

Abstract
Glutathionylation is generally a reversible posttranslational modification that occurs to cysteine residues that have been exposed to reactive oxygen species (P-SSG). This cyclical process can regulate various clusters of proteins, including those involved in critical cellular signaling functions. However, certain conditions can favor the formation of dehydroamino acids, such as 2,3-didehydroalanine (2,3-dehydroalanine, DHA) and 2,3-didehydrobutyrine (2,3-dehydrobutyrine), which can act as Michael acceptors. In turn, these can form Michael adducts with glutathione (GSH), resulting in the formation of a stable thioether conjugate, an irreversible process referred to as nonreducible glutathionylation. This is predicted to be prevalent in nature, particularly in more slowly turning over proteins. Such nonreducible glutathionylation can be distinguished from the more facile cycling signaling processes and is predicted to be of gerontological, toxicological, pharmacological, and oncological relevance. Here, we compare reversible and irreversible glutathionylation.
1. INTRODUCTION
Glutathione is a tripeptide (L-γ-glutamyl-L-cysteinyl-glycine, Fig. 5.1) with multiple biological functions (Lushchak, 2012; Meister & Anderson, 1983; Sies, 1999). It is an abundant low-molecular-mass thiol antioxidant, which either interacts directly with reactive oxygen and nitrogen species (ROS and RNS, respectively) or serves as a cofactor for many antioxidant and associated enzymes such as peroxidases and transferases (Foster, Hess, & Stamler, 2009). In addition, glutathione is (1) a storage form of cysteine; (2) a storage form and transporter of nitric oxide (as GSNO); (3) involved in the metabolism of estrogens, leukotrienes, and prostaglandins, reduction of ribonucleotides to deoxyribonucleotides, and maturation of iron–sulfur clusters of proteins; (4) involved in the regulation of certain transcription factors; and (5) involved in the detoxification of many endogenous compounds and xenobiotics (the mercapturate pathway). Glutathione also can be used even for the detoxification of ions of transition metals such as chromium (Giustarini et al., 2005; Holland & Avery, 2011; Lushchak, Kubrak, Nykorak, Storey, & Lushchak, 2008). Free glutathione exists in vivo mostly as two forms—reduced (GSH) and oxidized (glutathione disulfide; GSSG). Its biological activity is primarily related to the active thiol group of the cysteine residue. In the intracellular milieu, glutathione is relatively stable due to the presence of an unusual γ-peptide bond between glutamate and cysteine residues. Intracellular peptidases specifically cleave peptide bonds formed from the α-carboxyl group, but not from the γ-carboxyl group. Recent attention has been drawn to the importance of the glutathione pool that is utilized in the posttranslational modification of cysteine residues, S-glutathionylation.
Figure 5.1.

Chemical structure of glutathione in reduced (A) and oxidized (disulfide) forms (B).
Glutathione is synthesized in a two-step process catalyzed by the consecutive action of γ-glutamyl-L-cysteine ligase (γGLCL, EC 6.3.2.2) and glutathione synthetase (GLS, EC 6.3.2.3). The first enzyme in the pathway is generally considered to be a regulatory enzyme in the overall synthesis and is feedback-inhibited by glutathione (Richman & Meister, 1975). Glutathione is consumed through reactions involving oxidation, conjugation, and hydrolysis. Oxidation can take place nonenzymatically through direct interaction with ROS and RNS and via enzymatic reactions catalyzed by glutathione-dependent peroxidases (Fig. 5.2). Diverse glutathione S-transferases (GSTs) catalyze conjugation of glutathione to endogenous and exogenous electrophiles. Finally, a portion of the intracellular glutathi-one pool may be released to the extracellular environment in either reduced or oxidized forms (Fig. 5.2). Extracellular glutathione may be hydrolyzed by the ectoenzyme γ-L-glutamyl transpeptidase (GGT, EC 2.3.2.2) to cysteinylglycine, which in turn may be hydrolyzed by dipeptidases to cysteine and glycine (Meister, 1983). Cells can take up the products liberated by glutathione hydrolysis as individual amino acids or dipeptides. Thus, a balance between production, consumption, hydrolysis, and transport determines the concentrations of intra- and extracellular glutathione pools. These processes are finely regulated and, under normal conditions, are well balanced. Regulation of glutathione levels occurs at the levels of transcription and translation and by posttranslational modifications of the enzymes involved in its synthesis (Lushchak, 2012).
Figure 5.2.

Involvement of glutathione in the elimination of reactive oxygen and nitrogen species. Hydroxyl radical and nitric oxide (after oxidation to the NO+ form (nitrosyl cation)) or peroxynitrite (ONOO−) may interact directly with GSH leading to GSSG formation. Hydrogen peroxide may be removed by catalase or by glutathione peroxidase (GPx). The latter requires glutathione to reduce peroxide. GR, glutathione reductase; G6PDH, glucose-6-phosphate dehydrogenase; G6P, glucose-6-phosphate; 6PGL, 6-phosphogluconolactone.
Since glutathione plays a pivotal role as an antioxidant and participates in many regulatory and metabolic processes, the glutathione biosynthetic pathway has attracted attention from pharmacologists and biomedical scientists as a possible target for medical interventions. These strategies are directed toward decreasing or increasing glutathione levels either at the whole body level or in specific tissues. One mechanism for depleting glutathione reserves is to use inhibitors of the first step in the pathway (i.e., γGLCL), such as L-buthionine sulfoximine (BSO) (Griffith & Meister, 1979). Another strategy is to use externally added electrophiles that will react with glutathione either nonenzymatically or via reactions catalyzed by GSTs (Tew, 2007). A common strategy for increasing glutathione concentrations is to use compounds such as glutathione ethyl esters and N-acetyl L-cysteine each readily converted to glutathione or incorporated into glutathione in vivo. Dietary supplementation with cysteine-enriched peptides (some whey proteins) although less commonly considered does increase tissue glutathione levels (Droge, 2005). Glutathione levels can also be enhanced through increased expression of the enzymes involved in its biosynthesis. The upregulation of γGLCL and glutathione synthetase and increased activities of these enzymes can be induced by low-intensity ROS via redox-sensitive transcription factors, such as SoxR in bacteria, Yap1 in yeasts, and Nrf2/Keap1 in mammals (Lushchak, 2011).
Cancer cells differ from their normal counterparts in processes related to ROS metabolism because (i) ROS are frequently seen as potential inducers of transformation of normal cells to malignant cells and (ii) cancer cells possess substantially imbalanced free radical processes (Ralph, Rodriguez-Enriquez, Neuzil, Saavedra, & Moreno-Sanchez, 2010). Therefore, antioxidants are considered as potential prophylactic tools and frequently are included in diverse therapeutic procedures. Antioxidants may also be useful in preventing side effects from anticancer therapies such as radiation and drugs (Nakayama, Alladin, Igbokwe, & White, 2011). Such approaches are dependently linked with the utility of glutathione in modifying thiol groups of proteins, that is, glutathionylation. This posttranslational modification affects the operation of many proteins involved in intermediary metabolism (Cooper, Pinto, & Callery, 2011; Townsend, 2007). However, prominent effects of protein glutathionylation may be through its influence on different regulatory signaling pathways (Mieyal & Chock, 2012; Tew & Townsend, 2011).
GSH efficiently interacts with HO•, HOCl, RO•, RO2•, 1O2, peroxynitrite (ONOO•−), and numerous other ROS, leading to the formation of a thiyl radical (GS•) (Fig. 5.2). Glutathione also helps the cell to neutralize many products of ROS-promoted oxidation of lipids, including malondialdehyde and 4-hydroxy-2-nonenal. The thiyl radical can also combine with another glutathione thiyl radical leading to the production of GSSG. GSSG is also generated in the reactions catalyzed by glutathione peroxidases (GPx, EC1.11.1.9) (Eq. 5.1):
| (5.1) |
and by glutaredoxins (GRX, EC 1.20.4.1) (Eq. 5.2):
| (5.2) |
Clearly, as a result of neutralization of reactive species and formation of GSSG, the potential exists for (reduced) glutathione levels to be decreased. There are two major mechanisms for maintaining the glutathione pool in a reduced state: de novo synthesis (briefly covered in the preceding text) and reduction of GSSG. Reduction of GSSG in most organisms is carried out by glutathione reductase using NADPH as reductant. NADPH is maintained in the reduced state by several enzymatic reactions/pathways. Especially important is the pentose phosphate shunt (PPS), particularly the first and limiting step of this pathway catalyzed by glucose-6-phosphate dehydrogenase (G6PDH, EC 1.1.1.49) (Eq. 5.3):
| (5.3) |
Another important source of NADPH is the next enzymatic step of the PPS, catalyzed by 6-phosphogluconate dehydrogenase (6PGDH, EC 1.1.1.43) (Eq. 5.4):
| (5.4) |
In some tissues, particularly in the brain, malic enzyme (malate dehydrogenase (oxaloacetate-decarboxylating) (NADP+); EC 1.1.1.40) is thought to be an important source of NADPH. The enzyme catalyzes the following reaction (Eq. 5.5):
| (5.5) |
Finally, NADP+-dependent isocitrate dehydrogenase (isocitrate dehydrogenase (NADP+); EC 1.1.1.42) may also be an efficient supplier of NADPH:
| (5.6) |
Recently, substantial attention has been paid to detoxification of reactive carbonyl species, such as glyoxal (ethane-1,2-dione) and methylglyoxal (2-oxopropanal), and their detoxification by a pathway that involves glutathione (Inoue, Maeta, & Nomura, 2011; Li, Maloney, Circu, Alexander, & Aw, 2013). Cellular methylglyoxal is produced in vivo mainly by the spontaneous decomposition of glyceraldehyde-3-phosphate and possibly other triosephosphates. The toxicity of methylglyoxal is based on its covalent interaction with arginine, lysine, or cysteine residues in proteins and guanine bases of nucleic acids. Methylglyoxal is detoxified by the combined action of two enzymes collectively known as the glyoxalase pathway. The first enzyme in this pathway, glyoxalase I (Glo I, EC 4.4.1.5), catalyzes the following conjugation reaction (Eq. 5.7):
| (5.7) |
In the second reaction, glyoxalase II (Glo II, EC 3.12.6) hydrolyzes the product of the glyoxalase I reaction:
| (5.8) |
The glyoxalase pathway represents the main route of methylglyoxal elimination in yeast (Penninckx, Jaspers, & Legrain, 1983) and mammals (Xue et al., 2012).
2. REVERSIBLE PROTEIN GLUTATHIONYLATION REACTIONS
Most frequently, protein glutathionylation is a reversible (oxidative) posttranslational modification where GSH forms disulfide linkage with cysteine residues (P-SSG), affecting certain groups of proteins (Fig. 5.3; Grek, Zhang, Manevich, Townsend, & Tew, 2013; Townsend, 2007). P-SSG increases the net negative charge of proteins and has the potential to impact structure and function of redox-sensitive targets. Redox signal transduction is influenced by temporal fluxes in ROS/RNS homeostasis and is transmitted through forward and reverse glutathionylation of redox sensors accompanied by altered GSH/GSSG ratios. GSTP1 can catalyze the forward reaction of the glutathionylation cycle, while glutaredoxin (Grx) mediates the removal of GSH through direct thiol disulfide exchange reactions leading to the formation of GSSG, which is subsequently reduced by glutathione reductase. Sulfiredoxin (Srx) is an antioxidant protein with oxidoreductase activity and a primary function in the metabolism and reactivation of Prdx. Overexpression of Srx in mammalian cells demonstrated that the enzyme has additional functions, including deglutathionylase activity (Abbas, Riquier, & Drapier, 2013; Moon et al., 2013). Srx contains a single cysteine residue in the active site, whereas Grx has a CXXC motif, suggesting distinct molecular mechanism of deglutathionylation. Park, Mieyal, Rhee, and Chock (2009) evaluated the specificity of the two enzymes toward Prdx1 and determined that Srx has a greater binding affinity toward Prdx-SSG than does Grx. Grx and Srx may have distinct roles in the S-glutathionylation cycle perhaps influenced by substrate specificity and/or by tissue and subcellular distribution. Grx is a generalist deglutathionylase with a broad spectrum of substrates that may be dysregulated in some human pathologies (Gallogly, Starke, Leonberg, Ospina, & Mieyal, 2008).
Figure 5.3.

S-Glutathionylation cycle. Proteins in which cysteine residues possess unusually low pKa values (redox sensors) are targets for oxidative or nitrosative stress. Cysteine residues within redox sensors can be oxidized to form protein sulfenic (P-SOH) and sulfinic (P-SOOH) acids. Some protein glutathionylation (P-SSG) reactions are mediated by GGT, Grx, or GSTP. P-SSG proteins have a wide variety of functions in cellular physiology/pathology, summaries of which can be found in Townsend (2007); the categories are depicted in the pie chart.
3. IRREVERSIBLE GLUTATHIONYLATION
Here, we first briefly discuss the formation of dehydroamino acids catalyzed by enzymatic and nonenzymatic procedures. This background sets the stage for a discussion of the formation of dehydroamino acids in peptides and proteins. Dehydroamino acids, such as 2,3-didehydroalanine (2,3-dehydroalanine, DHA) and 2,3-didehydrobutyrine (2,3-dehydrobutyrine), are excellent Michael acceptors. Thus, when present in proteins, dehydroamino acid residues are predicted to form Michael adducts with glutathione (GSH), resulting in the formation of a stable thioether (a process known as nonreducible glutathionylation). Thus far, this phenomenon has been observed in mammals in vivo only in human cataractous lens but is predicted to be prevalent in nature, particularly in more slowly turning over proteins. We also discuss recent findings showing that proteins can be irreversibly glutathionylated by means of a double electrophile that reacts covalently with both a protein residue and the cysteine residue of GSH. Nonreducible glutathionylation is predicted to be of gerontological, toxicological, pharmacological, and oncological relevance.
4. 2,3-DEHYDROALANINE AND 2,3-DIDEHYDROBUTYRINE FORMATION IN ENZYME-CATALYZED REACTIONS
2,3-Dehydroalanine (aminoacrylate) is the end product of enzyme-catalyzed β-elimination reactions with amino acids containing a leaving group in the β position (Eq. 5.9). These reactions are catalyzed by pyridoxal 5′-phosphate (PLP)-containing enzymes. The aminoacrylate tautomerizes to the corresponding α-iminopropionate (Eq. 5.10), which is then hydrolyzed to pyruvate and ammonia (Eq. 5.11). The net reaction is shown in Eq. (5.12):
| (5.9) |
| (5.10) |
| (5.11) |
| (5.12) |
An example of this type of reaction is that catalyzed by mammalian serine/threonine dehydratase. When serine is the substrate, X=OH, the enamine formed is aminoacrylate and the eliminated fragment (XH) in Eq. (5.9) is H2O. A similar sequence of reactions occurs when threonine is the substrate (Eqs. 5.13–5.16). In this case, the eliminated fragment is also H2O, but the enamine formed is 2,3-dehydrobutyrine. For a detailed analysis of the reaction mechanism of rat serine/threonine dehydratase, see Zhao and Liu (2008). Another example of a PLP-dependent enzyme that catalyzes a β-elimination reaction is cystathionine β-lyase. In this example, the eliminated molecule is homocysteine:
| (5.13) |
| (5.14) |
| (5.15) |
| (5.16) |
When X (Eq. 5.9) is an exceptionally good leaving group, PLP-containing enzymes that do not normally catalyze a β-elimination reaction may catalyze a nonphysiological β-elimination (β-lyase) reaction. Such nonphysiological β-elimination reactions may be catalyzed, for example, by the PLP-dependent cytosolic and mitochondrial isozymes of aspartate aminotransferase. Thus, both enzymes catalyze the β-elimination of chloride from β-chloroalanine with the concomitant formation of pyruvate and ammonia. In another example, both enzymes catalyze a β-elimination reaction with the cysteine conjugates derived from halogenated alkenes. For a recent review, see Cooper, Krasnikov, et al. (2011).
Aminoacrylate released from the active site during enzyme-catalyzed β-elimination reactions involving β-substituted alanines has a half-life of up to a few minutes in vitro. This time is long enough for it to be trapped if a suitable nucleophile is included in the reaction mixture. For example, aminoacrylate formed as a result of β-elimination reactions catalyzed by aspartate aminotransferase can be trapped with thiosulfate (Cavallini, Federici, Bossa, & Granata, 1973) or β-mercaptoethanol (Adams, Lowpetch, Thorndycroft, Whyte, & Young, 2005). Because aminoacrylate (an enamine) released from β-substituted alanines and the corresponding enamine derived from threonine (2,3-dehydrobutyrine) are very reactive, they have the potential to interact with and damage nearby macromolecules. Additionally, many PLP-containing enzymes that catalyze nonphysiological β-elimination reactions are syncatalytically inactivated (i.e., the damage is self-inflicted) by covalent adduct formation of the enamine/imine with PLP cofactor and/or with a susceptible residue (Cooper, Krasnikov, et al., 2011).
It used to be thought that tautomerization of the enamine released from the active site of a β-lyase to the imine followed by hydrolysis occurs strictly by a nonenzymatic route. However, it has recently been shown that in the case of a bacterial threonine/serine deaminase, an associated protein known as RidA (reactive intermediate deiminase A) greatly accelerates the rate at which the released enamine/imine is converted to pyruvate and ammonia (Lambrecht, Flynn, & Downs, 2012; Lambrecht, Schmitz, & Downs, 2013), thus minimizing the potential for self-inflicted damage and damage to nearby macromolecules. The RidA proteins are highly conserved and present in all domains of life (Lambrecht et al., 2013). It is possible that this protective mechanism is less effective with aging and that an accumulation of damaged proteins may contribute to cancer progression.
The reactions mentioned in this section serve as a prelude to the following discussion of the unsaturated amino acids in peptides and proteins.
5. HISTORICAL CHARACTERIZATION OF DEHYDROPEPTIDES
Greenstein and colleagues in the 1940s prepared a number of acylated and peptide-linked dehydroamino acids and described some of their physical and enzymatic properties (Greenstein & Leuthardt, 1947; Levintow, Fu, Price, & Greenstein, 1950). They found that although 2,3-didehydroamino acids rapidly decompose to α-keto acids, 2,3-didehydroamino acids are relatively stable if the amino group is acylated or if the 2,3-didehydroamino acid is in peptide linkage. This work appears to have been largely forgotten but is important in any discussion of the biological importance of dehydroamino acids.
6. EXAMPLES OF ENZYME-CATALYZED FORMATION OF DEHYDROAMINO ACIDS IN PEPTIDE LINKAGE
A widely studied example of peptide-linked dehydroamino acids relates to the formation of a class of ribosome-directed polypeptide-based antibiotics known as lantibiotics (Asaduzzaman & Sonomoto, 2009) The synthesis of lantibiotics requires intramolecular addition of cysteine residues to DHA residues in the lantibiotic peptide precursor. DHA residues are formed by the enzymatic dehydration of serine residues. Michael attack on a DHA residue by the side group of a cysteine residue gives rise to a peptide covalently cross-linked by a thioether-containing lanthionine residue [ ]. In contrast to the easily reduced disulfide in glutathione disulfide (GSSG), the sulfide in lanthionine is stable to reduction under physiological conditions. Another interesting example of the formation of a DHA residue occurs during thyroid hormone biosynthesis. A di-iodinated aromatic ring in thyroglobulin is transferred from part of a di-iodinated tyrosine residue to an adjacent di-iodinated tyrosine residue forming a DHA residue and an adjacent thyroxine (T4) residue, respectively; upon proteolysis of the mature protein, T4 is released (Gavaret, Cahnmann, & Nunez, 1981; Gavaret, Nunez, & Cahnmann, 1980).
An example of a dehydrobutyrine intermediate is that involved in the formation of the N-acyl terminus of the complex bioactive polypeptides polytheonamides A and B derived from the marine sponge Theonella swinhoei (Freeman et al., 2012). Another example is the intramolecular addition of a cysteine residue to a 2,3-didehydrobutyrine residue generating a 3-methyl lanthionine link during the synthesis of the antimicrobial peptide nisin, a peptide used extensively in the food industry as a preservative (Li & van der Donk, 2007).
DHA, 2,3-didehydrobutyrine, lanthionine, and methyl lanthionine residues in proteins can be detected by automated Edman degradation coupled to direct detection by electrospray-ionization mass spectrometry (Walk et al., 1999). The formation of lanthionine (and methyl lanthionine) residues is discussed here because similar Michael chemistry is involved in the irreversible (nonreducible) glutathionylation of proteins/peptides discussed in the succeeding text.
7. NONENZYMATIC METHODS FOR THE INTRODUCTION OF DHA RESIDUES INTO GLUTATHIONE AND PROTEINS
Under certain conditions, the cysteine residue of glutathione (nucleophile) can be converted to a DHA residue (electrophile)—an umpolung conversion. An interesting example involves busulfan. Busulfan is a bifunctional alkylating agent used for the treatment of hematologic and other malignancies prior to stem cell transplantation (Iwamoto et al., 2004). The compound is converted in vivo to a glutathione S-conjugate (L-γ-glutamyl-β-(S-tetrahydrothiophenium)-L-alanylglycine; GS+THT) by direct interaction with GSH and enzymatic catalysis by GSTs, especially GST A1-1 (Ritter, Bohnenstengel, Hofmann, Kroemer, & Sperker, 1999). GS+THT undergoes a base-catalyzed β-elimination reaction to yield γ-glutamyldehydroalanylglycine (EdAG) and tetrahydrothiophene (Cooper et al., 2008). This reaction, however, occurs readily in vitro at physiological pH values and temperature, and EdAG was identified as a metabolite of busulfan in a human liver cytosol fraction (Younis et al., 2008). EdAG condenses with glutathione in a Michael addition reaction to produce a lanthionine-containing thioether (GSG) (Fig. 5.4), which is a nonreducible analog of GSSG (Younis et al., 2008). EdAG has recently been shown to be an excellent hydroxyl radical scavenging agent (Peer et al., 2012) as noted previously for several antioxidant N-acyl dehydrolanines (Buc-Calderon, Sipe, Flitter, Mason, & Roberfroid, 1990). The authors noted that “observation of the hydroxyl trapping properties of EdAG suggests that the busulfan metabolite EdAG may contribute to or mitigate redox-related cytotoxicity associated with the therapeutic use of busulfan, and reaffirms indicators that support a role in free radical biology for dehydroalanine-containing peptides and proteins” (Buc-Calderon et al., 1990).
Figure 5.4.

Formation of γ-glutamyldehydroalanylglycine (EdAG) from glutathione and busulfan. Glutathione reacts with busulfan [CH3S(O)2OCH2CH2CH2CH2OS(O)2CH3] in a reaction accelerated by GSTs. Two equivalents of CH3S(O)2H are released with the formation of the corresponding glutathione conjugate (GS+THT). This conjugate contains a cyclic sulfonium moiety, which is an excellent leaving group. The elimination of thiophene results in the formation of a glutathione analog (EdAG) in which the cysteine residue is converted to a dehydroalanine residue. EdAG can then participate in a Michael addition reaction with glutathione to generate the Michael adduct GSG that contains a stable thioether bond. From Younis et al. (2008) with permission.
Cysteine (and serine) residues in proteins can also be converted to DHA residues. Koshland’s group carried out some of the earliest work in this area in the 1960s (Strumeyer, White, & Koshland, 1963; Weiner, White, Hoare, & Koshland, 1966). The authors tosylated the active site serine of chymotrypsin. Under alkaline conditions, the tosylated serine residue was converted to a DHA residue. Since that time, a number of procedures have been described to convert a serine or a cysteine residue to a dehydroalanine residue. These procedures are discussed in Chalker, Bernardes, and Davis (2011), Cooper, Pinto, et al. (2011), Cooper et al. (2008), Griffin, Srinivasan, Zheng, Huang, and Coughlin (2001), Chalker, Gunnoo, et al. (2011), and Younis et al. (2008). As discussed by Chalker, Bernardes, et al. (2011), the DHA residue can readily react with a number of RSH compounds to generate S-substituted cysteine residues, where R may be, for example, –PO32−, glycosyl group, alkyl group, aminoalkyl group, or lipid side chain.
8. REDUCIBLE GLUTATHIONYLATION OF LENS PROTEINS
GSH is present at high concentrations in the human lens (~6 mM) where, as an antioxidant, it is essential for maintaining transparency (Craghill, Cronshaw, & Harding, 2004). The size of the GSH pool diminishes with age, and loss of GSH is associated with cataract formation. Cataractous lenses exhibit a decreased GSH/GSSG ratio compared with clear lenses (Zhang, Chai, Yan, Guo, & Harding, 2008). As discussed by Harding and colleagues (Craghill et al., 2004 and references cited therein), the unique development and structure of the lens may explain the need for high levels of GSH. During cataractogenesis, especially in the nucleus part of the lens, the lens proteins unfold and thiols that were buried become exposed and reactive. Some of these thiols become oxidized and react to form (a) mixed disulfides with GSH and cysteine and (b) disulfide cross-linked aggregates. With increasing severity of the cataract, total protein thiol decreases with a concurrent increase in protein disulfide content, including disulfide linkages to GSH. A major glutathionylated protein in the lens was found to have a molecular mass of about 47 kDa, which was shown to be composed of βB1-, βB2-, and γS-crystallins (Craghill et al., 2004). In theory, the disulfides, including glutathionylated cysteine residues, in the lens are reducible. Thus, the possibility has been considered that maintaining the GSH status may be helpful in preventing damaging protein disulfide formation and in ameliorating cataract formation (Craghill et al., 2004; Zhang et al., 2008). In a streptozotocin model of diabetes in rats, the administration of eye drops containing N-acetyl-L-cysteine and glutathione ethyl ester (compounds readily converted to GSH in vivo) showed significant inhibition of the progression of diabetic cataracts at early (but not late) stages of the diabetes-associated cataractogenesis (Zhang et al., 2008).
9. IRREVERSIBLE, NONREDUCIBLE GLUTATHIONYLATION OF LENS PROTEINS
The lens nucleus contains proteins that are present from birth. Moreover, the outer fibrous cells no longer make proteins after the lens has been fully shaped (Zhang et al., 2008 and references therein). Thus, it is not surprising that after many decades, some nonenzymatic posttranslational protein modifications occur in the lens. One such modification is the loss of H2S or H2O from cysteine or serine residues, respectively, resulting in the formation of DHA residues (Fig. 5.5). Michael addition by cysteine, histidine, or lysine residues will result in proteins cross-linked internally (or to other proteins) by lanthionine bridges, histidinoalanine linkages, or lysylalanine cross-links, respectively (Linetsky, Hill, LeGrand, & Hu, 2004; Linetsky & LeGrand, 2005). Linetsky and colleagues showed that lanthionine and histidinoalanine residues in hydrolyzed proteins are present at higher concentrations in cataractous lenses than in normal lenses; lysylalanine could only be detected in cataractous lenses (Linetsky et al., 2004).
Figure 5.5.
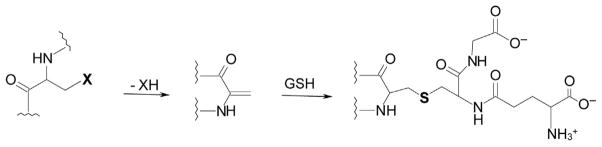
Formation of a dehydroalanyl (DHA) residue in proteins and peptides followed by irreversible glutathionylation. A protein/peptide containing a leaving group (X) in the β position of an amino acid residue may undergo spontaneous β-elimination to generate a protein/peptide containing a DHA residue (center). Michael addition of the sulfhydryl group of glutathione will generate an adduct containing a nonreducible thioether bond, resulting in irreversible glutathionylation. Residues such as cysteine (X=–SH) and serine (X =–OH) are expected to be relatively stable. However, for proteins with very long turnover times, such as those found in the lens, β-elimination reactions may occur as a consequence of aging. Moreover, other residues such as selenocysteine, serine O-phosphate, and serine O-sulfate are expected to be more labile and amenable to β-elimination. Modified from Cooper, Pinto, et al. (2011).
Lanthionine residues may be formed in lens tissues not only via Michael addition of a protein cysteine residue to a DHA residue but also by attack of either free cysteine or the cysteine moiety of GSH. Linetsky and LeGrand (Linetsky & LeGrand, 2005) showed that the increased lanthionine residues occurring in the aged and cataractous human lens result mainly from the addition of GSH to DHA residues, resulting in irreversible, nonreducible glutathionylation.
EdAG may be present at low concentrations as a result of naturally occurring umpolung transformations involving GSH. Thus, we suggest that another possibility for irreversible glutathionylation of proteins is the addition of EdAG to cysteine residues. This process is expected to be of minor consequence for most proteins but may become more prevalent with time especially with slowly turning over proteins or when individuals are exposed to xenobiotics that can promote the formation of EdAG from GSH. In this context, it is interesting to note that patients systemically treated with busulfan are at increased risk of cataractous changes to the posterior cortex of the lens (Li, Tripathi, & Tripathi, 2008). It is possible that increased formation of EdAG contributes to the damage to the lens.
10. NONREDUCIBLE GLUTATHIONYLATION INVOLVING COVALENT TETHERING
The polyunsaturated lipid peroxidation product 4-oxo-2-nonenal (ONE) is a highly reactive protein cross-linking agent (Zhu, Gallogly, Mieyal, Anderson, & Sayre, 2009). The compound also adds to GSH, resulting in the formation of a reactive 4-ketoaldehyde that has the potential to covalently add to proteins (Cooper, Pinto, et al., 2011). Zhu et al. (2009) showed that almost every lysine residue was modified when 0.25 mM bovine β-lactoglobulin was incubated in 100 mM potassium phosphate buffer containing 0.25–2 mM ONE and 1 mM GSH (an antielectrophile) at 37 °C for 24 h. The GSH is effectively bound to the protein via a thioether tether. The authors suggested that “stable antielectrophile-ONE-protein cross-links may serve as biomarkers of oxidative stress and may represent a novel mechanism of irreversible protein glutathionylation” (Zhu et al., 2009).
In another recent article, the arylated monodiazeniumdiolate JS-K was compared to the related bis-diazeniumdiolate (Double JS-K) for their ability to conjugate GSH to proteins (Holland et al., 2012). The results revealed a previously unrecognized mechanism of protein modification displayed by certain members of the arylated diazeniumdiolate family. GSH was shown to be irreversibly tethered to proteins in lung cancer cells and leukemia cells via reaction of the –SH of GSH and a protein –SH via a bivalent electrophile spacer, and the authors suggested that it is likely that other bivalent electrophiles, including some current chemotherapeutic agents, may support a similar type of cross-linking glutathionylation (Holland et al., 2012).
Finally, the anticancer drug piperlongumine has recently been shown to covalently tether GSH to proteins (Adams et al., 2012). The authors suggested a sequence of events involving first a Michael addition of GSH to the more electrophilic C2–C3 olefin in piperlongumine, followed by the formation of a noncovalent complex between the piperlongumine–GSH adduct and a GSH-binding protein, and finally a Michael addition of a nucleophilic residue of the GSH-binding protein to the less electrophilic C7–C8 olefin of piperlongumine that is accelerated by the formation of the complex (Adams et al., 2012).
11. CONCLUSIONS
In contrast to the cyclical reactions that are characterized by the addition/removal of GSH to reactive cysteines (Townsend, 2007; Townsend et al., 2009), irreversible glutathionylation of proteins in which the covalent linkage is via a lanthionine residue has thus far been detected in vivo only in lens proteins. The irreversible glutathionylation is more prevalent in cataractous lenses. We suggest, however, that irreversible glutathionylation may be more widespread in body tissues than is currently appreciated. Slowly turning over proteins throughout the body may become “damaged” in such a way that unfolding exposes and activates cysteine and serine residues making them more prone to conversion to DHA residues followed by Michael addition of GSH. Such processes are expected to increase with oxidative/nitrosative stress and with increasing age. Irreversible glutathionylation may lead to loss of function and/or enhanced removal of the protein. In addition to a cysteine residue of GSH reacting with a DHA residue, it is possible that irreversible glutathionylation may occur by reaction of a protein cysteine residue with EdAG. To our knowledge, irreversible protein glutathionylation via the addition of EdAG to a cysteine residue in a protein has not yet been demonstrated directly. However, our finding that GSH can add to EdAG suggests that if EdAG is generated in vivo, the possibility exists that it will react with protein cysteine residues. Certain drugs (and other xenobiotics) that can interact with GSH in vivo to generate EdAG (e.g., busulfan) may be toxic in part through irreversible glutathionylation. Finally, several examples have recently been described of bivalent xenobiotic electrophiles that can act as a tether covalently anchoring GSH to proteins.
In summary, there is currently only limited information on irreversible protein glutathionylation via lanthionine linkages or via bivalent electrophile linkages. However, the scope of these reactions, particularly in regard to disease, aging, drug metabolism, and disposition of anticancer drugs, is relatively large. Irreversible protein glutathionylation is a fruitful area for future research.
Acknowledgments
This work was supported by grants from the National Center for Research Resources (5P20RR024485-02) and the National Institute of General Medical Sciences (8 P20 GM103542-02) from the National Institutes of Health and by CA08660, CA117259, and R56 ES017453 and support from the South Carolina Centers of Excellence program.
References
- Abbas K, Riquier S, Drapier JC. Peroxiredoxins and sulfiredoxin at the crossroads of the NO and H2O2 signaling pathways. Methods in Enzymology. 2013;527:113–128. doi: 10.1016/B978-0-12-405882-8.00006-4. [DOI] [PubMed] [Google Scholar]
- Adams DJ, Dai M, Pellegrino G, Wagner BK, Stern AM, Shamji AF, et al. Synthesis, cellular evaluation, and mechanism of action of piperlongumine analogs. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:15115–15120. doi: 10.1073/pnas.1212802109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams B, Lowpetch K, Thorndycroft F, Whyte SM, Young DW. Stereochemistry of reactions of the inhibitor/substrates L- and D-beta-chloroalanine with beta-mercaptoethanol catalysed by L-aspartate aminotransferase and D-amino acid aminotransferase respectively. Organic and Biomolecular Chemistry. 2005;3:3357–3364. doi: 10.1039/b508199h. [DOI] [PubMed] [Google Scholar]
- Asaduzzaman SM, Sonomoto K. Lantibiotics: Diverse activities and unique modes of action. Journal of Bioscience and Bioengineering. 2009;107:475–487. doi: 10.1016/j.jbiosc.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Buc-Calderon P, Sipe HJ, Jr, Flitter W, Mason RP, Roberfroid M. N-acyl dehydroalanines scavenge oxygen radicals and inhibit in vitro free radical mediated processes. Chemico-Biological Interactions. 1990;73:77–88. doi: 10.1016/0009-2797(90)90109-z. [DOI] [PubMed] [Google Scholar]
- Cavallini D, Federici G, Bossa F, Granata F. The protective effect of thiosulfate upon the inactivation of aspartate aminotransferase by aminoacrylic-acid-producing substrates. European Journal of Biochemistry. 1973;39:301–304. doi: 10.1111/j.1432-1033.1973.tb03127.x. [DOI] [PubMed] [Google Scholar]
- Chalker JM, Bernardes GJ, Davis BG. A “tag-and-modify” approach to site-selective protein modification. Accounts of Chemical Research. 2011;44:730–741. doi: 10.1021/ar200056q. [DOI] [PubMed] [Google Scholar]
- Chalker JM, Gunnoo SB, Boutureira O, Gerstberger SC, Fernández-González M, Bernardes GJL, et al. Methods for converting cysteine to dehydroalanine on peptides and proteins. Chemical Science. 2011;2:1666–1676. [Google Scholar]
- Cooper AJL, Krasnikov BF, Niatsetskaya ZV, Pinto JT, Callery PS, Villar MT, et al. Cysteine S-conjugate beta-lyases: Important roles in the metabolism of naturally occurring sulfur and selenium-containing compounds, xenobiotics and anticancer agents. Amino Acids. 2011;41:7–27. doi: 10.1007/s00726-010-0552-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AJL, Pinto JT, Callery PS. Reversible and irreversible protein glutathionylation: Biological and clinical aspects. Expert Opinion on Drug Metabolism & Toxicology. 2011;7:891–910. doi: 10.1517/17425255.2011.577738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AJL, Younis IR, Niatsetskaya ZV, Krasnikov BF, Pinto JT, Petros WP, et al. Metabolism of the cysteine S-conjugate of busulfan involves a beta-lyase reaction. Drug Metabolism and Disposition. 2008;36:1546–1552. doi: 10.1124/dmd.108.020768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craghill J, Cronshaw AD, Harding JJ. The identification of a reaction site of glutathione mixed-disulphide formation on gammaS-crystallin in human lens. The Biochemical Journal. 2004;379:595–600. doi: 10.1042/BJ20031367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droge W. Oxidative stress and ageing: Is ageing a cysteine deficiency syndrome? Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 2005;360:2355–2372. doi: 10.1098/rstb.2005.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster MW, Hess DT, Stamler JS. Protein S-nitrosylation in health and disease: A current perspective. Trends in Molecular Medicine. 2009;15:391–404. doi: 10.1016/j.molmed.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman MF, Gurgui C, Helf MJ, Morinaka BI, Uria AR, Oldham NJ, et al. Metagenome mining reveals polytheonamides as posttranslationally modified ribosomal peptides. Science. 2012;338:387–390. doi: 10.1126/science.1226121. [DOI] [PubMed] [Google Scholar]
- Gallogly MM, Starke DW, Leonberg AK, Ospina SM, Mieyal JJ. Kinetic and mechanistic characterization and versatile catalytic properties of mammalian glutaredoxin 2: Implications for intracellular roles. Biochemistry. 2008;47:11144–11157. doi: 10.1021/bi800966v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavaret JM, Cahnmann HJ, Nunez J. Thyroid hormone synthesis in thyro-globulin. The mechanism of the coupling reaction. The Journal of Biological Chemistry. 1981;256:9167–9173. [PubMed] [Google Scholar]
- Gavaret JM, Nunez J, Cahnmann HJ. Formation of dehydroalanine residues during thyroid hormone synthesis in thyroglobulin. The Journal of Biological Chemistry. 1980;255:5281–5285. [PubMed] [Google Scholar]
- Giustarini D, Milzani A, Aldini G, Carini M, Rossi R, Dalle-Donne I. S-nitrosation versus S-glutathionylation of protein sulfhydryl groups by S-nitrosoglutathione. Antioxidants and Redox Signaling. 2005;7:930–939. doi: 10.1089/ars.2005.7.930. [DOI] [PubMed] [Google Scholar]
- Greenstein JP, Leuthardt FM. Presence of dehydropeptidases I and II activity in plant materials. Journal of the National Cancer Institute. 1947;8:35–37. [PubMed] [Google Scholar]
- Grek CL, Zhang J, Manevich Y, Townsend DM, Tew KD. Causes and consequences of cysteine S-glutathionylation. The Journal of Biological Chemistry. 2013;288:26497–26504. doi: 10.1074/jbc.R113.461368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin CT, Srinivasan Y, Zheng YW, Huang W, Coughlin SR. A role for thrombin receptor signaling in endothelial cells during embryonic development. Science. 2001;293:1666–1670. doi: 10.1126/science.1061259. [DOI] [PubMed] [Google Scholar]
- Griffith OW, Meister A. Glutathione: Interorgan translocation, turnover, and metabolism. Proceedings of the National Academy of Sciences of the United States of America. 1979;76:5606–5610. doi: 10.1073/pnas.76.11.5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland SL, Avery SV. Chromate toxicity and the role of sulfur. Metallomics. 2011;3:1119–1123. doi: 10.1039/c1mt00059d. [DOI] [PubMed] [Google Scholar]
- Holland RJ, Maciag AE, Kumar V, Shi L, Saavedra JE, Prud’homme RK, et al. Cross-linking protein glutathionylation mediated by O2-arylated bis-diazeniumdiolate “Double JS-K”. Chemical Research in Toxicology. 2012;25:2670–2677. doi: 10.1021/tx3003142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Maeta K, Nomura W. Glyoxalase system in yeasts: Structure, function, and physiology. Seminars in Cell & Developmental Biology. 2011;22:278–284. doi: 10.1016/j.semcdb.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Hiraku Y, Oikawa S, Mizutani H, Kojima M, Kawanishi S. DNA intrastrand cross-link at the 5′-GA-3′ sequence formed by busulfan and its role in the cytotoxic effect. Cancer Science. 2004;95:454–458. doi: 10.1111/j.1349-7006.2004.tb03231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrecht JA, Flynn JM, Downs DM. Conserved YjgF protein family deaminates reactive enamine/imine intermediates of pyridoxal 5′-phosphate (PLP)-dependent enzyme reactions. The Journal of Biological Chemistry. 2012;287:3454–3461. doi: 10.1074/jbc.M111.304477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrecht JA, Schmitz GE, Downs DM. RidA proteins prevent metabolic damage inflicted by PLP-dependent dehydratases in all domains of life. MBio. 2013;4:e00033–00013. doi: 10.1128/mBio.00033-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levintow L, Fu SC, Price VE, Greenstein JP. Preparation and enzymatic hydrolysis of three new homologous dehydropeptides. The Journal of Biological Chemistry. 1950;184:633–640. [PubMed] [Google Scholar]
- Li W, Maloney RE, Circu ML, Alexander JS, Aw TY. Acute carbonyl stress induces occludin glycation and brain microvascular endothelial barrier dysfunction: Role for glutathione-dependent metabolism of methylglyoxal. Free Radical Biology and Medicine. 2013;54:51–61. doi: 10.1016/j.freeradbiomed.2012.10.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Tripathi RC, Tripathi BJ. Drug-induced ocular disorders. Drug Safety. 2008;31:127–141. doi: 10.2165/00002018-200831020-00003. [DOI] [PubMed] [Google Scholar]
- Li B, van der Donk WA. Identification of essential catalytic residues of the cyclase NisC involved in the biosynthesis of nisin. The Journal of Biological Chemistry. 2007;282:21169–21175. doi: 10.1074/jbc.M701802200. [DOI] [PubMed] [Google Scholar]
- Linetsky M, Hill JM, LeGrand RD, Hu F. Dehydroalanine crosslinks in human lens. Experimental Eye Research. 2004;79:499–512. doi: 10.1016/j.exer.2004.06.026. [DOI] [PubMed] [Google Scholar]
- Linetsky M, LeGrand RD. Glutathionylation of lens proteins through the formation of thioether bond. Molecular and Cellular Biochemistry. 2005;272:133–144. doi: 10.1007/s11010-005-6908-1. [DOI] [PubMed] [Google Scholar]
- Lushchak OV, Kubrak OI, Nykorak MZ, Storey KB, Lushchak VI. The effect of potassium dichromate on free radical processes in goldfish: Possible protective role of glutathione. Aquatic Toxicology. 2008;87:108–114. doi: 10.1016/j.aquatox.2008.01.007. [DOI] [PubMed] [Google Scholar]
- Lushchak VI. Adaptive response to oxidative stress: Bacteria, fungi, plants and animals. Comparative Biochemistry and Physiology. Toxicology & Pharmacology. 2011;153:175–190. doi: 10.1016/j.cbpc.2010.10.004. [DOI] [PubMed] [Google Scholar]
- Lushchak VI. Glutathione homeostasis and functions: Potential targets for medical interventions. Journal of Amino Acids. 2012;2012:736837. doi: 10.1155/2012/736837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister A. Selective modification of glutathione metabolism. Science. 1983;220:472–477. doi: 10.1126/science.6836290. [DOI] [PubMed] [Google Scholar]
- Meister A, Anderson ME. Glutathione. Annual Review of Biochemistry. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- Mieyal JJ, Chock PB. Posttranslational modification of cysteine in redox signaling and oxidative stress: Focus on s-glutathionylation. Antioxidants and Redox Signaling. 2012;16:471–475. doi: 10.1089/ars.2011.4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JC, Kim GM, Kim EK, Lee HN, Ha B, Lee SY, et al. Reversal of 2-Cys peroxiredoxin oligomerization by sulfiredoxin. Biochemical and Biophysical Research Communications. 2013;432:291–295. doi: 10.1016/j.bbrc.2013.01.114. [DOI] [PubMed] [Google Scholar]
- Nakayama A, Alladin KP, Igbokwe O, White JD. Systematic review: Generating evidence-based guidelines on the concurrent use of dietary antioxidants and chemotherapy or radiotherapy. Cancer Investigation. 2011;29:655–667. doi: 10.3109/07357907.2011.626479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Mieyal JJ, Rhee SG, Chock PB. Deglutathionylation of 2-Cys peroxiredoxin is specifically catalyzed by sulfiredoxin. The Journal of Biological Chemistry. 2009;284:23364–23374. doi: 10.1074/jbc.M109.021394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peer CJ, Younis IR, Leonard SS, Gannett PM, Minarchick VC, Kenyon AJ, et al. Glutathione conjugation of busulfan produces a hydroxyl radical-trapping dehydroalanine metabolite. Xenobiotica. 2012;42:1170–1177. doi: 10.3109/00498254.2012.696740. [DOI] [PubMed] [Google Scholar]
- Penninckx MJ, Jaspers CJ, Legrain MJ. The glutathione-dependent glyoxalase pathway in the yeast Saccharomyces cerevisiae. The Journal of Biological Chemistry. 1983;258:6030–6036. [PubMed] [Google Scholar]
- Ralph SJ, Rodriguez-Enriquez S, Neuzil J, Saavedra E, Moreno-Sanchez R. The causes of cancer revisited: “Mitochondrial malignancy” and ROS-induced oncogenic transformation—Why mitochondria are targets for cancer therapy. Molecular Aspects of Medicine. 2010;31:145–170. doi: 10.1016/j.mam.2010.02.008. [DOI] [PubMed] [Google Scholar]
- Richman PG, Meister A. Regulation of gamma-glutamyl-cysteine synthetase by nonallosteric feedback inhibition by glutathione. The Journal of Biological Chemistry. 1975;250:1422–1426. [PubMed] [Google Scholar]
- Ritter CA, Bohnenstengel F, Hofmann U, Kroemer HK, Sperker B. Determination of tetrahydrothiophene formation as a probe of in vitro busulfan metabolism by human glutathione S-transferase A1-1: Use of a highly sensitive gas chromatographic-mass spectrometric method. Journal of Chromatography. B, Biomedical Sciences and Applications. 1999;730:25–31. doi: 10.1016/s0378-4347(99)00170-x. [DOI] [PubMed] [Google Scholar]
- Sies H. Glutathione and its role in cellular functions. Free Radical Biology and Medicine. 1999;27:916–921. doi: 10.1016/s0891-5849(99)00177-x. [DOI] [PubMed] [Google Scholar]
- Strumeyer DH, White WN, Koshland DE., Jr Role of serine in chymotrypsin action. Conversion of the active serine to dehydroalanine. Proceedings of the National Academy of Sciences of the United States of America. 1963;50:931–935. doi: 10.1073/pnas.50.5.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tew KD. Redox in redux: Emergent roles for glutathione S-transferase P (GSTP) in regulation of cell signaling and S-glutathionylation. Biochemical Pharmacology. 2007;73:1257–1269. doi: 10.1016/j.bcp.2006.09.027. [DOI] [PubMed] [Google Scholar]
- Tew KD, Townsend DM. Regulatory functions of glutathione S-transferase P1-1 unrelated to detoxification. Drug Metabolism Reviews. 2011;43:179–193. doi: 10.3109/03602532.2011.552912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DM. S-glutathionylation: Indicator of cell stress and regulator of the unfolded protein response. Molecular Interventions. 2007;7:313–324. doi: 10.1124/mi.7.6.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DM, Manevich Y, He L, Hutchens S, Pazoles CJ, Tew KD. Novel role for glutathione S-transferase pi. Regulator of protein S-Glutathionylation following oxidative and nitrosative stress. The Journal of Biological Chemistry. 2009;284:436–445. doi: 10.1074/jbc.M805586200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walk TB, Sussmuth R, Kempter C, Gnau V, Jack RW, Jung G. Identification of unusual amino acids in peptides using automated sequential Edman degradation coupled to direct detection by electrospray-ionization mass spectrometry. Biopolymers. 1999;49:329–340. doi: 10.1002/(SICI)1097-0282(19990405)49:4<329::AID-BIP7>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Weiner H, White WN, Hoare DG, Koshland DE., Jr The formation of anhydrochymotrypsin by removing the elements of water from the serine at the active site. Journal of the American Chemical Society. 1966;88:3851–3859. doi: 10.1021/ja00968a033. [DOI] [PubMed] [Google Scholar]
- Xue M, Rabbani N, Momiji H, Imbasi P, Anwar MM, Kitteringham N, et al. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress-responsive defence against dicarbonyl glycation. The Biochemical Journal. 2012;443:213–222. doi: 10.1042/BJ20111648. [DOI] [PubMed] [Google Scholar]
- Younis IR, Elliott M, Peer CJ, Cooper AJ, Pinto JT, Konat GW, et al. Dehydroalanine analog of glutathione: An electrophilic busulfan metabolite that binds to human glutathione S-transferase A1-1. The Journal of Pharmacology and Experimental Therapeutics. 2008;327:770–776. doi: 10.1124/jpet.108.142208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Chai FY, Yan H, Guo Y, Harding JJ. Effects of N-acetylcysteine and glutathione ethyl ester drops on streptozotocin-induced diabetic cataract in rats. Molecular Vision. 2008;14:862–870. [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Liu H. A quantum mechanical/molecular mechanical study on the catalysis of the pyridoxal 5′-phosphate-dependent enzyme L-serine dehydratase. The Journal of Physical Chemistry. B. 2008;112:13091–13100. doi: 10.1021/jp802262m. [DOI] [PubMed] [Google Scholar]
- Zhu X, Gallogly MM, Mieyal JJ, Anderson VE, Sayre LM. Covalent cross-linking of glutathione and carnosine to proteins by 4-oxo-2-nonenal. Chemical Research in Toxicology. 2009;22:1050–1059. doi: 10.1021/tx9000144. [DOI] [PMC free article] [PubMed] [Google Scholar]