

Abstract
Survivin is a tumor-associated antigen with significant potential as a cancer vaccine target. We have identified a survivin peptide mimic containing human MHC class I epitopes and a potential class II ligand that induces a potent antitumor response in C57BL/6 mice with GL261 cerebral gliomas. This peptide is able to elicit both CD8+ CTL and T helper cell responses in C57BL/6 mice. The corresponding region of the human survivin molecule represented by peptide SVN53-67 is 100% homologous to the murine protein, but SVN53-67 is weakly immunogenic in man. We evaluated several amino acid substitutions in putative human MHC I anchor positions in SVN53-67 to identify potential peptide mimics that could provide an enhanced antitumor immune response against human glioma and primary central nervous system lymphoma (PCNSL) cells in culture. We evaluated survivin peptides with predicted binding to human HLA-A*0201 antigen using peptide-loaded dendritic cells from PBMC of patients with these malignancies. One alteration (M57) led to binding to HLA-A*0201 with significantly higher affinity. We compared the ability of autologous dendritic cells loaded with SVN53-67 peptide and SVN53-67/M57 in CTL assays against allomatched and autologous, survivin-expressing, human malignant glioma and PCNSL cells. Both SVN53-67 and SVN53-67/M57 produced CTL-mediated killing of malignant target cells; however, SVN53-67/M57 was significantly more effective than SVN53-67. Thus, SVN53-67/M57 may act as a peptide mimic to induce an enhanced antitumor CTL response in tumor patients. The use of SVN53-67/M57 as a cancer vaccine might have application for cancer vaccine therapy.
Keywords: Antigen, Glioma, Peptide, Survivin, Tumor, Vaccine
Introduction
Survivin is one of the most specific cancer antigens identified to date because it is expressed in a large percentage of tumors and is rarely detectable in normal tissue [1, 2]. Survivin expression occurs commonly in malignant gliomas [3–5] where it is associated with a poor prognosis [6, 7]. Because many different cancer types express survivin, its use as a tumor vaccine target might have broad application for cancer vaccine therapy. Evidence suggests that patients with cancer, including those with malignant gliomas, are auto-immunized to survivin epitopes [8]. Antibodies to survivin and specific T cells that are reactive to the survivin molecule have been detected in cancer patients. Although the process by which this auto-immunization occurs is not well defined [8].
Epitopes of intracellular proteins, including those of survivin, appear on the surface of tumor cells in association with MHC class I molecules. CD8+ CTLs are activated by tumor-derived peptides presented in context with MHC class I antigens expressed on the surface of antigen presenting cells (APC’s). The tumor antigen-MHC class I complex binds to the antigen-specific T-cell receptor on CD8+ cells and stimulates T-cell activation. Certain survivin epitopes that are correctly presented by tumor cells, and are recognized by these activated, specific CTL, may elicit antitumor responses [5, 9]. Therefore, identification of survivin peptides that bind to MHC class I molecules and can elicit strong cellular immune responses is an important aspect of anti-survivin cancer immunotherapy.
Generation of an effective and specific cell-mediated immune response may require the activation of both CD8+ T lymphocyte subsets and CD4+ helper T cells. To activate a CD4+ T-cell response, antigens must be presented to CD4+ T cells in conjunction with MHC class II molecules. Once CD4+ T cells have been activated, they proliferate and produce cytokines (e.g. IFN-γ, IL-2, and IL-4) that enhance the immune response. These cytokines are essential to provide a fully activated and sustained CD8+ antitumor CTL response. Consequently, a cancer vaccine strategy that activates both specific CD8+ CTL responses and CD4+ helper cell support may be more effective than one that activates the CD8+ CTL arm alone.
The current study is based on our preliminary observations that survivin cDNA and peptide vaccines inhibit the growth of glioblastoma in rats and mice [5, 9]. CTL responses can be generated using either the whole survivin molecule, or smaller domains of survivin [5]. Responses generated from such vaccines can have an antitumor effect against cerebral gliomas in an orthotopic murine glioma model [5, 9]. In the current study, we designed a survivin peptide mimic with strong MHC class I binding properties that can also elicit T-helper cell support. The purpose of this approach was to identify survivin peptides that might be suitable for clinical application. The peptide mimic described below incorporates multiple class I epitopes with predicted binding to HLA-A*0201 and potential class II ligand activity.
Methods
Peptides
Peptide synthesis was performed using Fmoc chemistry and a solid support resin (Sigma-Genosys, The Woodlands, TX). Each peptide was stored at −70°C until use and was initially diluted in DMSO. The following peptide sequences were synthesized: SVN53-67 peptide DLAQCFFCFKELEGW, SVN53-67/M57 peptide DLAQCFFMFKELEGW, Flu peptide GILGFVFTL, HPV18-27 FLPSDKFPSV (FITC-K23), and GP100 209-217 ITDQVPFSV. An ovalbumin peptide (OVA258-265) with the sequence SIINFEKL was used as a negative control in some experiments. Keyhole Limpet Hemocyanin (KLH) was conjugated to SVN53-67/M57 by maleimide coupling to N-terminal cysteine.
Detection of survivin in tumors
Human and murine tumors were removed and immediately snap frozen in liquid nitrogen. RNA was extracted from frozen human gliomas and purified with an RNAeasy kit (Qiagen, Valencia, CA, USA). Oligodeoxyribonucleotide primers to the human survivin gene (GCATGGGTGCCCCGACGTG as a forward primer and TCCGGCCAGAGGCCTCAATCCAT as a reverse primer) were used in reverse transcription polymerase chain reaction (RT–PCR) detection of transcript using a Superscript III RT-PCR kit (Invitrogen, Carlsbad, CA, USA). For immunohistochemistry of murine tumors, frozen tissues were sectioned at 12 μ and sections were air-dried and fixed in acetone/chloroform for 10 min. Non-specific binding was blocked with sera specific for secondary antibodies and stained using antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) to survivin (FL-142) overnight at 4°C. Sections were washed 3 times with PBS. Labeled secondary antibodies (BioLegend, San Diego, CA, USA) were incubated with sections for 1 h at 27°C, followed by three washes with PBS. Detection was performed utilizing a Nikon Eclipse fluorescence microscope.
Intracerebral GL261 tumor cell injection and survival analysis
Male C57BL/6 mice were anesthetized with isofluorane and fixed in a stereotactic head frame (David Kopf Instruments, Tujunga, CA). A midline scalp incision was made and the bregma was identified. Stereotactic coordinates were measured (2.0 mm lateral, and 1.2 mm anterior to the bregma) for implantation of cells into the deep frontal white matter. A burr hole was drilled at this point and 1 × 105 GL261 cells suspended in 5 μl of DMEM were injected through a Hamilton syringe with a fixed, 25-gauge needle at a depth of 3.0 mm relative to the dura mater. Injections were performed at 2.5 μl/min. The needle was withdrawn and the incision sutured. Kaplan–Meier survival plots were drawn and median survival times were determined for all groups. Survival differences were assessed for significance using the Mantel–Cox method.
Immunization of mice
Proof of principle studies in mice were performed with SVN53-67/M57-KLH in incomplete Freund’s adjuvant (IFA) with 100 ng GM-CSF both given s.c. locally and with a peptide loaded DC2.4 dendritic cells as well. DC2.4 cells were maintained via in vitro cell culture. After 4 days of tumor implantation, 1 × 107 DC2.4 cells were pulsed with 100 μg of peptide (SVN53-67/M57) at 37°C for 2 h. Cells were washed and re-suspended in PBS, 1 × 106 peptide-loaded DCs were injected s.c. into C57BL/6 mice. This inoculation was repeated as a booster vaccination 7 and 14 days later. Mice receiving SVN53-67/M57 as a KLH conjugate were injected at the same time point as DC treated mice, receiving 100 μg of peptide emulsified in IFA with 100 ng of GM-CSF also injected s.c. in close proximity to the vaccination site.
Magnetic resonance imaging
Experimental imaging studies were carried out in a 4.7 T, 33-cm horizontal bore magnet (GE NMR Instruments, Fremont, CA) as described [10]. T2-weighted fast spin echo images were acquired on coronal and axial planes to determine the presence and size of established cerebral tumors [10].
Peptide binding
Computer predictions of peptide binding were determined using the “SYFPEITHI” program [11]. Binding predictions are based on published motifs (pool sequencing, natural ligands) which take into consideration the amino acids in the anchor and auxiliary anchor positions, as well as other frequent amino acids. Peptides were chosen for studies based upon predicted strong binding to HLA-A*0201. Binding affinity determinations of altered survivin peptides were performed as described by Dionne et al. [12]. The affinity determination of SVN 53-67/M57 for HLA-A*0201 was performed utilizing competitive peptide displacement assays. As much as 1 × 107 T2 cells were cultured in the absence of serum for 16 h at 27°C. The peptide HPV18-27/FITC-23 (1 μg/ml), which was used as a standard MHC class I binding reference peptide, was added to 2.5 × 105 T2 cells with recombinant β2m (2.0 μg/ml) in 200 μl RPMI and incubated with varying concentrations of SVN 53-67/M57, SVN 53-67, Flu or gp100 peptides at 27°C in a 5% CO2 incubator for 16–18 h. Cells were washed and analyzed by flow cytometry using a FACScan flow cytometer (BD Biosciences, Mountain View, CA). Mean Fluorescence Intensity (MFI) was used to measure the displacement of HPV-FITC bound to HLA-A*0201 molecules on T2 cells by individual peptides. Percent displacement was calculated as: [1−{(MFI T2 + HPV-FITC + SVN) – (MFI T2 +β2m)}/{(MFI T2 + HPV-FITC) – (MFI T2 + β2m)}] × 100. The IC50 of each peptide was determined by calculating the concentration of peptide required to displace HPV-FITC from sites on T2 cells by 50%.
Autologous and allogeneic target cells
Autologous primary glioma, PCNSL, and CLL cells were used as targets in CTL assays. Other established cell lines were used as allogeneic targets, including U87 human glioma cells (HLA-A*0201), which were obtained from ATTC. Primary cultures of human glioma cells with HLA-A*0301, HLA-A*0201 alleles were derived from human glioma surgical specimens. Primary cell cultures were HLA-typed using high-resolution PCR (A locus SSP Unitray) (Invitrogen, Carlsbad, CA, USA). Fresh tumor specimens were washed with sterile PBS, cut in to small pieces using sterile blades and treated with 0.25% trypsin for 10 min to obtain a cell suspension. Cells were seeded in plastic flasks containing culture medium and maintained in a humidified atmosphere of 5% CO2 at 37°C. All cell cultures were maintained in Dulbecco’s modified Eagle’s minimal essential medium (DMEM) with 10% fetal calf serum (FCS) in a humidified atmosphere of 5% CO2 at 37°C.
Dendritic cells and cytotoxic T lymphocytes
Human and murine PBMC were collected from whole blood and isolated via Accuspin Ficoll-Paque gradient column (Sigma) centrifugation. A portion of the human PBMC was cultured and used for dendritic cell differentiation. A second portion was cryopreserved for CTL production. Human dendritic cell cultures were differentiated with 20 ng/ml GM-CSF (Genscript, NJ) and 10 ng/ml IL-4 (RND systems) for 5 days, after which specific peptide and CD40L was added to induce maturation overnight. DC production was monitored by immunophenotyping for CD11b, CD11c, CD80, CD86, CD40, MHC class I, and class II expression. Cryopreserved human PBMC were thawed and added to DC cultures to differentiate into CTL in the presence of human IL-2 (20 ng/ml; RND systems). In contrast, PBMC from vaccinated mice were directly re-stimulated with specific peptide the presence of murine IL-2 (20 ng/ml) (RND Systems, Minneapolis, MN, USA) in vitro. After 10 days in culture, PBMC were harvested and utilized as described below.
CTL assays
Measurements of cytotoxic T-cell activity were performed on human and murine cultured PBMC using similar methods. After 10 days, CTLs were added to target (glioma, PCNSL, or CLL) cells in culture to assess cell killing ability. Target cells were labeled with DiOC18 (green fluorescence), washed and resuspended in DMEM. Next, target cells (DiOC18-labeled) were added to CTL in ratios ranging from 5:1 to 40:1 (effector:target cells) for 2 h. Conditions were also established to assay background, spontaneous membrane integrity loss and maximal lysis, as well as controls. Propidium iodide was added to each tube to label cells with permeated cell membranes red. Cells were analyzed by flow cytometry (FACScan flow cytometer, BD Biosciences, Mountain View, CA) and analysis was based upon gating on labeled target cells to eliminate background from effector cells. Spontaneous cytotoxicity represents target cells incubated in the absence of effector cells. Flu and gp100 peptide stimulation are positive controls. Maximum release is determined by ethanol treatment and percentage cytotoxicity is calculated using the formula: (experimental PI-DiOC18 cells − spontaneous)/(maximum PI-DiOC18 − spontaneous PI-DiOC18) × 100. Data were analyzed using FCS Express software. Data represent mean percent specific lysis ± S.E.M. of triplicate samples.
Direct HLA-A*0201 pentamer binding
We used MHC class I pentamers specific to wild type and mimic survivin peptide epitopes to assess the presence of survivin-specific T cells generated ex vivo by peptide-loaded DC stimulation, as described above. CD8 + T cells were expanded following ex vivo stimulation with SVN53-67/M57 of PBMC from tumor patients. Pro5® MHC class I pentamers (ProImmune, Bradenton, FL, USA) were designed to recognize T-cell receptors that bind the core CTL epitopes SVN56-64wt and SVN56-64/M57. Survivin-specific R-PE-labeled pentamers were incubated with PBMC for 10 min at 22°C. Samples were incubated with anti-CD8 FITC (clone LT8) for 20 min at 4°C. Cells were fixed for analysis by flow cytometry. CD8+ cells that were doubly labeled were considered positive for pentamer if >5% over baseline. Data were analyzed using FCS Express software. Analysis was based upon gating on CD8+ T cells only.
Cytokine production
CTL of glioma patients were derived as described above and assessed for specific cytokine production in response to SVN53-67/M57 and SVN53-67 stimulation in vitro. An IFNγ ELISArray kit (SA Biosciences, Frederick, MD) was used to analyze culture medium samples from days 2 to 14, according to the manufacturer’s instructions. Intracellular cytokine measurement and analysis in murine cells were performed as previously described [9]. Mouse Cytokine Intracellular Control Cells (eBioscience, San Diego, CA, USA) were used as positive controls for IFNγ expression.
Results
Survivin target antigen expression
We examined 25 human surgical glioblastoma specimens using RT-PCR for detection of survivin transcripts. Survivin was expressed in over 90% of the tumors. Figure 1a shows a representative sample of 12 human gliomas examined by RT-PCR. Of the 12 tumors screened, all but one (lane 7) had detectable survivin expression. In addition, immunofluorescence studies (Fig. 1b–c) of GL261 glioma cells growing in the brains of mice demonstrate that survivin is expressed throughout the tumor (Fig. 1c).
Fig. 1.

a RT–PCR of human glioma specimens for survivin expression. Frozen sections from intracranial GL261 tumors were analyzed for survivin expression by direct immunofluorescence. b Isotype control mAb; c anti-survivin mAb on glioma showing high survivin expression levels (Images were captured at 400× magnification)
Peptide selection for survivin vaccine
To select candidate peptides for a survivin vaccine, we searched the survivin molecule for sequences that would be potentially immunogenic in humans with HLA-A*0201 phenotype (Fig. 2). In order to provide a relevant mouse model for tumor immunotherapy, we preferentially considered peptides with epitopes that could also bind H2-Kb. Next, we tested candidate peptides spanning the survivin molecule for antitumor effects against GL261 murine gliomas grown orthotopically in immunocompetent, syngeneic C57BL/6 mice. Of 15 candidates, one peptide (SVN57-64) provided a distinct survival advantage [10]. Therefore, we chose to focus on this site in the molecule for further investigation. Because the SVN57-64 peptide does not exhibit strong HLA-A*0201 binding in humans it might be expected to be weakly immunogenic. Therefore, we used reverse immunology to consider the effects of various peptide alterations on binding and immunogenicity. We designed 12 peptides with various amino acid substitutions at anchor positions in an effort to optimize both factors. One peptide, containing a C-to-M (cysteine to methionine) amino acid substitution at position 57 (i.e. M57) resulted in greatest median survival in the orthotopic murine glioma model (data not shown).
Fig. 2.

Survivin peptide mimic vaccine amino acid sequence and subsequently derived MHC class I binding epitopes. Additionally, the SVN53-67/M57 15-mer is predicted to bind the following MHC class II alleles: HLA-DRB1*0301 (DR17), HLA-DRB1*0401 (DR4Dw4), HLA-DRB1*0701 and HLA-DRB1*1501 (DR2b)
We expanded upon this strategy based on the M57 substitution by synthesizing a larger peptide (15 AA) to encompass multiple MHC class I-binding epitopes for CD8+ T-cell (CTL) generation (Fig. 2). This peptide mimic retains the altered amino acid position in each of the CTL epitopes. Importantly, the resulting peptide mimic (SVN53-67/M57) was designed to be large enough so that it could also serve as an MHC class II binding ligand (Fig. 2) to stimulate survivin-specific CD4+ helper support.
Peptide binding to MHC class I molecules
We performed indirect MHC class I binding analysis using the core peptide epitope contained within the SVN53-67/M57 peptide mimic (Fig. 3a). To assess binding affinity of the mimic peptides, we used human T2 cells, which present empty HLA-A*201 molecules at their cell surface. We loaded the HLA-A*201 molecules on these cells with a fluorescently labeled HPV-18-27 MHC class I ligand for use in competitive displacement assays. Figure 3a shows that the survivin mimic peptides (SVN56-64/M57 and SVN53-67/M57) are capable of displacing HPV at low concentrations, similar to that observed with other known MHC class I ligands, such as gp100/209-217 and Flu58-66. In contrast, the wild-type peptides (SVN56-64 and SVN53-67) displaced HPV peptide at 556 and 1,176-fold (respectively) higher concentration than the mimics.
Fig. 3.

a MHC class I peptide competitive displacement assays. HLA-A*0201 binding of survivin peptides based upon the core SVN56-64 peptide. IC50 represents concentration of survivin peptide required to displace HPV 18-27 (known MHC I ligand) pre-loaded on human T2 cells. Control peptides (Flu and gp100) are used as known immunogenic MHC class I ligands. b Pentamer binding assay of SVN56-64 wild type and SVN56-64/M57 mimic peptides showing differential binding affinity of each peptide for MHC class I
We also used a second specific assay with synthetic pentamers to compare direct binding of the mimic and wild-type peptides to HLA-A*0201 (Fig. 3b). We found that the core peptide mimic (SVN56-64/M57) had 74-fold higher binding to HLA-A*0201 than the wild-type SVN56-64 core peptide. This would be expected to result in a longer association time between the mimic epitopes and the MHC I molecules.
Survival and immune response of immunized mice with gliomas
We examined the efficacy of the SVN53-67/M57 peptide mimic in an immunocompetent, syngeneic, murine intracerebral glioma model. In the present study C57BL/6 mice were immunized with SVN53-67/M57 either as a DC-loaded peptide vaccine, or as a KLH-conjugate, beginning 4 days after tumor implantation. Vaccinations were repeated (booster) every 7 days for a total of 3 doses. Kaplan–Meier survival plots were drawn and median survival times were determined for all groups. Figure 4a provides a graphical summary of data showing survival of mice with intracerebral GL261 gliomas. Mice vaccinated with the survivin peptide mimic survived significantly longer than control mice, with some long-term survivors living beyond 120 days (Fig. 4a). MeST: Control DC = 19.5 days; Ova DC = 21 days; SVN53-67/M57-KLH = 52.5 days; SVN53-67/M57-DC = 55 days. Survival of SVN53-67/M57-KLH and SVN53-67/M57-DC vaccinated mice was significantly different from control DC vaccinated mice, p < 0.0001, by Mantel–Cox analysis. Survival of SVN53-67/M57-KLH and SVN53-67/M57-DC vaccinated mice was not significantly different between vaccine delivery modalities, p = 0.6740. Long-term survivors were confirmed to be tumor-free by high field strength MRI (Fig. 4b). CTL responses against GL261 glioma cells were observed in culture using splenocytes from mice vaccinated with SVN53-67/M57; however, the core peptide mimic SVN56-64/M57 did not generate as robust CTL response (Fig. 4c). Intracellular IFNγ production specifically by CD4+ cells also increased significantly (Fig. 4d) following SVN53-67/M57 immunization, indicative of a class II response with the larger peptide only.
Fig. 4.

a Survival of C57BL6 mice with intracerebral GL261 glioma implants. Mice received intracerebral implants of 1 × 105 GL261 cells, followed by vaccination beginning 4 days after tumor implantation to simulate a therapeutic setting. Vaccinations were repeated every 7 days up to a total of 3 immunizations. b High field strength MRI of glioma-implanted C57BL/6 mice without (left) and with (right) peptide mimic vaccination. c CTL responses against GL261 glioma cells obtained in vitro using the splenocytes of mice vaccinated with SVN53-67/M57. d Intracellular IFN-γ production in vitro of CD4+ cells from mice in response to SVN53-67/M57 immunization. Mouse cytokine intracellular control cells (ebiosciences) were used as positive controls for IFNγ expression
Human studies of the SVN53-67/M57 peptide mimic
Using fresh PBMC from glioma and PCNSL patients to produce dendritic cells (DC) in vitro, we assessed the ability of survivin peptides to stimulate CTL responses against allogeneic and autologous human tumor cells ex vivo. The survivin peptide mimic produces a more potent tumor lytic response than the wild type survivin peptide. An HLA mismatch of PBMC and tumor led to an abrogation of cell-mediated cytotoxicity (Fig. 5a). As observed in H2-Kb mice with GL261 gliomas, lymphocytes from HLA-A*0201 tumor patients were able to be stimulated to lyse allogeneic HLA-A*0201 glioma cells (Fig. 5b). In addition, we observed that the SVN53-67/M57 vaccine stimulated CTL lytic responses in samples from HLA-A*0301 patients (Fig. 5c). Patient PBMC also effectively lysed autologous tumor cells (Fig. 5d–f). In addition to observing cytotoxic activity against malignant gliomas, we also detected CTL activity induced by the mimic peptide against autologous (HLA-A*2901/A*3002) CNS lymphoma cells (Fig. 5e) and chronic lymphocytic leukemia (CLL) cells (HLA-A*01/A*24) (Fig. 5f). Thus, SVN53-67/M57-stimulated DC not only produced a lytic response to several different MHC I haplotypes, but it also produced a lytic response to a variety of different tumor types. One HLA-A*11 patient’s cells were not reactive to the peptide mimic (data not shown).
Fig. 5.

Ex vivo cytotoxic T lymphocyte (CTL) responses against allogeneic and autologous human glioma, PCNSL and CLL cells to assess specificity and efficacy of cell killing toward tumor cells displaying MHC class I contextualized survivin peptides. a HLA-A*0301 patient effector cells versus allogeneic-mismatched HLA-A*0201 human U87 target glioma cells; b HLA-A*0201 patient effector cells versus allogeneic-matched U87 glioma cells; c HLA-A*0301 patient effector cells versus allogeneic-matched patient-derived tumor cells; d HLA-A*0301/HLA-A*2901 patient effector cells versus autologous patient-derived glioma cells. e HLA-A*2901/HLA-A*3002 patient effector cells versus autologous patient-derived PCNSL target cells; f HLA-A*01/HLA-A*24 patient effector cells versus autologous patient-derived CLL target cells. Legend: Control DC = Flu/58-66; Positive control = gp100/209-217; SVN wt = SVN53-67; SVN Mimic = SVN53-67/M57
Cytokine support in mice and humans
IFNγ was measured in murine and human PBMC derived cells (Fig. 6). Initial murine studies of CD4+ T cells stimulated by SVN53-67/M57 was performed using intracellular cytokine FACS analysis (Fig. 4d). Subsequent human IFNγ secretion was measured via ELISA of cell culture supernatant from HLA-A*0201 glioma patient PBMC’s stimulated with SVN53-67 or SVN53-67/M57 in vitro, over 14 days (Fig. 6). IFNγ secretion progressively increased in cultures stimulated with the SVN53-67/M57 peptide mimic relative to unstimulated PBMC, indicative of cytokine support. Wild type SVN53-67 peptide resulted in only modest IFNγ secretion.
Fig. 6.

IFNγ accumulation in culture medium as measured by ELISA. PBMC were stimulated by indicated peptide-loaded DC in culture. Medium was analyzed on days 2, 10 and 14 and results are displayed as pg/ml detected in culture medium. Control DC are non-specific control stimulated DC and T-cell control represents wells of non-stimulated T cells used to assess background. Results are mean values of triplicate wells ± SEM. The figure shown is representative of three patient experiments
T-cell specificity
We used HLA-A*0201 pentamers loaded with the core CTL epitope of SVN 53-67 (either SVN56-64 or SVN56-64/M57) to examine T-cell specificity (Fig. 7). Using fresh PBMC from an HLA-A*0201 glioma patient to produce DC in vitro, we assessed the ability of survivin peptides to stimulate specific T cells. FACS analysis of SVN53-67/M57-stimulated PBMC show a significant increase in the number of survivin-specific CD8+ T cells (Fig. 7a). In addition, the survivin-specific CD8+ T cells were cross-reactive to both SVN56-64 wild type and SVN56-64/M57 pentamers (Fig. 7a). The cross-reactivity observed is indicative that T cells stimulated with SVN56-64/M57 also recognize wild-type survivin epitopes. Corresponding CTL assay data for patient T cells used in Fig. 7a are shown in Fig. 7b confirming lytic activity toward autologous glioma target cells.
Fig. 7.
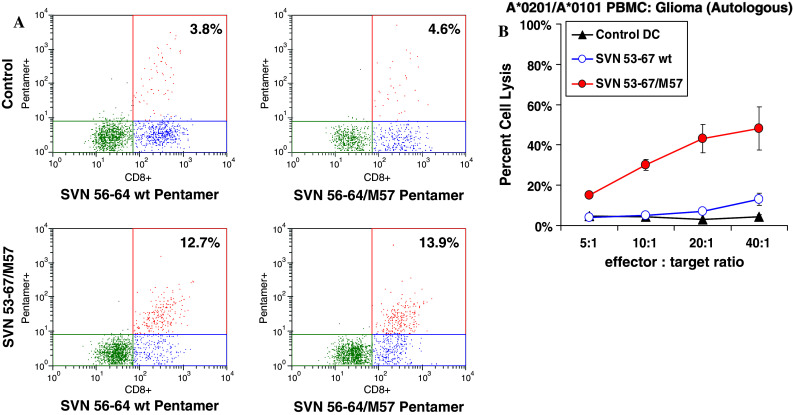
Pentamer binding from patient-derived CTL generation showing that SVN53-67/M57 peptide mimic can produce CD8+ T cells that are reactive to wild-type peptide. a HLA-A*0201 pentamer binding CD8+ T cells specific for SVN56-64/M57 peptide mimic as a result of indicated antigen stimulation. Percentage shown is relative to all cells analyzed from restimulated cultures. b Corresponding CTL assay data for patient cells used in (a)
Discussion
Survivin is an intracellular protein that belongs to the inhibitor of apoptosis protein (IAP) family. It has been shown to modulate the function of a number of terminal effector cell death proteases leading to an inhibition of apoptosis [1, 3, 13]. Although expressed during fetal development, survivin is rarely detectable in the normal tissues of adult organisms [4]. Malignant gliomas express survivin at high levels; whereas, normal glial cells do not [5–7]. Survivin is highly expressed in many other tumors as well, including prostate [13], endometrial [14], ovarian [15] and breast [16] carcinoma, and in multiple myeloma [17] and leukemias [18]. Consequently, survivin is recognized as a specific tumor target for cancer immunotherapy.
Circulating anti-survivin antibodies have been detected in patients with cancer, but not in normal volunteers [8]. Since survivin is an intracellular protein, anti-survivin antibodies are not likely to be clinically beneficial in treating cancer patients. We have also detected anti-survivin antibodies in a number of glioma patients (unpublished data). Thus, many cancer patients may already be primed to survivin antigens. In addition to anti-survivin antibodies, some cancer patients have survivin-specific CTL [19]. For example, monitoring of T-cell reactivity against survivin-derived peptides in one melanoma patient in complete remission following non-specific IL-2 based immunotherapy identified naturally occurring functional CTL capable of recognizing survivin [20]. Low levels of MHC class I-restricted CTL directed against survivin peptides have also been identified in patients with breast cancer, leukemia and melanoma [19, 21]. Thus, the immune system of many cancer patients is often primed to recognize survivin, perhaps as a result of exposure to the survivin protein liberated by dying tumor cells. This may enable a specific immunotherapeutic approach to enhance and re-direct the existing immune response against the tumor.
T-cell clones with the capacity to be activated by self-proteins are frequently preserved following negative selection of higher affinity, self-recognizing clones in the thymus [22]. These potentially self-reactive cells ordinarily remain tolerized [23]. Altered peptide ligands (mimics) can provide a way to break tolerance to the natural self-epitope [24, 25]. Altered peptide ligands generated by substituting single amino acids within a peptide epitope can markedly affect immune responses. Specifically, substitution of binding anchor residues may be used to increase the affinity of the peptide mimic for MHC-I molecules [26–29]. Effects of this manipulation can range from the induction of TCR antagonism, to T-cell anergy, to enhancement of T-cell responses [30, 31]. Based on its strong binding and immunogenicity relative to the wild-type survivin peptide (SVN53-67), the survivin peptide mimic (SVN53-67/M57) should stimulate lower affinity T-cell receptors, which have not been deleted during development. This would be expected to induce the proliferation of cross-reactive T-cell clones that recognize wild type survivin epitopes presented by tumor cells [5, 9, 22, 23, 25, 29].
The antigen binding groove of MHC class I molecules normally accept peptide epitopes that are 8–10 amino acids in length. Occasionally, longer peptide epitopes can be accommodated, resulting in peptide bulging from the MHC cleft. The bulging peptide may facilitate an interaction between receptors that might not otherwise occur [32, 33]. It is possible that one or more processed forms of the survivin peptide mimic could undergo this type of interaction, in part accounting for enhanced immunogenicity of the mimic.
The generation of an effective and specific cell-mediated immune response may require the activation of both highly specific CD8+ lymphocytes and CD4+ helper T cells. The presence of MHC class II-restricted CD4+ T cells that are specific for tumor-associated antigens has been recognized to provide helper factors in eliciting and sustaining cytotoxic CD8+ T-cell responses to tumors [34, 35]. Attempts to maximize the immune response to tumors may be more effective if they include efforts to elicit T-helper cell support. A peptide vaccination strategy combining CTL and tumor-specific helper support provides an avenue of investigation that is distinct from previous efforts. We believe that this is an approach that has not been given sufficient attention previously in anticancer peptide vaccine trials. The survivin peptide mimic outlined here makes use of this strategy to induce anti-tumor CTL and specific T helper cell support as well.
While the SVN53-67/M57 peptide was designed to bind HLA-A*0201, SYPEITHI analysis and empirical data indicate that the peptide mimic binds to a number of other MHC class I molecules and stimulates a CTL response. Collectively, this represents a highly diverse patient population in which the vaccine might prove immunogenic (Table 1). Also, our studies have revealed no significant advantage to using either peptide-loaded DC or a peptide-KLH conjugate, provided that GM-CSF is used with the conjugate as an attractant and maturant for DC. This is a potentially important observation since it would simplify the development of the peptide mimic for clinical use.
Table 1.
Class I alleles predicted to be capable of binding the survivin peptide mimic via computer analysis (SYPEITHI)
| MHC class I | |
|---|---|
| *HLA-A*02 | HLA-B*0702 |
| *HLA-A*03 | HLA-B*08 |
| HLA-A*1101 | HLA-B*1402 |
| HLA-A*24 | HLA-B*1501 |
| HLA-A*26 | HLA-B*1510 |
| *HLA-A*29 | HLA-B*18 |
| *HLA-A*30 | HLA-B*2705 |
| HLA-A*6801 | HLA-B*2709 |
| HLA-B*3901 | |
| HLA-B*4402 | |
| HLA-B*5101 | |
* Indicates alleles confirmed empirically to be associated with antitumor immune responses
As compared to the wild-type peptide, SVN53-67/M57 elicits a 3- to 5-fold increase in CTL-mediated killing against allogeneic HLA-matched human glioma cells, autologous human glioma cells and autologous human CNS lymphoma cells. SVN53-67/M57 can elicit a cell-mediated immune response that is significantly greater than that induced by the wild-type peptide. Thus, SVN53-67/M57 can elicit a cell-mediated immune response against different types of survivin-expressing human cancer cells. Importantly, tumor cell killing is achieved through SVN53-67/M57-stimulated T-cell recognition of naturally processed survivin epitopes presented by tumor cells.
Several recent clinical immunotherapy trials have shown promising immunological and clinical responses in cerebral glioma patients. A phase I clinical trial of DCVax®-Brain showed 9 out of 19 patients still alive 8–82 months after initial surgery, most with no evidence of tumor recurrence. Phase II studies for this group have already indicated 5 immune responses associated with stable disease in one arm of that study [36, 37]. In a phase II clinical trial of 18 patients receiving CDX-110® (EGFRvIII-KLH conjugated peptide vaccine), median overall survival (OS) was 26 months and median time to tumor progression (TTP) was 14.2 months. In addition, 3 patients remained relapse-free for more than 5 years. In another phase II study of 23 subjects, CDX-110® was given with chemotherapy and median TTP reached 16.6 months with median OS of 33.1 months. Both CDX-110® trials were single arm studies compared to historical controls having median OS of 15.0 months and median TTP of 6.3 months. After CDX-110® vaccination, gliomas that progressed no longer expressed EGFRvIII, indicating that the initial immune response had selected for glioma cells that did not express the target [38, 39].
These examples suggest that immunotherapy has potential utility for the treatment of gliomas. The prospect of having an additional specific and highly expressed, immunological target, such as survivin, for peptide-based immunotherapy could facilitate the development of a multivalent vaccine strategy for these difficult-to-treat tumors.
Acknowledgments
This work was supported by NIH 5R21 NS049309-02 (RAF), NIH 5P30 CA16056-29, the Linda Scime Fund of the Roswell Park Alliance Foundation, and the Glioblastoma Multiforme Grant awarded to MJC by the National Brain Tumor Foundation.
References
- 1.Conway EM, Pollefeyt S, Cornelissen J, DeBaere I, Steiner-Mosonyi M, Ong K, Baens M, Collen D, Schuh AC. Three differentially expressed survivin cDNA variants encode proteins with distinct antiapoptotic functions. Blood. 2000;95:1435–1442. [PubMed] [Google Scholar]
- 2.Islam A, Kageyama H, Takada N, Kawamoto T, Takayasu H, Isogai E, Ohira M, Hashizume K, Kobayashi H, Kaneko Y, Nakagawara A. High expression of Survivin, mapped to 17q25, is significantly associated with poor prognostic factors and promotes cell survival in human neuroblastoma. Oncogene. 2000;19:617–623. doi: 10.1038/sj.onc.1203358. [DOI] [PubMed] [Google Scholar]
- 3.Shin S, Sung BJ, Cho YS, Kim HJ, Ha NC, Hwang JI, Chung CW, Jung YK, Oh BH. An anti-apoptotic protein human survivin is a direct inhibitor of caspase-3 and -7. Biochemistry. 2001;40:1117–1123. doi: 10.1021/bi001603q. [DOI] [PubMed] [Google Scholar]
- 4.Adida C, Crotty PL, McGrath J, Berrebi D, Diebold J, Altieri DC. Developmentally regulated expression of the novel cancer anti-apoptosis gene survivin in human and mouse differentiation. Am J Pathol. 1998;152:43–49. [PMC free article] [PubMed] [Google Scholar]
- 5.Ciesielski MJ, Apfel L, Barone TA, Castro CA, Weiss TC, Fenstermaker RA. Antitumor effects of a xenogeneic survivin bone marrow derived dendritic cell vaccine against murine GL261 gliomas. Cancer Immunol Immunother. 2006;55:1491–1503. doi: 10.1007/s00262-006-0138-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chakravarti A, Noll E, Black PM, et al. Quantitatively determined survivin expression levels are of prognostic value in human gliomas. J Clin Oncol. 2002;20:1063–1068. doi: 10.1200/JCO.20.4.1063. [DOI] [PubMed] [Google Scholar]
- 7.Kajiwara Y, Yamasaki F, Hama S, et al. Expression of survivin in astrocytic tumors: correlation with malignant grade and prognosis. Cancer. 2003;97:1077–1083. doi: 10.1002/cncr.11122. [DOI] [PubMed] [Google Scholar]
- 8.Rohayem J, Diestelkoetter P, Weigle B, et al. Antibody response to the tumor-associated inhibitor of apoptosis protein survivin in cancer patients. Cancer Res. 2000;60:1815–1817. [PubMed] [Google Scholar]
- 9.Ciesielski MJ, Kozbor D, Castanaro CA, Barone TA, Fenstermaker RA. Therapeutic effect of a T helper cell supported CTL response induced by a survivin peptide vaccine against murine cerebral glioma. Cancer Immunol Immunother. 2008;57:1827–1835. doi: 10.1007/s00262-008-0510-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seshadri M, Ciesielski M. MRI-based characterization of vascular disruption by 5, 6-dimethylxanthenone-4-acetic acid in gliomas. J Cereb Blood Flow Metab. 2009;29:1373–1382. doi: 10.1038/jcbfm.2009.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rammensee HG, Bachmann J, Emmerich NN, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 12.Dionne SO, et al. Functional characterization of CTL against gp100 altered peptide ligands. Cancer Immunol Immunother. 2003;52:199–206. doi: 10.1007/s00262-002-0358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krajewska M, Krajewski S, Banares S, et al. Elevated expression of inhibitor of apoptosis proteins in prostate cancer. Clin Cancer Res. 2003;9:4914–4925. [PubMed] [Google Scholar]
- 14.Takai N, Miyazaki T, Nishida M, Nasu K, Miyakawa I. Survivin expression correlates with clinical stage, histological grade, invasive behavior and survival rate in endometrial carcinoma. Cancer Lett. 2002;184:105–116. doi: 10.1016/S0304-3835(02)00190-8. [DOI] [PubMed] [Google Scholar]
- 15.Takai N, Miyazaki T, Nishida M, Nasu K, Miyakawa I. Expression of survivin is associated with malignant potential in epithelial ovarian carcinoma. Int J Mol Med. 2002;10:211–216. [PubMed] [Google Scholar]
- 16.Yamashita S, Masuda Y, Kurizaki T, et al. Survivin expression predicts early recurrence in early-stage breast cancer. Anticancer Res. 2007;27:2803–2808. [PubMed] [Google Scholar]
- 17.Andersen MH, Svane IM, Becker JC, Straten PT. The universal character of the tumor-associated antigen survivin. Clin Cancer Res. 2007;13:5991–5994. doi: 10.1158/1078-0432.CCR-07-0686. [DOI] [PubMed] [Google Scholar]
- 18.Oto OA, Paydas S, Tanriverdi K, Seydaoglu G, Yavuz S, Disel U. Survivin and EPR-1 expression in acute leukemias: prognostic significance and review of the literature. Leuk Res. 2007;31:1495–1501. doi: 10.1016/j.leukres.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 19.Andersen MH, Pedersen LO, Capeller B, Brocker EB, Becker JC, thor Straten P. Spontaneous cytotoxic T-cell responses against survivin-derived MHC class I-restricted T-cell epitopes in situ as well as ex vivo in cancer patients. Cancer Res. 2001;61:5964–5968. [PubMed] [Google Scholar]
- 20.Hadrup SR, Gehl J, Sorensen RB, Geertsen PF, Straten PT, Andersen MH. Persistence of survivin specific T cells for seven years in a melanoma patient during complete remission. Cancer Biol Ther. 2006;5:480–482. doi: 10.4161/cbt.5.5.2652. [DOI] [PubMed] [Google Scholar]
- 21.Altieri DC. Validating survivin as a cancer therapeutic target. Nat Rev Cancer. 2003;3:46–54. doi: 10.1038/nrc968. [DOI] [PubMed] [Google Scholar]
- 22.Overwijk WW, Restifo NP. Autoimmunity and the immunotherapy of cancer: targeting the “self” to destroy the “other”. Crit Rev Immunol. 2000;20:433–450. [PMC free article] [PubMed] [Google Scholar]
- 23.Lohr J, Knoechel B, Nagabhushanam V, Abbas AK. T-cell tolerance and autoimmunity to systemic and tissue-restricted self-antigens. Immunol Rev. 2005;204:116–127. doi: 10.1111/j.0105-2896.2005.00241.x. [DOI] [PubMed] [Google Scholar]
- 24.Fikes JD, Sette A. Design of multi-epitope, analogue-based cancer vaccines. Expert Opin Biol Ther. 2003;3:985–993. doi: 10.1517/14712598.3.6.985. [DOI] [PubMed] [Google Scholar]
- 25.Guevara-Patino JA, Turk MJ, Wolchok JD, Houghton AN. Immunity to cancer through immune recognition of altered self: studies with melanoma. Adv Cancer Res. 2003;90:157–177. doi: 10.1016/S0065-230X(03)90005-4. [DOI] [PubMed] [Google Scholar]
- 26.Trojan A, Witzens M, Schultze JL, et al. Generation of cytotoxic T lymphocytes against native and altered peptides of human leukocyte antigen-A*0201 restricted epitopes from the human epithelial cell adhesion molecule. Cancer Res. 2001;61:4761–4765. [PubMed] [Google Scholar]
- 27.Keogh E, Fikes J, Southwood S, Celis E, Chesnut R, Sette A. Identification of new epitopes from four different tumor-associated antigens: recognition of naturally processed epitopes correlates with HLA-A*0201-binding affinity. J Immunol. 2001;167:787–796. doi: 10.4049/jimmunol.167.2.787. [DOI] [PubMed] [Google Scholar]
- 28.Valmori D, Fonteneau JF, Lizana CM, et al. Enhanced generation of specific tumor-reactive CTL in vitro by selected Melan-A/MART-1 immunodominant peptide analogues. J Immunol. 1998;160:1750–1758. [PubMed] [Google Scholar]
- 29.Parkhurst MR, Salgaller ML, Southwood S, et al. Improved induction of melanoma-reactive CTL with peptides from the melanoma antigen gp100 modified at HLA-A*0201-binding residues. J Immunol. 1996;157:2539–2548. [PubMed] [Google Scholar]
- 30.Evavold BD, Sloan-Lancaster J, Allen PM. Tickling the TCR: selective T-cell functions stimulated by altered peptide ligands. Immunol Today. 1993;14:602–609. doi: 10.1016/0167-5699(93)90200-5. [DOI] [PubMed] [Google Scholar]
- 31.Loftus DJ, Squarcina P, Nielsen MB, et al. Peptides derived from self-proteins as partial agonists and antagonists of human CD8+ T-cell clones reactive to melanoma/melanocyte epitope MART1(27-35) Cancer Res. 1998;58:2433–2439. [PubMed] [Google Scholar]
- 32.Tynan FE, Burrows SR, Buckle AM, et al. T cell receptor recognition of a ‘super-bulged’ major histocompatibility complex class I-bound peptide. Nat Immunol. 2005;6:1114–1122. doi: 10.1038/ni1257. [DOI] [PubMed] [Google Scholar]
- 33.Tynan FE, Reid HH, Kjer-Nielsen L, et al. A T cell receptor flattens a bulged antigenic peptide presented by a major histocompatibility complex class I molecule. Nat Immunol. 2007;8:268–276. doi: 10.1038/ni1432. [DOI] [PubMed] [Google Scholar]
- 34.Pardoll DM. Inducing autoimmune disease to treat cancer. Proc Natl Acad Sci USA. 1999;96:5340–5342. doi: 10.1073/pnas.96.10.5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The central role of CD4(+) T cells in the antitumor immune response. J Exp Med. 1998;188:2357–2368. doi: 10.1084/jem.188.12.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim W, Liau LM. Dendritic cell vaccines for brain tumors. Neurosurg Clin N Am. 2010;21:139–157. doi: 10.1016/j.nec.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wheeler CJ, Black KL. DCVax-Brain and DC vaccines in the treatment of GBM. Expert Opin Investig Drugs. 2009;18:509–519. doi: 10.1517/13543780902841951. [DOI] [PubMed] [Google Scholar]
- 38.Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Herndon JE, 2nd, Lally-Goss D, McGehee-Norman S, Paolino A, Reardon DA, Friedman AH, Friedman HS, Bigner DD. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol Cancer Ther. 2009;8:2773–2779. doi: 10.1158/1535-7163.MCT-09-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heimberger AB, Sampson JH. The PEPvIII-KLH (CDX-110) vaccine in glioblastoma multiforme patients. Expert Opin Biol Ther. 2009;9:1087–1098. doi: 10.1517/14712590903124346. [DOI] [PMC free article] [PubMed] [Google Scholar]