

Abstract
Background
The genus Brachyspira currently encompasses seven valid species that colonize the intestines of mammals and birds. In a previous study a group of strongly haemolytic isolates from pigs and mallards was provisionally described as a new species within genus Brachyspira, “B. suanatina”, and enteropathogenic properties were demonstrated in a porcine challenge model.
Methods
In the current study characterization of B. suanatina was performed on the basis of cell morphology, growth characteristics, enzyme profiles, DNA-DNA hybridization (DDH) and whole genome comparisons. The draft genome sequence of B. suanatina strain AN4859/03 was determined and compared with the available genomes of all valid species of Brachyspira.
Results
According to morphological traits, growth characteristics and enzymatic profiles, B. suanatina was similar to the type strain of B. hyodysenteriae, but using the recommended threshold value of 70 % similarity by DDH it did not belong to any of the recognized Brachyspira species (range 16–64 % similarity). This was further supported by average nucleotide identity values. Phylogenetic analysis performed using housekeeping genes and core genomes of all valid Brachyspira sp. and “B. hampsonii” revealed that B. suanatina and B. intermedia formed a clade distinct from B. hyodysenteriae. By comparing the genomes of the three closely related species B. intermedia, B. hyodysenteriae and B. suanatina similar profiles of general genomic features and distribution of genes in different functional categories were obtained. However, the genome size of B. hyodysenteriae was smallest among the species, suggesting the possibility of reductive evolution in the divergence of this species. A bacteriophage region and a putative plasmid sequence were also found in the genome of B. suanatina strain AN4859/03.
Conclusions
The results of our study suggest that despite being similar to B. hyodysenteriae phenotypically, B. suanatina should be regarded as a separate species based on its genetic characteristics. Based on characteristics presented in this report we propose that strains AN4859/03, AN1681:1/04, AN2384/04 and Dk12570-2 from pigs in Sweden and Denmark, and strains AN3949:2/02 and AN1418:2/01 isolated from mallards in Sweden, represent a unique species within genus Brachyspira. For this new species we propose the name B. suanatina for which the type strain is AN4859/03T (=ATCC® BAA-2592™ = DSM 100974T).
Electronic supplementary material
The online version of this article (doi:10.1186/s12866-015-0537-y) contains supplementary material, which is available to authorized users.
Keywords: Brachyspira suanatina sp. nov., Core genome, DNA-DNA hybridization, Enteropathogen, Housekeeping genes phylogeny, Spirochaete, Mallard, Phenotype, Pig, Whole genome sequence
Background
The advent of next generation sequencing technologies with their low cost, high throughput and rapid speed has strongly influenced modern taxonomy. Many taxonomists therefore turn towards integrating genome sequence analysis for formal proposals of new bacterial taxa or reclassification of old taxa [1–3]. Early classification methods were based solely on phenotypic characteristics, but the introduction of DNA-DNA hybridization (DDH) [4, 5] and 16S ribosomal RNA (rRNA) gene phylogeny [6] in taxonomy changed the species definition [7–9]. However, due to the high level of conservation of the 16S rRNA gene [10] and the labour intensive and error prone nature of DDH experiments, there has been a constant demand for advanced methods at complete genome level. Average nucleotide identity (ANI) [11–13], multi locus sequence analysis of conserved genes [8, 14] and phylogenetic analysis of core genomes [15] are some of the suggested methods.
Brachyspira species are gGram-negative, anaerobic but oxygen-tolerant spirochaetes that colonize the intestines of many mammals and birds [16]. It forms a clade within phylum Spirochaetes that has undergone repeated taxonomic changes over the past few decades. Initially the isolates colonizing pigs were allocated to genus Treponema [17, 18], however they were later transferred to genus Serpula and then Serpulina [19, 20]. In 1997, the genera Serpulina and Brachyspira were unified leading to the reclassification of Serpulina species [21]. Genus Brachyspira now encompasses seven valid species: B. aalborgi, B. alvinipulli, B. hyodysenteriae, B. intermedia, B. innocens, B. murdochii and B. pilosicoli with B. aalborgi as a type species [22]. These species vary in terms of their host range and enteropathogenicity. Brachyspira hyodysenteriae and B. pilosicoli are well known porcine pathogens [16]. Furthermore, B. pilosicoli can cause gastrointestinal disease and egg-production losses in chickens [23], and it can also colonize the human intestine leading to human intestinal spirochaetosis [24]. However, most cases in humans are associated with B. aalborgi, at least in industrialized countries [25]. Moreover, B. alvinipulli and B. intermedia cause diarrhoea in chickens [23, 26]. However, B. intermedia, is in general considered as a harmless commensal in pigs similar to B. innocens and B. murdochii [16]. There are several proposed species without current standing in nomenclature, including “B. suanatina” [27].
Some members of the genus Brachyspira have low interspecies 16S rRNA gene variability that does not always support species delineations, especially among the indole test-positive species B. hyodysenteriae, B, intermedia and B. suanatina that require additional methods for species confirmation [27, 28]. The proposed new species B. suanatina was first isolated from pigs with dysentery-like disease in Sweden and Denmark, and the enteropathogenicity of strain AN4859/03 was confirmed by experimental challenge in pigs [27]. The phenotypic characterization based on the intensity of β-haemolysis and enzyme activities showed that these isolates were indistinguishable from B. hyodysenteriae, but detection by a species-specific PCR was unsuccessful [27]. Based on 16S rRNA and NADH oxidase (nox) gene similarities, these Brachyspira isolates from pigs were similar to some isolates from mallards [29] and were distinct from all currently recognized Brachyspira species, suggesting that they were members of a putatively unrecognized species. Available data suggest that B. suanatina most likely is a rare genotype in both pigs and mallards, and it has so far only been isolated in northern Europe. Identification of B. suanatina currently relies on nox gene sequencing and/or nox-RFLP [30]. The objective of the present study was to identify the taxonomical position of B. suanatina based on phenotypic characteristics and complete genome analyses. We have used a “taxono-genomics” approach that is based on the polyphasic combination of phenotypic and genomic characteristics [31].
Methods
Bacterial cultures and growth conditions
Type and reference strains B. hyodysenteriae B78T (ATCC 27164), B204R (ATCC 31212), B. intermedia PWS/AT (ATCC 51140), B. pilosicoli P43/6/78T (ATCC 51139), B. innocens B256T (ATCC 29796), B. murdochii 56-150T (ATCC 51284), B. alvinipulli C1T (ATCC 51933) and B. aalborgi 513AT (ATCC 43994) were originally obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and stored in liquid nitrogen in the Brachyspira strain collection of the National Veterinary Institute (SVA). Brachyspira suanatina strains AN4859/03, AN1681:1/04, AN2384/04 and Dk12570-2 from pigs, and AN3949:2/02 and AN1418:2/01 from mallards were isolated in Sweden and Denmark as previously described [27] and stored in liquid nitrogen. The bacteria were retrieved from storage and were grown anaerobically (atmosphere generated by Anaerogen sachets, Oxoid) on fastidious anaerobe agar (FAA) (LabM) plates with 10 % equine blood at 42 °C for 72 h and subcultured once for 48 h before use. Cells for electron microscopy were harvested from broth cultures grown anaerobically on a shaker at 37 °C to mid log phase in brain heart infusion culture broth (BHI) (Difco) with 10 % foetal calf serum (FCS) (Biochrome AG) (BHIS). FAA plate cultures and broth culture were used to test the capability of all B. suanatina strains to grow at 32, 37, 42 and 46 °C. For the generation of growth curves, broth cultures of B. suanatina strains AN4859/03 and AN3949:2/02 and B. hyodysenteriae B78T were prepared in triplicates in 10 mL BHIS at 106 cells/mL and grown at 37 °C. Population densities were determined every 4 h between 11 and 70 h of incubation by colony forming unit (CFU) counts obtained from serial dilutions on FAA plates in duplicates and by spectrophotometry (OD620). Comparisons were made against an in-house standard curve for B. hyodysenteriae B78T. Purity was confirmed throughout the studies by phase contrast microscopy.
Biochemical phenotype and substrate utilization
The API-ZYM test (bioMérieux) was applied to all B. suanatina strains with B. hyodysenteriae B78T and Pseudomonas aeruginosa (ATCC 27853) as control strains. Bacterial cells were harvested from FAA plates incubated at 42 °C for 3 days, suspended in 2 mL sterile ddH2O to a density of 5–6 McFarland and the API-ZYM test was performed as described by the manufacturer. The test strips were incubated for 4 h in aerobic atmosphere.
Brachyspira suanatina strains AN4859/03, AN3949:2/02 and B. hyodysenteriae strain B78T were used for growth substrate utilization studies. All six B. suanatina strains were tested for growth in D-mannitol because of diverging results obtained with strains AN4859/03 and AN3949:2/02. Potential substrates were added at 0.4 wt/v in heart infusion culture broth (HI) (Bacto™) and autoclaved or sterile filtered. The FCS concentration in the HI was selected at 7 % (v/v) from test runs at 5, 7 and 10 % FCS as described previously [26, 32]. Triplicate flasks with 10 mL HI and 7 % FCS culture broth (HIS) were used for each isolate and substrate, and were inoculated with 106 cells from a stock solution made from 48 h old FAA cultures. Cultures were incubated anaerobically at 37 °C on a shaking platform. Glucose was used as positive control and HIS without added substrate as negative control. Growth was determined spectrophotometrically (OD620) at 3, 6 and 9 days post inoculation for all cultures, and the purity, cell density and the condition of the cells were examined by phase contrast microscopy. Compounds producing ≤0.3 OD620 were not considered to be growth substrates. These cultures contained degenerated and/or non-motile cells as determined by phase contrast microscopy. Cultures producing ≥0.4 OD620 values and moderate to profuse growth of motile spirochaetes on at least one occasion were considered to be growth substrates. The following compounds were investigated: D-glucose, D-fructose, D-fucose, D-ribose, D-mannose, sucrose, lactate, D-xylose, D-trehalose, D-galactose, D-melobiose, D-cellobiose, L-arabinose, L-fucose, L-rhamnose, D-raffinose, D-galactosamine, N-acetyl-D-glucosamine, N-acetyl-D-galactosamine, mannitol, pyruvate, esculin, porcine gastric mucin, 50x MEM amino acids, pectin, hyaluronic acid, glutathione, poly-galacturonic acid, cellulose, glycerol, succinate and glycogen.
Electron microscopy
Strain AN4859/03 was grown in BHIS to mid-log phase, centrifuged at 14,000 × g for 3 min and washed twice in 1 mL 0.01 M sodium phosphate buffer at pH 7.0. The pellet was resuspended in 0.1 mL each of sterile ddH2O and negatively stained with 0.1 mL 2 % phosphotungistic acid at pH 7.0. The cells were dropped on grids and examined. For scanning electron microscopy the cells were harvested and washed as described above, dropped on grids and coated with gold-palladium with standard procedures. The samples were examined with a JSM 820 scanning electron microscope (JEOL Skandinaviska AB, Sweden). Ten cells or more were examined to determine cell dimensions, the location of attachment sites of periplasmatic flagella and the number of flagella per cell.
DNA-DNA hybridization
The DDH of B. suanatina strain AN4859/03 was performed with all the seven recognized Brachyspira species. Cells were grown to mid-log phase in BHIS, checked for purity and centrifuged at 4 °C at 14,000 g followed by washing in PBS twice before mixing with 1:1 v/v iso-propanol (Sigma). DNA isolation and DDH were performed by DSMZ GmbH Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH DNA (DSMZ GmbH, Braunschweig, Germany). DNA was extracted using a French pressure cell (Thermo Spectronic) and was purified by chromatography on hydroxyapatite as described [33]. DNA-DNA hybridization was performed as previously described [34] using a model Cary 100 Bio UV/VIS-spectrophotometer equipped with a Peltier-thermostatted 6x6 multicell changer and a temperature controller with in situ temperature probe (Varian).
DNA isolation and complete genome sequencing
Genomic DNA for sequencing of strain AN4859/03 was obtained from 60 mL mid-log BHIS cultures using the DNeasy Blood and Tissue Kit (Qiagen) as described by the manufacturer. The genome sequence was obtained using a combination of Ion torrent and Illumina MiSeq sequencing techniques performed at Uppsala Genome Centre (http://www.igp.uu.se/facilities/genome_center/). Single end reads were generated with the read size 300 bp on the Ion Torrent platform and paired end reads were generated using the Illumina MisSeq platform producing reads of 300 bp. Genomic sequences of available strains of currently valid Brachyspira species and two strains of the proposed species “B. hampsonii” were obtained from GenBank (https://www.ncbi.nlm.nih.gov/). Strain names and accession numbers are provided in Additional file 1: Table S1. Draft genome scaffolds for B. aalborgi were downloaded from the MetaHit website (http://www.sanger.ac.uk/resources/downloads/bacteria/metahit/).
Genome assembly and average nucleotide identity
Single end reads generated by the Ion torrent platform were assembled using the Roche GS assembler. The resulting contigs were oriented using the B. hyodysenteriae WA1 genome as a reference and joining was performed manually. Reads obtained from Illumina MiSeq paired end sequencing were assembled using Mira version 4 [35]. Before performing the assembly, the reads were trimmed and filtered based on quality, using PRINSEQ [36] and further reduction in the coverage was performed using a custom perl script. Scaffolds were obtained using SSPACE [37]. The scaffolds were aligned to Ion torrent assembly using MAUVE [38] to find possible overlaps. Overlapping scaffolds were manually joined in Consed [39]. A putative plasmid sequence was predicted by blasting the B. hyodysenteriae WA1 plasmid’s (pBHWA1) sequence [40] against the B. suanatina contigs.
The average nucleotide identity (ANI) between B. suanatina and type strains of all the seven valid Brachyspira species was calculated using Jspecies v1.2.1 [13] using BLAST (ANIb) and MUMMER (ANIm) algorithms with default parameters.
Genome annotation and core genome analysis
B. suanatina scaffolds were annotated using the GenDB platform [41]. Predictions made by the GenDB pipeline were also manually reviewed and curated. Prophages were predicted using PHAST [42] and by manual curation in the annotated genome. Clusters of orthologous group (COG) categories were assigned to genes by performing blast search against the COG database (http://www.ncbi.nlm.nih.gov/COG) using the threshold of > =30 % amino acid identity and e-value < = 0.00005. In order to obtain annotations based on the latest versions of search databases, all the genomes used in the analysis were also annotated with GenDB. Comparative analysis of the B. suanatina genome with other Brachyspira genomes including pan and core genome calculations was performed using the EDGAR software framework for prokaryotic genomes [43]. Whole genome alignment of B. suanatina with the complete genomes of B. hyodysenteriae WA1 and B. intermedia PWS/AT was performed using the Artemis comparison tool (ACT). Phylogenetic analysis based on core genomes of all Brachyspira species including B. suanatina was also performed as a part of the EDGAR comparative analysis. For phylogenetic analysis, multiple sequence alignments of amino acid sequences of the core genome were obtained in MUSCLE [44], unaligned blocks were removed using GBLOCKS [45] and a phylogenetic tree of concatenated alignment was generated in PHYLIP using the neighbour joining method. Bootstrap analysis was performed on the tree using 200 (bootstrap) replicates.
Phylogenetic analysis of housekeeping genes
For phylogenetic analysis of housekeeping genes, 31 protein coding genes that are universally conserved among bacteria [46] were selected (dnaG, frr, infC, nusA, pgk, pyrG, rplA, rplB, rplC, rplD, rplE, rplF, rplK, rplL, rplM, rplN, rplP, rplS, rplT, rpmA, rpoB, rpsB, rpsC, rpsE, rpsI, rpsJ, rpsK, rpsM, rpsS, smpB, and tsf). Nucleotide sequences of these genes for B. hyodysenteriae B78T, B. hyodysenteriae WA1, B. intermedia PWS/AT, B. murdochii 56-150T, B. pilosicoli 95/1000, B. pilosicoli B2904, B. pilosicoli WesB, B. pilosicoli P43/6/78T, Treponema denticola aT and Borrelia Burgdorferi B31T were obtained from the NCBI Gene database (http://www.ncbi.nlm.nih.gov/gene). Nucleotide sequences for B. aalborgi 513AT, B. alvinipulli C1T, B. innocens B256T, “B. hampsonii” 30446, “B. hampsonii” 30599 and B. suanatina AN4859/03 were predicted by performing homology searches against their gene sequences with B. hyodysenteriae strain WA1’s genes as reference. Sequences were aligned using MUSCLE [44] and conserved blocks were obtained using Gblocks [45]. Genes with < 50 % alignment conservation were removed leaving 26 genes. Phylogenetic inference using the concatenated alignment of those 26 genes was drawn using the maximum likelihood method (ML) [47] with 1000 bootstrap replicates. The general time reversible (GTR) [48] model with discrete gamma distribution allowing some proportion of invariable sites (+G + I) as predicted by JModelTest [49] was used to model the evolutionary rates among sites. Phylogenetic analysis was performed in MEGA6, an evolutionary analysis tool [50].
Results
Morphology and growth characteristics
Cells of strain AN4859/03 were ultrastructurally similar in appearance but slightly shorter and thinner and possessed fewer flagella than those of B. hyodysenteriae B78T and B. intermedia PWS/AT (Table 1). The cells were shaped as spirochaetes with tapered ends and periplasmic flagella that were inserted on a line subterminally at both cell ends.
Table 1.
Morphologic comparison of indole-positive species of genus Brachyspira a, b
| Species | Strains used | Cell length (μm) | Cell diameter (μm) | No. of flagella/cell |
|---|---|---|---|---|
| B. hyodysenteriae | B78T | 9.78 ± 1.87 | 0.35 ± 0.02 | 16 – 24 |
| 8 – 10 | 0.3 – 0.4 | 14 – 18 | ||
| 6 – 8.5 | 0.35 – 0.4 | 24 – 28 | ||
| B. intermedia | PWS/AT | 7.5 – 10 | 0.35 – 0.45 | 24 – 28 |
| B. suanatina | AN4859/03 | 5.5 ± 1.0 | 0.3 ± 0.0 | 14 – 16 |
Strains AN4859/03 and AN3949:2/02 that were cultured anaerobically in BHIS broth at 37 °C entered the log-phase at approximately 31 h of incubation and reached maximum population densities at 36–38 h post inoculation. Maximum CFU/mL was 2 × 108 cells/mL at 36 h. At mid log-phase, the population doubling time was approximately 2 h. Both strains grew equally well at 37 and 42 °C, but growth was inhibited at 32 and 46 °C.
Biochemical tests
The enzymatic profiles are shown in Table 2. All six B. suanatina isolates had a similar enzymatic profile to B. hyodysenteriae strain B78T, but reactions were generally weaker.
Table 2.
Enzymatic profile of B. hyodysenteriae type strain (B78T) and six strains of B. suanatina
| Strain | Brachyspira panel a | api-ZYM enzyme activitiesb | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Haem | IS | Hipp | α-gal | β-glu | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | |
| B78T | s | + | – | – | + | 0 | 4 | 3 | 1 | 0 | 0 | 0 | 0 | 1 | 2 | 4 | 1 | 1 | 5 | 2 | 4 | 4 | 0 | 0 | 0 |
| AN4859/03 | s | + | – | – | + | 0 | 2 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 1 | 0 | 5 | 0 | 2 | 4 | 0 | 0 | 0 |
| AN1681:1/04 | s | + | – | – | + | 0 | 2 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 1 | 0 | 4 | 0 | 2 | 3 | 0 | 0 | 0 |
| AN2384/04 | s | + | – | – | + | 0 | 2 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 0 | 5 | 0 | 2 | 4 | 0 | 0 | 0 |
| Dk12570-2 | s | + | – | – | + | 0 | 3 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 1 | 0 | 5 | 0 | 2 | 3 | 0 | 0 | 0 |
| AN3949:2/02 | s | + | – | – | + | 0 | 3 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 1 | 0 | 5 | 0 | 2 | 3 | 0 | 0 | 0 |
| AN1418:2/01 | s | + | – | – | + | 0 | 2 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 1 | 0 | 5 | 0 | 3 | 4 | 0 | 0 | 0 |
a Brachyspira panel according to Fellström et al., 1999. Results according to Råsbäck et al., 2007. Haem = intensity of β-haemolysis; IS = indole spot test; Hipp = hippurate cleavage capacity; α-galactosidase; β-glucosidase; s = strong reaction; + = positive reaction, − = negative reaction
bAPI-ZYM test: 1 control; 2 alkaline phosphatase; 3 C4 esterase; 4 C8 esterase lipase; 5 C14 lipase; 6 leucine arylamidase; 7 valine arylamidase; 8 cystine arylamidase; 9 trypsin; 10 chymotrypsin; 11 acid phosphatase; 12 naphtolphosphohydrolase; 13 α-galactosidase; 14 β-galactosidase; 15 β-glucoronidase; 16 α-glucosidase; 17 β-glucosidase; 18 N-acetyl-β-glucosaminidase; 19 α-mannosidase; 20 α-fucosidase; values 0–5 assigned according to the manufacturer’s instruction 0 = negative reaction, 5 = maximum intensity
Growth substrate utilization
The maximum growth (median OD620 values of three replicates) of strains B. hyodysenteriae B78T and B. suanatina strains AN4859/03 and AN3949:2/02 in HIS but without added growth substrate was 0.2–0.3. The following compounds were utilized by all three strains as growth substrates: D-glucose, D-fructose, D-galactose, D-mannose, sucrose, D-trehalose, lactose, N-acetyl-D-glucosamine, pyruvate (OD620 ≥ 0.4–1.0). Three out of six tested B. suanatina-strains (AN3949:2/02, AN14 18:2/01 and Dk12570) metabolized D-mannitol, but B. hyodysenteriae B78T did not. Substrates that were not metabolized by the three tested strains included: D-cellobiose, D-fucose, D-melobiose, D-ribose, D-raffinose, D-xylose, L-arabinose, L-fucose, L-rhamnose, N-acetyl-D-galactosamine, soluble starch, galactosamine, Na-acetate, lactate, esculin, succinate, pectin, 50x MEM amino acids, cellulose, glutathione, glycerol, glycogen, poly-galacturonic acid, hyaluronic acid and hog gastric mucin. The control strain B78T showed results consistent with an earlier study [32].
DNA-DNA hybridization
The percent DNA-DNA similarities of B. suanatina strain AN4859/03 to other Brachyspira species type strains are given in Table 3.
Table 3.
Percent DNA-DNA similarity of B. suanatina AN4859/03 to type strains (duplicated results) and percent ANIb/ANIm values
| Brachyspira sp. | Strain designation | ATCC no. | % DNA-DNA similaritya | ANIb/ANIm values |
|---|---|---|---|---|
| B. hyodysenteriae | B78T | 27164 | 62.9/64.0 | 92.12/92.61 |
| B. intermedia | PWS/AT | 51140 | 59.0/64.0 | 92.69/93.19 |
| B. pilosicoli | P43/6/78T | 51139 | 22.1/27.9 | 79.29/86.36 |
| B. innocens | B256T | 29796 | 19.9/16.2 | 82.78/86.08 |
| B. murdochii | 56-150T | 51284 | 35.7/31.0 | 82.78/85.98 |
| B. alvinipulli | C1T | 51933 | 49.9/58.9 | 83.81/87.01 |
| B. aalborgi | 513AT | 43994 | 34.1/31.0 | 75.28/82.17 |
ain 2 X SSC at 59 °C
Assembly statistics and Average nucleotide identity
The final assembly of B. suanatina AN4859/03 consisted of 34 scaffolds comprising 3,258,009 bp with a GC content of 27 %. Three large scaffolds of sizes 2,243,936 bp, 499,175 bp and 433,837 bp covered 98 % of the assembled genome. The remaining scaffolds ranged in size from 450 bp to 30,000 bp. One of the scaffolds contained a putative plasmid sequence of 30,236 bp sharing 88 % identity over 51 % of its length with the B. hyodysenteriae strain WA1 plasmid (pBHWA1) sequence, see below. The assembled genome of B. suanatina strain AN4859/03 was larger than that of the available genomes of B. hyodysenteriae strains B78T and WA1, but smaller than the genome of B. intermedia strain PWS/AT. The ANI of B. suanatina to type strains of other Brachyspira species was lower than 95 %, with the highest value obtained between B. suanatina and B. intermedia (Table 3).
General genomic features and putative plasmid
General genomic features of B. suanatina strain AN4859/03, B. hyodysenteriae (strains B78T and WA1) and B. intermedia (PWS/AT) are depicted in Table 4. The draft genome assembly of B. suanatina AN4859/03 consisted of 2,892 open reading frames (ORFs), including 30 ORFs (BRSU_2794–BRSU_2823) belonging to a putative plasmid sequence. By comparing the plasmid genes of B. suanatina with that of pBHWA1, we found 19 CDSs that were conserved in both strains. These include four genes encoding for epimerases, six genes encoding for transferases, and one gene each encoding for radical sterile alpha motif (SAM) domain containing protein, plasmid partition protein, putative replicative DNA helicase, DNA primase-like protein, integrase, lipopolysaccharide biosynthesis protein, dehydratase, reductase and a hypothetical protein. Genes that were present in B. suanatina but were not found in the plasmid sequence of B. hyodysenteriae were one glycosyl transferase, one peptide ABC transporter substrate-binding protein, one radical SAM domain-containing protein, five hypothetical proteins, one conserved hypothetical protein and two of unknown function.
Table 4.
Genome assembly statisticsa, b: Summary of general genomic features of B. suanatina strain AN4859/03 and its comparison with genomes of B. hyodysenteriae strains B78T and WA1 and B. intermedia strain PWS/AT
| Genomic feature | B. suanatina AN4859/03 | B. hyodysenteriae B78 T | B. hyodysenteriae WA1 | B. intermedia PWS/A T |
|---|---|---|---|---|
| Total Assembly size (bp) | 3,258,009 | 3,046,380 | 3,036,634 | 3,308,048 |
| Number of contigs | 35 | 46 | 2 | 2 |
| Chromosome | ||||
| Size (bp) | 3,227,373 | 3,046,380 | 3,000,694 | 3,304,788 |
| DNA GC % | 27 | 27 | 27.06 | 27.22 |
| Protein coding genes | 2,826 | 2,593 | 2,613 | 2,870 |
| Plasmid | ||||
| Size (bp) | 30.236 | - | 35,940 | 3,260 |
| DNA GC % | 22.14 | - | 21.82 | 21.00 |
| Protein coding genes | 30 | - | 31 | 3 |
| Number of predicted tRNA genes | 33 | 33 | 33 | 33 |
| Number of predicted rRNA genes | 3 | 3 | 3 | 3 |
| Number of prophages | 2 | 0 | 0 | 2 |
| Genes assigned to COG categories | 2,207 | 2,105 | 2,104 | 2,224 |
aData from references [40, 53] and from the present study (BB. suanatina, strain AN4859/03)
bBB. hyodysenteriae B78T data taken from ncbi WGS assembly GCF_000383255 with accession NZ_ARSY00000000
Prophages and mobile genetic elements
The B. suanatina genome contained a 15 kb region similar to the core region of the prophage-like gene transfer agent (GTA) named VSH-1 (Virus of Serpulina hyodysenteriae) of B. hyodysenteriae (BRSU_2672 – BRSU_2703). By comparing this region with the GTA region of B. hyodysenteriae strain WA1 (GTA/Bh-WA1) [51], we found that all the late function genes were present in the same order in both strains. Of the twelve ORFs of unknown function described in B. hyodysenteriae WA1’s GTA region, orfA, orfB, orfE, orfF, orfG, orf1, orf5 and orf6 were conserved in B. suanatina, and orfC, orf2, orf3 and orf4 were not present in the B. suanatina genome. Instead six other ORFs of unknown function were found in the corresponding region in B. suanatina. Two of them were also conserved in the VSH-1 regions of B. hyodysenteriae B78T (ARSY01_298 and ARSY01_299) and B. intermedia PWS/AT (Bint_0120 and Bint_0123). One of them was homologous to a gene encoding for sulfatase (Bint_1870) outside the VSH-1 region in B. intermedia PWS/AT and three of them were unique to the B. suanatina genome. Besides these six ORFs, three additional ORFs were present between hvp45 and hvp19. Two of them were also present in the VSH-1 region of B. intermedia encoding for hypothetical proteins and one of them was unique to B. suanatina. Another additional ORF was present between hvp101 and lys gene in the B. suanatina genome and was homologous to hvp28 gene in VSH-1 region of B. intermedia PWS/AT.
A putative bacteriophage region, BSP was predicted in the B. suanatina genome, which has not been described in genomes of B. hyodysenteriae strain WA1 or B. intermedia strain PWS/AT. Analysis revealed that BSP was present approximately 33 kb upstream to the VSH-1 region. The BSP region spanned over a 31.2 kb sequence length and consisted of 31 CDSs (BRSU_2592–BRSU_2638) including one VSH-1 related gene. The BSP region was not present in the complete genome of B. hyodysenteriae strain WA1 genome, but a homologous bacteriophage region was found in scaffold number 2 of the B. hyodysenteriae B78T assembly (NZ_ARSY00000000) and consisted of 29 CDSs (Fig. 1). A bacteriophage region almost identical to BSP was found in the B. pilosicoli P43/6/78T genome (CP002873) spanning over 39 kb sequence and it contained 50 CDSs (BPP43_03795 to BPP43_04045) including one VSH-1 related gene. The organization of genes in BSP and its comparison with identical regions in B. hyodysenteriae B78T and B. pilosicoli P43/6/78T is shown in Fig. 1. Further analysis revealed that a significant portion of the BSP region was conserved and had been previously reported as pP1, pP2 and pP3, in B. pilosicoli 95/1000, B. pilosicoli strains B2904 and B. pilosicoli WesB respectively [52], and pM1, pM2 and pM3 in B. murdochii strain 56-150T [53]. Some of the BSP genes were also found in genomes of B. innocens B256T and B. alvinipulli C1T scattered in the genome either as single genes or in small clusters of phage related genes. These include ARQI01_47, ARQI01_48, ARQI01_86, ARQI01_102, ARQI01_109, ARQI01_110, ARQI01_347-ARQI01_356, ARQI01_462-ARQI01_466, ARQI01_469-ARQI01_472, ARQI01_477, ARQI01_478 in the B. innocens genome (accession: NZ_ARQI00000000) and JADF01_1734, JADF01_1737, JADF01_1738, JADF01_1749, JADF01_2178, JADF01_2181, JADF01_2185, JADF01_2187, JADF01_2568, JADF01_2575, JADF01_ 2578, JADF01_2975, JADF01_2976, JADF01_2980 – JADF01_2989, JADF01_2992, JADF01_3246, JADF01_3247, JADF01_3249, JADF01_3252, JADF01_3255, JADF01_3257, JADF01_3258 in the B. alvinipulli genome (accession: NZ_JADF00000000). Only two genes of BSP shared homology with B. intermedia strain PWS/AT genes. These were: Bint_1528 encoding hypothetical protein of B. intermedia phage and Bint_1547 encoding putative integrase in B. intermedia. A part from these bacteriophage regions, some phage related genes and mobile genetic elements were also found in the B. suanatina genome including putative integrases, terminases, tail protein, portal protein that were not found in B. hyodysenteriae genome except for the two reported phage related genes BHWA_01969 and BHWA_02688. Bacteriophage regions of B. intermedia PWS/AT [53] were not found in the B. suanatina genome except one gene (Bint_0072) that encodes for a hypothetical membrane spanning protein, and is also conserved in B. hyodysenteriae strains WA1 and B78T.
Fig. 1.
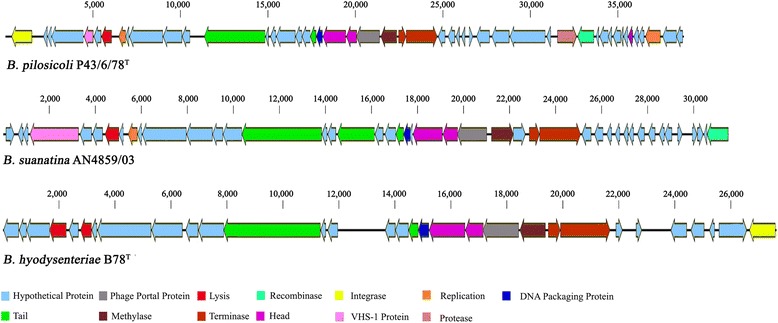
Bacteriophage region comparison: Comparison of the organization of the bacteriophage region BSP in B. suanatina AN4859/03 with the homologous bacteriophage regions in B. pilosicoli P43/6/78T and B. hyodysenteriae B78T genomes
Gene content and synteny conservation
The pan genome of B. suanatina AN4859/03, B. hyodysenteriae B78T, B. hyodysenteriae WA1 and B. intermedia PWS/AT consisted of 3,682 genes. Among them 2,219 genes were present in all four strains, 2,351 genes were shared between B. hyodysenteriae WA1 and B. suanatina, 2,386 genes were shared between B. hyodysenteriae B78T and B. suanatina, and 2,366 genes were shared between B. intermedia and B. suanatina (Fig. 2a). The remaining genes had their highest similarities with other Brachyspira species. There were 83 genes in B. suanatina that were not present in any of the other Brachyspira species, most of them consisted of hypothetical proteins and some had matches in Clostridium spp. and Bacillus spp.
Fig. 2.
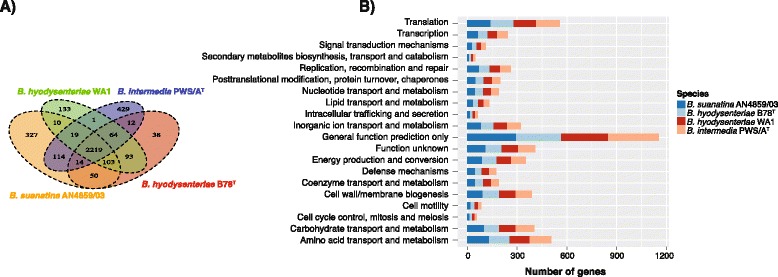
Global gene conservation and comparison of functional classification of genes: The figure shows the number of genes and their classification in different COG categories from B. suanatina AN4859/03, B. hyodysenteriae B78T, B. hyodysenteriae WA1 and B. intermedia PWS/AT (a) Venn diagram (b) The bar chart shows the distribution of genes in different functional categories. Genes were assigned to different categories using blastP against the COG database
By comparing the functional distribution of genes in different COG categories, the four strains B. suanatina AN4859/03, B. hyodysenteriae B78T, B. hyodysenteriae WA1 and B. intermedia PWS/AT contained similar numbers of genes for particular functions (Fig. 2b) (Additional file 2: Table S2).
In contrast to gene content, gene synteny was highly conserved between B. hyodysenteriae and B. suanatina with only two major genomic rearrangement events that were found in B. hyodysenteriae WA1 and B. suanatina. Some insertions were also found in the B. suanatina genome, when compared with B. hyodysenteriae WA1, comprising mainly of genes encoding for unknown proteins, bacteriophages and mobile genetic elements. In B. intermedia and B. suanatina, there was a low level of conservation of synteny as compared to the gene content. The alignment of genomic sequences for two isolates of B. suanatina and B. intermedia PWS/AT using ACT identified extensive genomic rearrangements, including at least eight major inversions (Additional file 3: Figure S1).
Phylogenetic inference based on the core genome and housekeeping genes
In order to infer the evolutionary relationship of B. suanatina with other Brachyspira species, phylogenetic analysis was performed on the amino acid sequences of core genomes and nucleotide sequences of housekeeping genes. In total 1,309 genes were found conserved in all Brachyspira species and their phylogenetic tree exhibited a close relationship between B. suanatina and B. intermedia whereas B. hyodysenteriae WA1 and B. hyodysenteriae B78T formed a distinct clade. The phylogenetic tree is shown in Fig. 3. Phylogenetic analysis of 26 housekeeping genes also showed a highly similar tree topology to the core genome analysis and confirmed a clear distinction between B. hyodysenteriae and B. suanatina. The confidence level for the tree topology using 1000 bootstrap replicates ranged from 60 % to 100 % (Fig. 4).
Fig. 3.
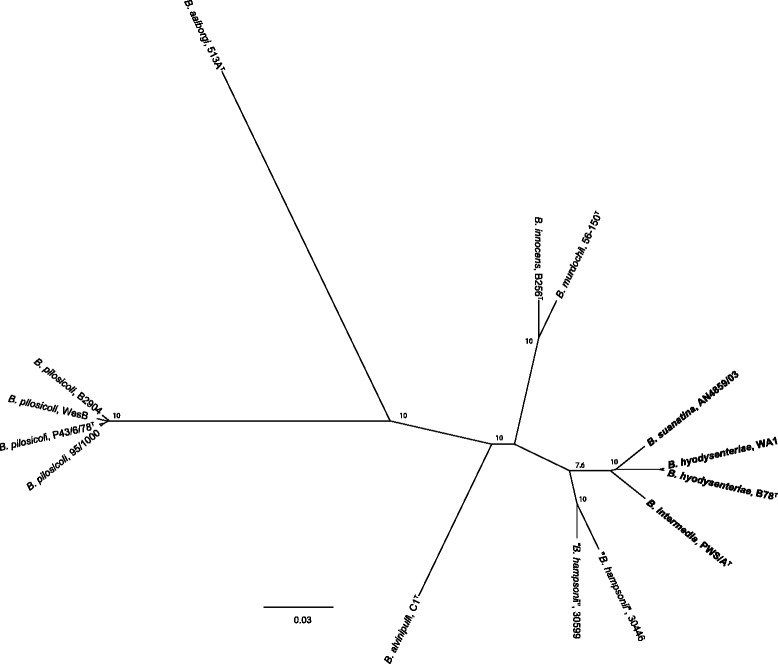
Phylogenetic tree of core genome: Radial unrooted phylogenetic representation based on concatenated amino acid sequences of 1,309 core genes of available genomes of genus Brachyspira and B. suanatina strain AN4859/03. The clade containing B. hyodysenteriae, B. intermedia and B. suanatina strains are shown in bold. The tree was constructed using the neighbour joining method. Numbers above the branches represent support values (>0.5) obtained from 200 bootstrap replicates. The tree is drawn to scale with branch lengths measured in the number of substitutions per site
Fig. 4.
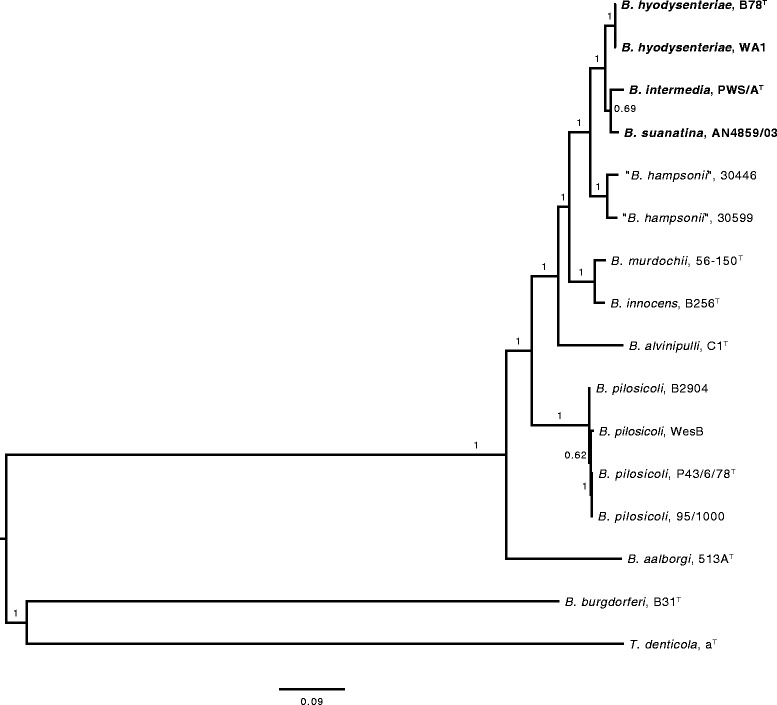
Phylogenetic tree of housekeeping genes: The evolutionary history of 26 housekeeping genes of a selection of strains representing phylum Spirochaetes was inferred by the maximum likelihood method [47] based on the general time reversible model [48]. A discrete Gamma distribution was used to model evolutionary rate differences among sites (+G, parameter). The model allowed some sites to be in variable (+I, parameter). The clade containing B. hyodysenteriae, B. intermedia and B. suanatina strains are shown in bold. Treponema denticola aT and Borrelia burgdorferi B31T were used as outgroup. Numbers above branches represent support values obtained from 1000 bootstrap replicates. The tree is drawn to scale with branch lengths measured in the number of substitutions per site. Evolutionary analysis was conducted in MEGA6 [50]
Discussion
Using the threshold value of 70 % DNA-DNA similarity for the delineation of bacterial species, as recommended by the ad hoc committee [9], B. suanatina strain AN4859/03 does not belong to any of the currently recognized seven Brachyspira spp. The highest observed % DNA-DNA similarities (in the range of 59.0-64.0) were to the type strains of B. hyodysenteriae and B. intermedia. Our finding was further confirmed by the ANI values, which are considered to correspond with the % DDH values [11]. Since the degree of robustness of ANIm is higher compared to ANIb for genomes having >60 % DDH values [13] we calculated the ANI values using both algorithms and the values obtained from them are in agreement to each other. Furthermore, the evolutionary history inferred from analysis of the 16S rRNA gene [27], housekeeping genes and the core genome (present study) suggests a common ancestor of the three related strains B78T, AN4859/03, and PWS/AT, which each represent indole test-positive phenotypes of B. hyodysenteriae, B. suanatina and B. intermedia, respectively. High bootstrap values supported the phylogram based on housekeeping genes and the assertion that B. suanatina and B. intermedia make a separate clade that is distinct from the clade formed by B. hyodysenteriae strains. Interestingly, the phenotypes of B. hyodysenteriae, B. suanatina and B. intermedia were very similar, with the strong β-haemolysis of the two former being strongly positive vs. the weak β-haemolysis in B. intermedia. Notably, in this study, no consistent diagnostic phenotypic feature was found to differentiate B. suanatina from B. hyodysenteriae. The strains of B. suanatina in this study showed varying capabilities of using D-mannitol as growth substrate, whereas strain B78T could not. Even though this phenotype could be further investigated it would not qualify as a reliable diagnostic feature.
Interestingly, among the three strains, the type strain of B. intermedia PWS/AT seems to have undergone the highest number of changes since its divergence from the common ancestor based on the housekeeping genes phylogram. When comparing the conservation of gene synteny of B. suanatina with B. hyodysenteriae WA1 and B. intermedia PWS/AT, very low conservation was found between B. suanatina and B. intermedia as compared to B. suanatina and B. hyodysenteriae WA1. These results also point to the high level of genomic rearrangements that have taken place in the genome of B. intermedia after these species diverged. Previous reports based on multi locus sequence typing have suggested that B. intermedia is a genetically diverse species that may possibly consist of several species [54, 55]. The same applies when strongly β-haemolytic indole-test positive isolates, to which B. hyodysenteriae and B. suanatina and other isolates of unknown species affiliation, are investigated [29]. Clearly, this group of Brachyspira isolates needs more detailed genomic and phenotypic study to elucidate evolutionary and taxonomic relationships.
General genomic features including the distribution of genes in similar functional classes in B. suanatina, B. hyodysenteriae and B. intermedia support the fact that they are highly similar species. However the genome size of B. hyodysenteriae is smaller as compared to B. suanatina and B. intermedia. Speculatively, the reason for the smaller genome size could be that B. hyodysenteriae has adapted to the rich environment of porcine intestinal tissues through reductive evolution. Reductive evolution is a process of reduction in genome size of a host associated bacterium by the loss of genes rendered non-essential [56]. The presence of intact bacteriophage region BSP in B. suanatina and its conservation in other Brachyspira genomes including B. hyodysenteriae B78T while absence in B. hyodysenteriae WA1 and B. intermedia PWS/AT also suggests the loss of this prophage in two later species due to the reductive evolution. Events of horizontal gene transfer (HGT) are also evident in the genome of B. suanatina when examining the genes that had no homology in the genomes of Brachyspira species with other bacteria. These include the presence of some genes that had their highest similarities in Clostridium spp. and Bacillus spp. Both these species are also inhabitants of the large intestine where the possible exchange of genetic material with B. suanatina could have taken place. Events of reductive evolution and HGT in the genomes of Brachyspira species have been described earlier [40, 52, 53].
The unusual phage-like gene transfer agent VSH-1 produced by B. hyodysenteriae is likely involved in natural gene transfer and recombination within different strains of the species [57, 58]. It spans over a 16.3 kb region of the genome of B. hyodysenteriae B204R and contains all the late function genes [59]. The region containing genes homologous to VSH-1 has also been identified in the genomes of different strains of B. hyodysenteriae, B. intermedia, B. pilosicoli and B. murdochii [51, 53, 60]. A region similar to VSH-1 was also found in the genome of B. suanatina in this study. Although it is not clear if this GTA in B. suanatina strain AN4859/03 is functional, the conservation of the late function genes and the presence of unknown genes in the GTA region suggest functional capabilities.
Conclusions
This study demonstrates the importance of integrating genomic data into the classification of different bacterial species. Based on genomic characteristics we conclude that B. suanatina does not belong to any of the currently validated seven Brachyspira species. The use of genomic data from more than one strain of B. suanatina species of the strain will facilitate future comparative genomic studies.
Description of Brachyspira suanatina sp. nov.
Brachyspira suanatina su.a.na.ti'na. L. masc n. suus swine/pig, L. fem. adj. anatina from/of a duck, N.L. fem. adj. suanatina of pig and duck, referring to the host animals (pigs and mallards) from which this spirochete was isolated.
Cells are oxygen-tolerant but anaerobic and Gram-negative of a typical spirochaetal ultrastructure. Cell dimensions are 5.5 ± 1.0 μm in length and 0.3 ± 0.0 μm in diameter. The number of periplasmic flagella are 14–18 (mean 16) per cell inserted subterminally at cells ends. Bacterial growth occurs at 37 and 42 °C but not at 32 and 46 °C. When grown anaerobically in BHIS at 37 °C cultures reach log-phase at approximately 31 h of inoculation and reach maximum population densities of approximately 2 × 108 CFU/mL at 36–38 h post inoculation. At mid log-phase, the population doubling time is approximately 2 h. Cells grow anaerobically on fastidious anaerobe agar with swarming colonies and a strong β-haemolysis on trypticase soy agar. Gives positive reactions in tests for tryptophanase activity, alkaline phosphatase, C4 esterase, C8 esterase lipase, acid phosphatase, naphtolphosphohydrolase, β-galactosidase. α-glucosidase, β-glucosidase. The following substrates can be utilized as single carbon, nitrogen and energy sources for growth: D-glucose, D-fructose, D-galactose, D-mannose, sucrose, D-trehalose, lactose, N-acetyl-D-glucosamine and pyruvate. D-mannitol sustain growth of some isolates. Growth was not detected with D-cellobiose, D-fucose, D-melobiose, D-ribose, D-raffinose, D-xylose, L-arabinose, L-fucose, L-rhamnose, N-acetyl-D-galactosamine, soluble starch, galactosamine, Na-acetate, lactate, esculin, succinate, pectin, 50x MEM amino acids, cellulose, glutathione, glycerol, glycogen, poly-galacturonic acid, hyaluronic acid and hog gastric mucin. The draft genomic DNA G + C content of the type strain is 27 % and the genome comprises 3,258,009 bp, and 2,892 ORFs. DNA-DNA hybridization of the type strain against all type strains of recognized Brachyspira spp. (<64.0 %) and ANIb/ANIm values (75.28-92.69 and 85.98-93.19 respectively) support the taxonomic classification of B. suanatina as a sp. nov. The evolutionary history of 16S rRNA and housekeeping genes, and the core genome shows a common ancestor of B. hyodysenteriae, B. intermedia and B. suanatina. A bacteriophage region and a phage-like GTA similar to VSH-1 in B. hyodysenteriae and a putative plasmid sequence related to B. hyodysenteriae strain WA1 plasmid (pBHWA1) are present in the genome of B. suanatina strain AN4859/03. Type strain is enteropathogenic in pigs with swine dysentery-like symptoms when using the type strain, and mild diarrhoea was associated with challenge with an isolate of mallard origin [27]. The type strain, AN4859/03T (=ATCC® BAA-2592™ = DSM 100974T) was obtained from a Swedish pig with swine dysentery-like disease.
Accession numbers
Draft genome sequences of B. suanatina AN4859/03 have been deposited in the European Nucleotide Archive (ENA) database with scaffold accession numbers: CVLB01000001-CVLB01000034 under the study accession number: PRJEB9032.
Acknowledgements
The authors wish to thank Leif Ljung, Uppsala University for assistance with electron microscopy and Lena Lundgren, SVA for laboratory assistance. Advice of Professor Hans G. Trüper regarding nomenclature of Brachyspira suanatina is gratefully acknowledged. We would also like to thank Jochen Blom for providing assistance with EDGAR framework. Funding was provided by the Higher Education Commission (HEC) of Pakistan, the Swedish Research Council Formas (grant 2004–1260), The Swedish Farmers’ Foundation for Agricultural Research (grant V0550134), the Royal Swedish Academy of Agriculture and Forestry (KSLA, Stiftelsen Carl-Fredrik von Horns fond, grant H-574) and the SVA Research Fund. This work was supported by EU FP7 AllBio project, contract 289452.
Abbreviations
- ACT
Artemis comparison tool
- ANI
Average nucleotide identity
- BHI
Brain heart infusion culture broth
- BHIS
Brain heart infusion culture broth with calf serum
- CDS
Coding sequence
- CFU
Colony forming unit
- COG
Clusters of orthologous group
- DDH
DNA-DNA hybridization
- FAA
Fastidious anaerobe agar
- FCS
Foetal calf serum
- GTA
Gene transfer agent
- HI
Heart infusion culture broth
- HIS
Heart infusion culture broth with foetal calf serum
- HGT
Horizontal gene transfer
- ML
Maximum likelihood
- Nox
NADH oxidase gene
- ORF
Open reading frame
Additional files
Strain names and accession numbers of available genome used for the study. (XLSX 10 kb)
Functional distribution of genes in different COG categories of strains B. suanatina AN4859/03, B. hyodysenteriae B78T, B. hyodysenteriae WA1 and B. intermedia PWS/AT. (XLSX 10 kb)
Pairwise genome alignment of B. hyodysenteriae WA1, B. intermedia PWS/AT and B. suanatina AN4859/03. The genomes were aligned using Artemis comparison tool (ACT). Alignment was performed according to the sequence start of B. hyodysenteriae WA1 and visualised in ACT with a cut-off set to blast scores > 250. Red bars indicate the similar regions in the same orientation, whereas blue bars show inverted regions. (PDF 662 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MM, EBR and DSJ designed the experiments. MM, SZ, DSJ and TR conducted experiments and data analyses. MM and DSJ drafted the manuscript. All authors contributed to the final manuscript. All authors read and approved the final manuscript.
Contributor Information
Mamoona Mushtaq, Email: memoonamushtaq@yahoo.com.
Saima Zubair, Email: saimazubair.mail@gmail.com.
Therese Råsbäck, Email: therese.rasback@sva.se.
Erik Bongcam-Rudloff, Email: erik.bongcam@slu.se.
Désirée S. Jansson, Email: desiree.jansson@sva.se
References
- 1.den Bakker HC, Manuel CS, Fortes ED, Wiedmann M, Nightingale KK. Genome sequencing identifies Listeria fleischmannii subsp. coloradonensis subsp. nov., isolated from a ranch. Int J Syst Evol Microbiol. 2013;63:3257–3268. doi: 10.1099/ijs.0.048587-0. [DOI] [PubMed] [Google Scholar]
- 2.Bennett JS, Jolley KA, Maiden MC. Genome sequence analyses show that Neisseria oralis is the same species as ‘Neisseria mucosa var. heidelbergensis’. Int J Syst Evol Microbiol. 2013;63:3920–3926. doi: 10.1099/ijs.0.052431-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Urbanczyk H, Ogura Y, Hayashi T. Taxonomic revision of Harveyi clade bacteria (family Vibrionaceae) based on analysis of whole genome sequences. Int J Syst Evol Microbiol. 2013;63:2742–2751. doi: 10.1099/ijs.0.051110-0. [DOI] [PubMed] [Google Scholar]
- 4.Brenner DJ, McWhorter AC, Knutson JK, Steigerwalt AG. Escherichia vulneris: a new species of Enterobacteriaceae associated with human wounds. J Clin Microbiol. 1982;15:1133–1140. doi: 10.1128/jcm.15.6.1133-1140.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colwell R. Polyphasic taxonomy of the genus Vibrio: numerical taxonomy of Vibrio cholerae, Vibrio parahaemolyticus, and related Vibrio species. J Bacteriol. 1970;104(1):410–433. doi: 10.1128/jb.104.1.410-433.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woese CR, Fox GE. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci. 1977;74:5088–5090. doi: 10.1073/pnas.74.11.5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stackebrandt E, Goebel BM. Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int J Syst Bacteriol. 1994;44:846–849. doi: 10.1099/00207713-44-4-846. [DOI] [Google Scholar]
- 8.Stackebrandt E, Frederiksen W, Garrity GM, Grimont PA, Kämpfer P, Maiden MC, et al. Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology. Int J Syst Evol Microbiol. 2002;52:1043–1047. doi: 10.1099/00207713-52-3-1043. [DOI] [PubMed] [Google Scholar]
- 9.Wayne LG, Brenner DJ, Colwell RR, Grimont PAD, Kandler O, Krichevsky MI, et al. Report of the ad hoc committee on reconciliation of approaches to bacterial systematics. Int J Syst Bacteriol. 1987;37:463–464. doi: 10.1099/00207713-37-4-463. [DOI] [Google Scholar]
- 10.Fox GE, Wisotzkey JD, Jurtshuk P., Jr How close is close: 16S rRNA sequence identity may not be sufficient to guarantee species identity. Int J Syst Bacteriol. 1992;42:166–170. doi: 10.1099/00207713-42-1-166. [DOI] [PubMed] [Google Scholar]
- 11.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57:81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 12.Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci U S A. 2005;102:2567–2572. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richter M, Rosselló-Móra R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A. 2009;106:19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mende DR, Sunagawa S, Zeller G, Bork P. Accurate and universal delineation of prokaryotic species. Nat Methods. 2013;10:881–884. doi: 10.1038/nmeth.2575. [DOI] [PubMed] [Google Scholar]
- 15.Daubin V, Gouy M, Perrière G. A phylogenomic approach to bacterial phylogeny: evidence of a core of genes sharing a common history. Genome Res. 2002;12:1080–1090. doi: 10.1101/gr.187002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hampson D. Brachyspiral colitis. In: Zimmerman JJ, Karriker LA, Ramirez A, Schwartz KJ, Stevenson GW, editors. Diseases of Swine. 10. Ames, Iowa, USA: Wiley Blackwell; 2012. pp. 680–696. [Google Scholar]
- 17.Harris DL, Glock RD, Christensen CR, Kinyon JM. Inoculation of pigs with Treponema hyodysenteriae (new species) and reproduction of the disease. Vet Med Small Anim Clin. 1972;67:61–64. [PubMed] [Google Scholar]
- 18.Kinyon JM, Harris DL. Treponema innocens, a new species of intestinal bacteria, and emended description of the type strain of Treponema hyodysenteriae Harris et al. Int J Syst Bacteriol. 1979;29:102–109. doi: 10.1099/00207713-29-2-102. [DOI] [Google Scholar]
- 19.Stanton TB, Jensen NS, Casey TA, Tordoff LA, Dewhirst FE, Paster BJ. Reclassification of Treponema hyodysenteriae and Treponema innocens in a new genus, Serpula gen. nov., as Serpula hyodysenteriae comb. nov. and Serpula innocens comb. nov. Int J Syst Bacteriol. 1991;41:50–58. doi: 10.1099/00207713-41-1-50. [DOI] [PubMed] [Google Scholar]
- 20.Stanton TB. Proposal to change the genus designation Serpula to Serpulina gen. nov. containing the species Serpulina hyodysenteriae comb. nov. and Serpulina innocens comb. nov. Int J Syst Bacteriol. 1992;42:189–190. doi: 10.1099/00207713-42-1-189. [DOI] [PubMed] [Google Scholar]
- 21.Ochiai S, Adachi Y, Mori K. Unification of the genera Serpulina and Brachyspira, and proposals of Brachyspira hyodysenteriae comb. nov., Brachyspira innocens comb. nov. and Brachyspira pilosicoli comb. nov. Microbiol Immunol. 1997;41:445–452. doi: 10.1111/j.1348-0421.1997.tb01877.x. [DOI] [PubMed] [Google Scholar]
- 22.Hovind-Hougen K, Birch-Andersen A, Henrik-Nielsen R, Orholm M, Pedersen JO, Teglbjaerg PS, et al. Intestinal spirochetosis: morphological characterization and cultivation of the spirochete Brachyspira aalborgi gen. nov., sp. nov. J Clin Microbiol. 1982;16:1127–1136. doi: 10.1128/jcm.16.6.1127-1136.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McLaren AJ, Trott DJ, Swayne DE, Oxberry SL, Hampson DJ. Genetic and phenotypic characterization of intestinal spirochetes colonizing chickens and allocation of known pathogenic isolates to three distinct genetic groups. J Clin Microbiol. 1997;35:412–417. doi: 10.1128/jcm.35.2.412-417.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oxberry SL, Trott DJ, Hampson DJ. Serpulina pilosicoli, waterbirds and water: potential sources of infection for humans and other animals. Epidemiol Infect. 1998;121:219–225. doi: 10.1017/S0950268898008863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mikosza AS, Hampson DJ. Human intestinal spirochetosis: Brachyspira aalborgi and/or Brachyspira pilosicoli? Anim Health Res Rev. 2001;2:101–110. [PubMed] [Google Scholar]
- 26.Stanton TB, Postic D, Jensen NS. Serpulina alvinipulli sp. nov., a new Serpulina species that is enteropathogenic for chickens. Int J Syst Bacteriol. 1998;48:669–676. doi: 10.1099/00207713-48-3-669. [DOI] [PubMed] [Google Scholar]
- 27.Rasback T, Jansson DS, Johansson K-E, Fellstrom C. A novel enteropathogenic, strongly haemolytic spirochaete isolated from pig and mallard, provisionally designated ‘Brachyspira suanatina’ sp. nov. Environ Microbiol. 2007;9:983–991. doi: 10.1111/j.1462-2920.2006.01220.x. [DOI] [PubMed] [Google Scholar]
- 28.Pettersson B, Fellstrom C, Andersson A, Uhlén M, Gunnarsson A, Johansson K-E. The phylogeny of intestinal porcine spirochetes (Serpulina species) based on sequence analysis of the 16S rRNA gene. J Bacteriol. 1996;178:4189–4199. doi: 10.1128/jb.178.14.4189-4199.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jansson DS, Johansson K-E, Olofsson T, Rasback T, Vagsholm I, Pettersson B, et al. Brachyspira hyodysenteriae and other strongly beta-haemolytic and indole-positive spirochaetes isolated from mallards (Anas platyrhynchos) J Med Microbiol. 2004;53:293–300. doi: 10.1099/jmm.0.05488-0. [DOI] [PubMed] [Google Scholar]
- 30.Rohde J, Habighorst-Blome K. An up-date on the differentiation of Brachyspira species from pigs with nox-RFLP-based restriction fragment length polymorphism. Vet Microbiol. 2012;158:211–215. doi: 10.1016/j.vetmic.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 31.Ramasamy D, Mishra AK, Lagier JC, Padhmanabhan R, Rossi M, Sentausa E, et al. A polyphasic strategy incorporating genomic data for the taxonomic description of novel bacterial species. Int J Syst Evol Microbiol. 2014;64:384–391. doi: 10.1099/ijs.0.057091-0. [DOI] [PubMed] [Google Scholar]
- 32.Trott DJ, Stanton TB, Jensen NS, Duhamel GE, Johnson JL, Hampson DJ. Serpulina pilosicoli sp. nov., the agent of porcine intestinal spirochetosis. Int J Syst Bacteriol. 1996;46:206–215. doi: 10.1099/00207713-46-1-206. [DOI] [PubMed] [Google Scholar]
- 33.Cashion P, Holder-Franklin MA, McCully J, Franklin M. A rapid method for the base ratio determination of bacterial DNA. Anal Biochem. 1977;81:461–466. doi: 10.1016/0003-2697(77)90720-5. [DOI] [PubMed] [Google Scholar]
- 34.Huss VA, Festl H, Schleifer KH. Studies on the spectrophotometric determination of DNA hybridization from renaturation rates. Syst Appl Microbiol. 1983;4:184–192. doi: 10.1016/S0723-2020(83)80048-4. [DOI] [PubMed] [Google Scholar]
- 35.Chevreux B, Wetter T, Suhai S. Genome sequence assembly using trace signals and additional sequence information. In: Proceedings of German Conference on Bioinformatics, 1999. http://www.bioinfo.de/isb/gcb99/talks/chevreux/. Accessed 4 April 2015.
- 36.Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27:578–579. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- 38.Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gordon D. Viewing and editing assembled sequences using Consed. Curr Protoc Bioinformatics. 2003;Chapter 11:Unit 11.2. doi: 10.1002/0471250953.bi1102s02. [DOI] [PubMed] [Google Scholar]
- 40.Bellgard MI, Wanchanthuek P, La T, Ryan K, Moolhuijzen P, Albertyn Z, et al. Genome sequence of the pathogenic intestinal spirochete Brachyspira hyodysenteriae reveals adaptations to its lifestyle in the porcine large intestine. PLoS One. 2009;4 doi: 10.1371/journal.pone.0004641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meyer F, Goesmann A, McHardy AC, Bartels D, Bekel T, Clausen J, et al. GenDB–an open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 2003;31:2187–2195. doi: 10.1093/nar/gkg312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39(Suppl 2):W347–W352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blom J, Albaum SP, Doppmeier D, Pühler A, Vorhölter F-J, Zakrzewski M, et al. EDGAR: a software framework for the comparative analysis of prokaryotic genomes. BMC Bioinformatics. 2009;10:154. doi: 10.1186/1471-2105-10-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 46.Wu M, Eisen JA. A simple, fast, and accurate method of phylogenomic inference. Genome Biol. 2008;9:R151. doi: 10.1186/gb-2008-9-10-r151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- 48.Tavaré S. Some Probabilistic and Statistical Problems in the Analysis of DNA Sequences. Lectures Math Life Sci (Amer Math Soc) 1986;17:57–86. [Google Scholar]
- 49.Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008;25(7):1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- 50.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Motro Y, La T, Bellgard MI, Dunn DS, Phillips ND, Hampson DJ. Identification of genes associated with prophage-like gene transfer agents in the pathogenic intestinal spirochaetes Brachyspira hyodysenteriae, Brachyspira pilosicoli and Brachyspira intermedia. Vet Microbiol. 2009;134:340–345. doi: 10.1016/j.vetmic.2008.09.051. [DOI] [PubMed] [Google Scholar]
- 52.Mappley LJ, Black ML, AbuOun M, Darby AC, Woodward MJ, Parkhill J, et al. Comparative genomics of Brachyspira pilosicoli strains: genome rearrangements, reductions and correlation of genetic compliment with phenotypic diversity. BMC Genomics. 2012;13:454. doi: 10.1186/1471-2164-13-454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hafstrom T, Jansson DS, Segerman B. Complete genome sequence of Brachyspira intermedia reveals unique genomic features in Brachyspira species and phage-mediated horizontal gene transfer. BMC Genomics. 2011;12:395. doi: 10.1186/1471-2164-12-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fellstrom C, Rasback T, Johansson K-E, Olofsson T, Aspán A. Identification and genetic fingerprinting of Brachyspira species. J Microbiol Methods. 2008;72:133–140. doi: 10.1016/j.mimet.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 55.Phillips ND, La T, Amin MM, Hampson DJ. Brachyspira intermedia strain diversity and relationships to the other indole-positive Brachyspira species. Vet Microbiol. 2010;143:246–254. doi: 10.1016/j.vetmic.2009.10.020. [DOI] [PubMed] [Google Scholar]
- 56.Wixon J. Featured Organism: Reductive evolution in bacteria: Buchnera sp., Rickettsia prowazekii and Mycobacterium leprae. Comp Funct Genomics. 2001;2:44–48. doi: 10.1002/cfg.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Humphrey SB, Stanton TB, Jensen NS, Zuerner RL. Purification and characterization of VSH-1, a generalized transducing bacteriophage of Serpulina hyodysenteriae. J Bacteriol. 1997;179:323–329. doi: 10.1128/jb.179.2.323-329.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matson EG, Zuerner RL, Stanton TB. Induction and transcription of VSH-1, a prophage-like gene transfer agent of Brachyspira hyodysenteriae. Anaerobe. 2007;13:89–97. doi: 10.1016/j.anaerobe.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 59.Matson EG, Thompson MG, Humphrey SB, Zuerner RL, Stanton TB. Identification of genes of VSH-1, a prophage-like gene transfer agent of Brachyspira hyodysenteriae. J Bacteriol. 2005;187:5885–5892. doi: 10.1128/JB.187.17.5885-5892.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pati A, Sikorski J, Gronow S, Munk C, Lapidus A, Copeland A, et al. Complete genome sequence of Brachyspira murdochii type strain (56-150T) Stand Genomic Sci. 2010;2:260–269. doi: 10.4056/sigs.831993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stanton TB, Fournié-Amazouz E, Postic D, Trott DJ, Grimont PAD, Baranton G, et al. Recognition of two new species of intestinal spirochaetes: Serpulina intermedia sp. nov. and Serpulina murdochii sp. nov. Int J Syst Bacteriol. 1997;47:1007–1012. doi: 10.1099/00207713-47-4-1007. [DOI] [PubMed] [Google Scholar]