

Abstract
Objective:
To evaluate the response to salbutamol and ephedrine in the treatment of congenital myasthenic syndromes due to CHRNE mutations causing severe acetylcholine receptor (AChR) deficiency.
Methods:
A cohort study of 6 patients with severe AChR deficiency, symptomatic despite optimal therapy with anticholinesterase and 3,4-diaminopyridine, were analyzed for their response to the addition of salbutamol or ephedrine to their medication. Baseline quantitative myasthenia gravis (QMG) (severity) scores were worse than 15 of 39. Patients were assessed in clinic with QMG and mobility scores. Pretreatment and 6- to 8-month follow-up scores were evaluated.
Results:
All 6 patients tolerated treatment well and reported no side effects. There was a strong positive response to treatment over the 6- to 8-month assessment period with significant improvement in QMG (p = 0.027) and mobility scores. The analysis of subcomponents of the QMG score revealed marked improvement in upper (p = 0.028) and lower (p = 0.028) limb raise times. All patients reported enhanced activities of daily living at 6 to 8 months.
Conclusions:
Oral salbutamol and ephedrine appear to be effective treatments in severe cases of AChR deficiency on pyridostigmine. They are well tolerated and improvement in strength can be dramatic.
Classification of evidence:
This study provides Class IV evidence that salbutamol or ephedrine improves muscle strength in patients with congenital myasthenia from severe AChR deficiency.
Congenital myasthenic syndromes (CMS) are disorders caused by mutations in genes encoding proteins essential for neuromuscular transmission.1 All patients share the clinical feature of fatigable weakness, but presentation, pattern of weakness, and therapy vary, depending on the molecular mechanism resulting from the genetic defect. Mutations in at least 20 different genes can cause CMS.2
The most common subtype is acetylcholine receptor (AChR) deficiency syndrome, which represents approximately 30% of the UK CMS cohort.3 The majority are due to either missense or frameshift mutations within the gene encoding the adult-specific ε subunit of the AChR (CHRNE).4 They decrease the response to acetylcholine by reducing the number of receptors at the muscle endplate and simplifying the postjunctional folds.5
The clinical features of patients with AChR deficiency vary from mild to severe.6 The most severely affected have ocular, bulbar, limb, and respiratory weakness from birth, while the mildly affected tend to have a marked limitation of ocular movements, but only mild limb weakness and fatigable ptosis. Subjects with AChR deficiency respond favorably to acetylcholinesterase inhibitors7 and 3,4-diaminopyridine (3,4-DAP).8,9 However, in the severe cases, the response is generally incomplete and the prolonged use may result in a relatively modest improvement. Anecdotal reports suggest that patients with CHRNE mutations may improve with salbutamol.10
Salbutamol and ephedrine are β2-adrenergic receptor agonists used with benefit in DOK7 CMS11,12 and congenital endplate acetylcholinesterase deficiency.13–16 We hypothesized that they would be beneficial for patients with severe AChR deficiency on pyridostigmine.
METHODS
We report a case series of 6 patients, from 4 different kinships, with severe AChR deficiency caused by CHRNE mutations. The condition was defined as severe by a baseline quantitative myasthenia gravis (QMG) (severity) score worse than 15 of 39.17 Despite optimal therapy sustained for several years, all were severely disabled. The patients were followed up over the past 2 years at the UK National Diagnostic and Advisory CMS Service in Oxford. Clinical details, objective strength measures, and functional scores were recorded routinely at each clinic visit. The study was designed to answer one primary research question: Can the addition of salbutamol or ephedrine lead to improvement in the muscle strength and function of patients with severe AChR deficiency due to CHRNE mutations? The study is rated Class IV because of the absence of a control group not treated with β2-adrenergic receptor agonists.
Standard protocol approvals, registrations, and patient consents.
Written informed consent for analysis and publication of genetic and clinical data was obtained for all patients (Oxfordshire Research Ethics Committees B, 04.OXB.017 and C, 09/H0606/740).
Medication dosages.
Medication was started on an outpatient basis with incremental dosage dependent on body weight and tolerability; the final dose ranged between 0.5 and 1 mg/kg/d for ephedrine and 0.05 and 0.2 mg/kg/d for salbutamol. In all patients, baseline therapy with pyridostigmine and 3,4-DAP or pyridostigmine alone remained unchanged for at least a year before adding salbutamol or ephedrine and during the follow-up period. Blood pressure, heart rate, and ECG were performed before treatment and at each dosage increment.
Outcomes.
Strength measures, which are routinely performed in patients with CMS during clinic visits, included the QMG score and mobility score using the 10-m timed walk test, before the initiation of treatment and 6 to 8 months later.17 The QMG score is made up of 13 components, each scored from 0 (normal) to 3 (severe weakness). Components for ptosis, diplopia, swallowing, speech, and facial muscles are based on clinical interpretation. Components for arm raise time, leg raise time, grip strength, head lift time, and forced vital capacity (FVC) are based on objective, continuous values assigned to a range (e.g., for arm raise time 0 ≥ 240 seconds, 1 = 91–240 seconds, 2 = 11–90 seconds, and 3 ≤ 11 seconds), and are amenable to subanalysis. Lifestyle improvements were also recorded.
Statistical analysis.
We report the QMG score, QMG components, and 10-m timed walk test difference between baseline and 6- to 8-month follow-up using paired samples. We used GraphPad Prism version 6.0e (GraphPad Software, La Jolla, CA) for the statistical analysis.
RESULTS
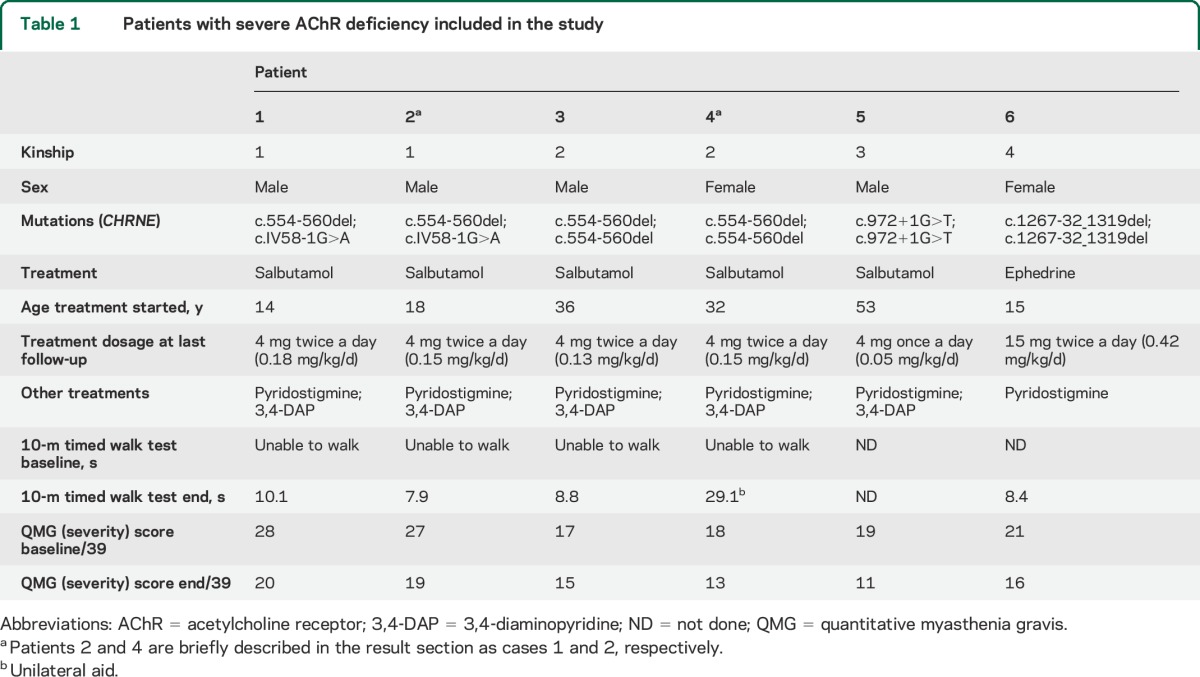
Six patients with severe AChR deficiency were included in the study (table 1). Five received salbutamol and one ephedrine, in addition to their previous therapy, which remained unchanged before and during the follow-up period. Median age was 25 years (range 14–53 years) and the male to female ratio was 4:2. All patients tolerated the additional medication without side effects. To illustrate the response to therapy, we briefly describe 2 cases.
Table 1.
Patients with severe AChR deficiency included in the study
Case 1 (patient 2).
An 18-year-old man was unable to walk or dress himself and required a wheelchair full time despite optimal treatment with pyridostigmine and 3,4-DAP. Initial examination revealed severe limb weakness with inability to produce movements against gravity. Treatment with salbutamol 4 mg twice a day produced a marked effect in muscle strength within weeks, especially in the limbs, and allowed him to become independent. Whereas he was previously nonambulant, 6 months later he was able to walk short distances and performed the 10-m timed walk test unaided in 7.9 seconds. At 6 months, the time for keeping his limbs outstretched had increased from 0 to 33 seconds in the arms and from 0 to 9 seconds in the legs. Distally, his handgrip had increased from 3 to 14 kg and the FVC had increased from 2.60 to 3.47 L. Of note, from being initially unable to rise up from the floor, at 6 months he was able to rise to stand within 6.3 seconds.
Case 2 (patient 4).
A 32-year-old woman with generalized muscle weakness was unable to walk and only able to stand for a few seconds despite optimal treatment. Examination revealed severe muscle weakness, especially in the neck muscles and lower limbs. She was prescribed 4 mg of salbutamol twice a day and she felt a significant improvement from the second week of treatment. She is now able to walk around the house and to drive a car, which is something she could not do before. By 6 months, the time for keeping her right leg outstretched increased from 3 to 15 seconds and from 21 to 33 seconds in the right arm. Timed head lift improved from 0 to 31 seconds and distally, right handgrip increased from 5 to 16 kg.
Medication dosage and side effects.
Patients 1 to 4 were started on 2 mg of salbutamol twice a day, approximately 0.05 mg/kg/d. During the course of 6 to 8 months from the initial visit, medication was increased progressively up to 4 mg of salbutamol twice a day, which corresponds to approximately 0.1 mg/kg/d. This was done in 2 steps by adding 2 mg of salbutamol in the morning (0.075 mg/kg/d), and subsequently 2 mg in the afternoon (0.1 mg/kg/d). Patient 5 was reluctant to increase his medication further from 4 mg of salbutamol once a day (0.05 mg/kg/d) as he already felt an improvement in muscle function. Patient 6 was started on ephedrine 15 mg twice a day, approximately 0.5 mg/kg/d, and chose not to increase it further. During the follow-up period, none of these patients reported adverse effects derived from the medication.
QMG (severity) score.
All patients reported a progressive improvement in muscle strength from the second week of treatment. The mean QMG scores (a higher score indicates greater disability) significantly improved from baseline to the 6- to 8-month follow-up from 21.67 ± 4.72 to 15.67 ± 3.45 (Wilcoxon signed rank test, p = 0.027; table 1).
QMG components.
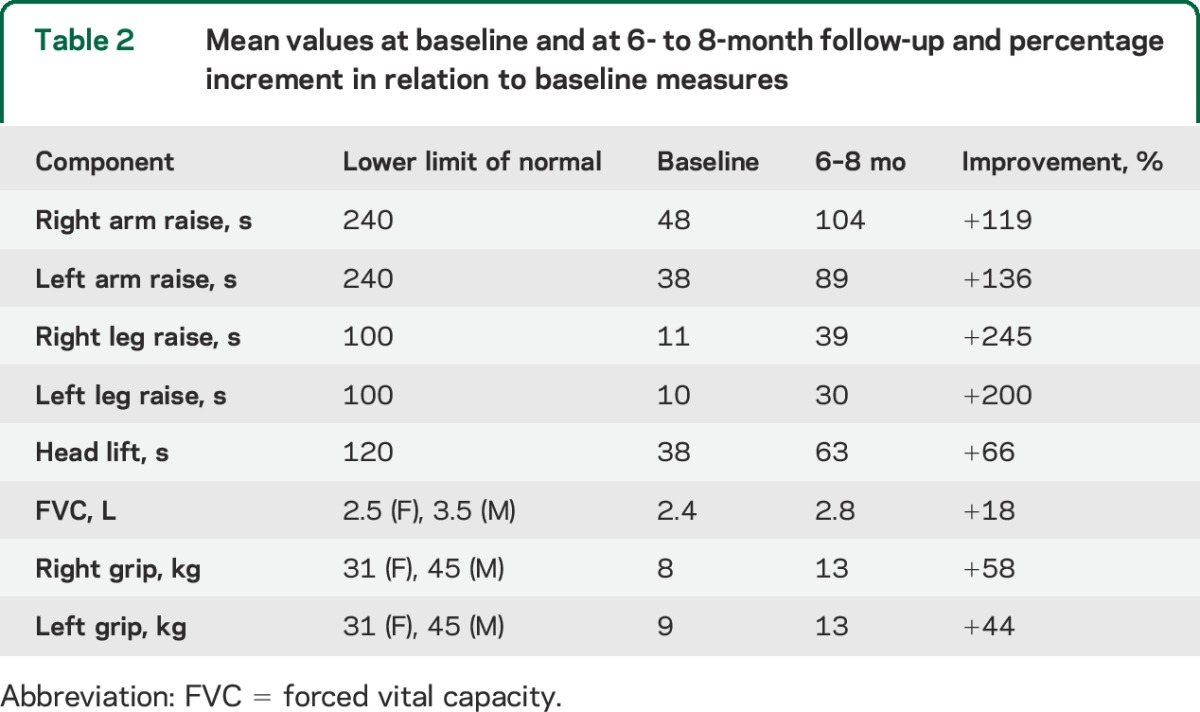
Scores for diplopia, swallowing, and counting were mainly normal at baseline, and thus had no scope for improvement. Treatment did not have an effect on ptosis, ophthalmoplegia, or facial weakness, which were present in all subjects. Improvements over the follow-up period occurred in QMG components for head lift, arm raise time, leg raise time, and less markedly for handgrip and FVC (figure, A). Times for arm and leg raise had an overall improvement from baseline of 127% and 222%, respectively (table 2; figure, B and C).
Figure. Change in performance for subcomponents of the QMG score.
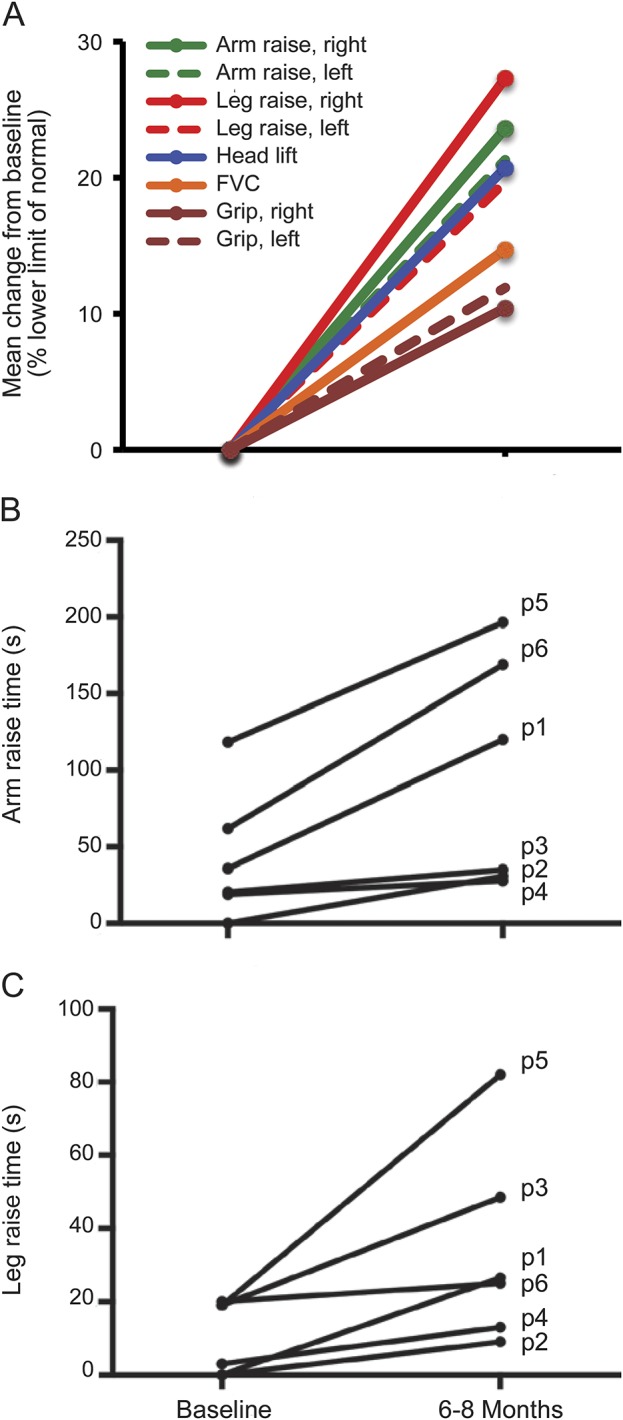
(A) Mean change in percentage of the lower limit of normal (minimum required to achieve 0 in the QMG scale). Times for individual patients for arm raise at 90° (B) and leg raise at 45° (C). Data points are the mean of right- and left-side scores. The mean values at 6 to 8 months were significantly increased in both arm raise (p = 0.028; Wilcoxon signed rank test) and leg raise (p = 0.028; Wilcoxon signed rank test) compared with baseline. FVC = forced vital capacity; p(n) = patient number; QMG = quantitative myasthenia gravis.
Table 2.
Mean values at baseline and at 6- to 8-month follow-up and percentage increment in relation to baseline measures
Mobility scores.
At baseline, 4 patients were unable to walk because of extreme weakness (patients 1–4). In the follow-up assessment, all 4 were able to complete the test. Patient 4 had developed severe fixed joint contractures of knees and hips, which made walking difficult even though she showed considerable improvement in strength. Preassessment 10-m walk time was not done for patients 5 and 6.
DISCUSSION
This study demonstrates that for patients with CMS who have severe AChR deficiency due to CHRNE mutations taking pyridostigmine, the addition of either salbutamol or ephedrine medication can substantially improve muscle strength, as confirmed by the QMG severity score and 10-m timed walk test, without notable adverse effects. These cases showed an unequivocal improvement in functional ability. Four patients who had been nonambulant for many years acquired the ability to walk independently.
In our experience, patients with milder phenotypes of AChR deficiency also report an increase in stamina and function with the addition of salbutamol or ephedrine to their treatment regimen. However, the improvement is more marked in the severe patients, probably because profound weakness provides a greater margin to record improved muscle function. We found that patients' perception of the positive treatment response was greater than the objective measurement scores revealed, especially handgrip. As reported in previous studies of ephedrine11 and salbutamol12,13 in DOK7 CMS, outcome measures may not fully reflect functional benefit. A beneficial response to albuterol in 2 adult female patients with CMS due to ε mutations has been reported previously,10 although quantitative assessment of improvement using pre- and posttreatment measures was not performed.
The commonly used maintenance dose for salbutamol was 4 mg twice a day although most of the patients reported a clear benefit within the first 2 weeks of starting at 2 mg twice a day. One of the subjects (patient 5) improved significantly despite only taking salbutamol 4 mg once a day. This differs from the slower response we observe in DOK7 CMS, where patients benefit slowly but progressively over a period of months before stabilizing at between 6 and 24 months.11 In general, we would recommend increasing salbutamol progressively up to 4 mg twice a day in the course of 6 months when side effects are not apparent. Medication can be increased further up to 8 mg twice a day in older children/adults if required. Patient 6 improved on ephedrine 15 mg twice a day although higher doses can be used. We have also reviewed 3 additional patients with severe AChR deficiency due to CHRNE mutations in clinic on long-term treatment with pyridostigmine, who have started ephedrine, where baseline formal strength measures were incomplete. All reported a clear benefit after the initiation of ephedrine with dosages between 15 mg once a day and 30 mg twice a day with no side effects. Ephedrine and salbutamol can be used interchangeably, although we currently use salbutamol more frequently because there are more safety data for its use in children12 and it is easier to prescribe. However, ephedrine is a good alternative for those in whom salbutamol causes side effects.
The present report applies to patients with severe AChR deficiency due to mutations in CHRNE. We have not assessed this treatment option on severe AChR deficiency due to mutations in other subunits of the AChR that are extremely rare.
In this quantitative study, we have seen a positive response to treatment in subjects of different ages (range 14–53 years). Although patients with AChR deficiency usually do not deteriorate markedly with time,18 it is possible that early introduction of salbutamol or ephedrine may improve mobility over the long term. A limitation of this study is the lack of a control group not treated with β2-adrenergic receptor agonist. This is attributable to the rare occurrence of severe AChR deficiency and thus the low patient numbers.
The molecular mechanisms for ephedrine and salbutamol at the neuromuscular junction are not known. We note that patients with mutations in the AGRN-LRP4-MUSK-DOK7 pathway (proteins that promote clustering of AChRs at the neuromuscular junction) improve with these treatments, and one of the plausible explanations is that β2-adrenergic agonists provide a compensatory mechanism to stabilize motor endplate structures and thus partially mitigate disruption of the MuSK signaling pathway.19 In our experience, patients with AChR deficiency report that the early beneficial response to cholinesterase inhibitors is less effective with its longer-term use. The cause may be an additional reduction in the number of endplate AChR sites as a result of disorganization of the postsynaptic muscle membrane resulting from the pyridostigmine.20–22 This would further decrease the safety margin of neuromuscular transmission and thus lead to additional muscle weakness. We hypothesize that β2-adrenergic agonists can optimize the beneficial response to pyridostigmine by increasing stability of the muscle endplate structures and prevent the additional loss of endplate AChRs that results from chronic anticholinesterase medication.
In conclusion, we report a robust improvement in all 6 patients assessed with severe AChR deficiency following the addition of oral salbutamol or ephedrine to their long-term treatment regimen that included pyridostigmine. Because ε-subunit AChR mutations causing AChR deficiency represent the most frequent CMS subtype in the United Kingdom, these results represent an important beneficial therapeutic step in treatment.
ACKNOWLEDGMENT
The authors thank the patients who participated in the study and Myaware (Myasthenia Gravis Association) for support.
GLOSSARY
- AChR
acetylcholine receptor
- CHRNE
cholinergic receptor, nicotinic, epsilon (muscle)
- CMS
congenital myasthenic syndrome
- 3,4-DAP
3,4-diaminopyridine
- FVC
forced vital capacity
- QMG
quantitative myasthenia gravis
AUTHOR CONTRIBUTIONS
Pedro M. Rodríguez Cruz: acquisition of data, analysis and interpretation, critical revision of the manuscript for important intellectual content. Jacqueline Palace: acquisition of data, study concept and design, analysis and interpretation, critical revision of the manuscript for important intellectual content, study supervision. Hayley Ramjattan: acquisition of data. Sandeep Jayawant: acquisition of data. Stephanie A. Robb: acquisition of data, critical revision of the manuscript for important intellectual content. David Beeson: study concept and design, analysis and interpretation, critical revision of the manuscript for important intellectual content, study supervision.
STUDY FUNDING
The UK NHS National Highly Specialised Service funded the Diagnostic and Advisory service for CMS in Oxford.
DISCLOSURE
P. Rodríguez Cruz, J. Palace, H. Ramjattan, S. Jayawant, and S. Robb report no disclosures relevant to the manuscript. D. Beeson holds MRC Programme Grant MR/M006824/1. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Palace J, Beeson D. The congenital myasthenic syndromes. J Neuroimmunol 2008;201–202:2–5. [DOI] [PubMed] [Google Scholar]
- 2.Cruz PM, Palace J, Beeson D. Congenital myasthenic syndromes and the neuromuscular junction. Curr Opin Neurol 2014;27:566–575. [DOI] [PubMed] [Google Scholar]
- 3.Finlayson S, Beeson D, Palace J. Congenital myasthenic syndromes: an update. Pract Neurol 2013;13:80–91. [DOI] [PubMed] [Google Scholar]
- 4.Beeson D, Newson-Davis J. Mutations affecting muscle nicotinic acetylcholine receptors and their role in congenital myasthenic syndromes. In: Lehmann-Horn F, Jurkat-Rott K, editors. Channelopathies—Common Mechanisms in Aura, Arrhythmia and Alkalosis. New York: Elsevier; 2000:85–114. [Google Scholar]
- 5.Vincent A, Cull-Candy SG, Newsom-Davis J, Trautmann A, Molenaar PC, Polak RL. Congenital myasthenia: end-plate acetylcholine receptors and electrophysiology in five cases. Muscle Nerve 1981;4:306–318. [DOI] [PubMed] [Google Scholar]
- 6.Engel AG. Congenital myasthenic syndromes in 2012. Curr Neurol Neurosci Rep 2012;12:92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engel AG. The therapy of congenital myasthenic syndromes. Neurotherapeutics 2007;4:252–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palace J, Wiles CM, Newsom-Davis J. 3,4-Diaminopyridine in the treatment of congenital (hereditary) myasthenia. J Neurol Neurosurg Psychiatry 1991;54:1069–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harper C, Engel A. Treatment of 31 congenital myasthenic syndrome patients with 3,4-diaminopyridine. Neurology 2000;54(suppl 3):A39. [Google Scholar]
- 10.Sadeh M, Xin-Ming S, Engel A. Beneficial effect of albuterol in CMS with epsilon subunit mutations. Muscle Nerve 2012;44:289–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lashley D, Palace J, Jayawant S, Robb S, Beeson D. Ephedrine treatment in congenital myasthenic syndrome due to mutations in DOK7. Neurology 2010;74:1517–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burke G, Hiscock A, Klein A, et al. Salbutamol benefits children with congenital myasthenic syndrome due to DOK7 mutations. Neuromuscul Disord 2013;23:170–175. [DOI] [PubMed] [Google Scholar]
- 13.Lorenzoni PJ, Scola RH, Kay CSK, et al. Salbutamol therapy in congenital myasthenic syndrome due to DOK7 mutation. J Neurol Sci 2013;331:155–157. [DOI] [PubMed] [Google Scholar]
- 14.Chan SHS, Wong VCN, Engel AG. Neuromuscular junction acetylcholinesterase deficiency responsive to albuterol. Pediatr Neurol 2012;47:137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bestue-Cardiel M, Sáenz de Cabezón-Alvarez A, Capablo-Liesa JL, et al. Congenital endplate acetylcholinesterase deficiency responsive to ephedrine. Neurology 2005;65:144–146. [DOI] [PubMed] [Google Scholar]
- 16.Mihaylova V, Müller JS, Vilchez JJ, et al. Clinical and molecular genetic findings in COLQ-mutant congenital myasthenic syndromes. Brain 2008;131:747–759. [DOI] [PubMed] [Google Scholar]
- 17.Jaretzki A, Barohn RJ, Ernstoff RM, et al. Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America. Neurology 2000;55:16–23. [DOI] [PubMed] [Google Scholar]
- 18.Burke G, Cossins J, Maxwell S, et al. Distinct phenotypes of congenital acetylcholine receptor deficiency. Neuromuscul Disord 2004;14:356–364. [DOI] [PubMed] [Google Scholar]
- 19.Glass DJ, Bowen DC, Stitt TN, et al. Agrin acts via a MuSK receptor complex. Cell 1996;85:513–523. [DOI] [PubMed] [Google Scholar]
- 20.Engel AG, Lambert EH, Santa T. Study of long-term anticholinesterase therapy: effects on neuromuscular transmission and on motor end-plate fine structure. Neurology 1973;23:1273–1281. [DOI] [PubMed] [Google Scholar]
- 21.Gillies JD, Allen J. Effects of neostigmine and pyridostigmine at the neuromuscular junction. Clin Exp Neurol 1977;14:271–279. [PubMed] [Google Scholar]
- 22.Chang CC, Chen TF, Chuang ST. Influence of chronic neostigmine treatment on the number of acetylcholine receptors and the release of acetylcholine from the rat diaphragm. J Physiol 1973;230:613–618. [DOI] [PMC free article] [PubMed] [Google Scholar]