: 10.1126/science.6259737. [DOI] [PubMed] [Google Scholar]
- Woodfin A, Reichel CA, Khandoga A, Corada M, Voisin MB, Scheiermann C, Haskard DO, Dejana E, Krombach F, Nourshargh S. JAM-A mediates neutrophil transmigration in a stimulus-specific manner in vivo: evidence for sequential roles for JAM-A and PECAM-1 in neutrophil transmigration. Blood. 2007 doi: 10.1182/blood-2006-09-047431. [DOI] [PubMed] [Google Scholar]