

Summary
The unfolded protein response (UPR) is a eukaryotic signaling pathway linking protein flux through the endoplasmic reticulum to transcription and translational repression. Herein, we demonstrate UPR activation in the leukodystrophy Pelizaeus-Merzbacher disease (PMD) as well as in three mouse models of this disease and transfected fibroblasts expressing mutant protein. The CHOP protein, widely known as a proapoptotic transcription factor, modulates pathogenesis in the mouse models of PMD; however, this protein exhibits antiapoptotic activity. Together, these data show that the UPR has the potential to modulate disease severity in many cells expressing mutant secretory pathway proteins. Thus, PMD represents the first member of a novel class of disparate degenerative diseases for which UPR activation and signaling is the common pathogenic mechanism.
Introduction
Of the myriad physiological processes ongoing in all living organisms, few are more important to survival at the levels of the cell and the organism than adaptation to stress. Primordial lifeforms contended with ever changing environmental conditions by rapid changes at the transcriptional and posttranscriptional levels, and since that time, virtually all species have retained and expanded such rudimentary signaling cascades to sustain life under adverse conditions. The unfolded protein response (UPR) is a stress-induced signaling cascade in eukaryotes, and recent appreciation of its importance as a central regulatory circuit in the secretory pathway has prompted broad interest in consequences to cells should the UPR be disrupted or overwhelmed (Travers et al., 2000). Indeed, correlative evidence implicates the UPR in a number of genetic diseases (Aridor and Balch, 1999). For example, mutations in the PRESENILIN gene from familial Alzheimer patients cause abnormal IRE1 processing (Katayama et al., 1999; Niwa et al., 1999). In this instance, the molecular defect alone does not compromise UPR induction (Sato et al., 2000) but, conceivably, may sensitize neurons to other insults.
We have investigated molecular pathogenesis of the leukodystrophy Pelizaeus-Merzbacher disease (PMD), a neurodegenerative disease causing diffuse hypomyelination of the central nervous system (CNS). PMD is an X-linked recessive pediatric disorder characterized by three common genetic forms of disease: coding region or splice site mutations, duplications of the wild-type PLP1 gene, and null alleles. These mutations yield a broad spectrum of disease phenotypes from severe, connatal disease to mild forms characterized by pure spastic paraparesis (reviewed in Garbern et al., 1999; Southwood and Gow, 2001). Mutant alleles that model all three of these genetic forms of PMD are available in mice, including (1) myelin synthesis-deficient (msd), an A242V missense mutation causing severe disease, also identified in patients (Gencic and Hudson, 1990; Yamamoto et al., 1998) and rumpshaker (rsh), an I186T missense mutation causing mild disease, also found in patients (Kobayashi et al., 1994; Schneider et al., 1992); (2) 4e-Plp, comprising supernumerary copies of a 40 kb wild-type Plp1 transgene causing overexpression (Kagawa et al., 1994); and (3) Plp1 null (Klugmann et al., 1997; Rosenbluth et al., 1996; Stecca et al., 2000). Disease in msd mice is apparent by 12 days postnatal (P12) as moderate tremors, which rapidly progress through severe tremors to seizures causing death by 3–4 weeks. On the other hand, rsh mice develop symptoms by P14–16, exhibit moderate tremors and ataxia, but rarely exhibit seizures and have a normal life span (Fanarraga et al., 1992; Griffiths et al., 1990). Disease severity caused by PLP1 gene duplication is proportional to the degree of overexpression (Ikenaka and Kagawa, 1995), and null alleles cause mild disease.
Molecular mechanisms underlying the phenotypes of most PLP1 mutations are virtually unknown. Morphological analyses of oligodendrocytes from animal models indicate that distinct cellular defects underlie pathogenesis of different genetic forms of PMD. For example, several missense mutations perturb endoplasmic reticulum ultrastructure (Duncan, 1990); PLP1 overexpression causes swelling of the Golgi apparatus and aberrant cholesterol trafficking (Kagawa et al., 1994; Readhead et al., 1994; Simons et al., 2002); and null alleles cause axonal abnormalities with late Wallerian degeneration and oligodendrocyte loss (Griffiths et al., 1998; Stecca et al., 2000).
Herein, we investigate involvement of the UPR in pathogenesis arising from PLP1 coding region mutations and demonstrate that several mutations activate this signaling cascade. CHOP is induced in and localized to the nuclei of transfected fibroblasts expressing mutant PLP1, cultured oligodendrocytes from msd mice, and oligodendrocytes from rsh and msd mice expressing mutant Plp1 gene products in vivo. Moreover, we show by Northern blotting and immunocytochemistry that CHOP is induced in oligodendrocytes from a PMD patient harboring a splice site mutation that excludes exon 6 from PLP1 gene products. Finally, we directly demonstrate that CHOP modulates disease severity in rsh mice and normally serves to protect oligodendrocytes from apoptosis. Together, these data provide compelling evidence that the UPR is relevant to pathogenesis in PMD, thereby attesting to the importance of this regulatory pathway in disease. More broadly, suppositions by others that mutant secretory pathway proteins cause disease in different cell types through activation of the UPR portends PMD as the first example of a novel class of disparate degenerative diseases that nevertheless arise from a common molecular mechanism.
Results
The UPR has been examined in yeast and cultured mammalian cells following induction by noxious stimuli such as tunicamycin to inhibit glycoprotein synthesis, thapsigargin to mobilize internal Ca2+ stores and expression of mutant secretory pathway proteins that accumulate in the endoplasmic reticulum (ER). Currently, the UPR is known to be activated through posttranslational processing of three ER-resident proteins, the Ire1 and PERK receptor kinases and the membrane-tethered transcription factor ATF6. These initiating proteins regulate translation of mRNAs encoding the XBP-1 and ATF-4 transcription factors and the expression of effector genes, including molecular chaperones and additional transcription factors such as CHOP and ATF3 (reviewed in Ma and Hendershot, 2001; Zinszner et al., 1998). Given the complexity of the UPR and our incomplete knowledge of this signaling cascade, we contend that the most reliable reporters of a UPR “in progress” are the effector molecules. In this regard, CHOP is downstream of PERK, BiP is downstream of Ire1, while Erp59 and Erp72 are downstream of ATF6 (Harding et al., 2000; Ma et al., 2002; Okada et al., 2002).
CHOP Is Expressed in Fibroblasts Transfected with Mutant PLP1 Constructs
To determine if the UPR is induced in vitro by mutant forms of PLP1 gene products, we transiently transfected COS-7 cells with plasmids encoding either wild-type PLP1 or the missense mutant from msd mice (PLP1msd). At 24 hr, essentially all transfected cells expressing PLP1msd localize this protein in the ER (green staining in Figure 1Aa) as demonstrated previously (Gow et al., 1994a, 1994b). Most transfected cells also express CHOP (white/pink staining, arrowheads), which colocalizes with the nuclear stain DAPI (blue). In contrast, the overwhelming majority of nontransfected cells are CHOP negative, indicating that the experimental procedure alone does not induce this gene. Transfected cells that express wild-type PLP1 transport this protein to all major compartments of the secretory and endocytic pathways (arrows), as shown previously (Gow et al., 1994a), but the vast majority of these cells are CHOP negative (Figure 1Ab).
Figure 1. The UPR Is Induced in Cells Expressing Mutant PLP1.

(A) Transfected COS-7 cells expressing mutant (Aa) but not wild-type (Ab) PLP1 (green) localize CHOP (red) in the nucleus (blue). Arrowheads show CHOP+ cells (pink/white nuclei). Scale bar, 20 μm.
(B) Morphometry of transfected COS-7 cells showing transfection efficiency (bars i and iv) and PLP1+/CHOP+ cells expressed as a proportion of transfected (ii and v ) or stressed (iii and vi) cells.
(C) Northern blots show induction of UPR effector genes in 293T cells untreated (NT), treated with tunicamycin (Tunic), or transfected with wild-type or mutant PLP1.
(D) Oligodendrocyte progenitors from wild-type mice (Da) elaborate cell processes, differentiate, and incorporate Plp1 gene products into the plasma membrane (green). Progenitors from msd mice also extend processes, which are subsequently resorbed (arrows) after the cells differentiate and express the Plp1 gene (Db). These cells localize CHOP (red) in the nucleus which appears as yellow/orange (arrowheads). Scale bar, 10 μm.
Cell counts demonstrate that CHOP expression strongly correlates with the expression of mutant but not wild-type PLP1. Transfected cells (n = 200) labeled with antibodies against PLP1 and CHOP in duplicate plates from two independent experiments are characterized in Figure 1B using several criteria. First, transfected cells are represented as a proportion of total cells (white bars i and iv). This transfection efficiency is similar for PLP1 and PLP1msd expressing cells (~20%) and rules out significant contributions from differences in plasmid toxicity. Second, dark bars (ii and v ) represent double-labeled cells (PLP1+/CHOP+) as a proportion of transfected cells (PLP1+). Only 10% of cells expressing wild-type PLP1 also express CHOP; however, 70% of cells expressing PLP1msd also express CHOP. Third, bars iii and vi show that most CHOP-positive cells express PLP1 (~85%), which indicates that transfection conditions alone do not induce CHOP expression. Consistent with this finding, few cells from cultures transfected without plasmid DNA or with an empty expression plasmid express CHOP (1.5%–4%). On the other hand, 93% of untransfected cells treated with 2 μg/ml of tunicamycin for 4 hr are strongly labeled by anti-CHOP antibodies. Together, these data clearly indicate that the UPR is activated by inhibiting N-glycosylation and by expressing mutant forms of PLP1 that accumulate in the ER.
From Northern blots, we find that several UPR effector genes are induced in tunicamycin-treated and PLP1msd-expressing 293T cells (Figure 1C). Steady-state levels of mRNAs encoding the transcription factors Chop and Atf3 as well as the molecular chaperones BiP, Erp72, and possibly Erp59 are induced in these cells compared to controls. Together, these data reflect activation of Ire1, PERK, and ATF6. On the other hand, the level of activation of downstream target genes differs between tunicamycin-treated and PLP1msd-expressing cells. For example, inhibiting glycoprotein synthesis induces CHOP ~5-fold more effectively than expressing PLP1msd on a per cell basis (we routinely obtain 50% transfection efficiency in 293T cells), and we obtain similar results using COS-7 cells (data not shown). Thus, these data suggest that UPR activation is a graded response rather than an on/off switch.
To confirm the transfection data in Figure 1A, we cultured oligodendrocyte progenitors from msd mouse brains and differentiated these cells to induce expression of Plp1 gene products. Primary transcripts from this gene are alternatively spliced in exon 3 to encode two protein isoforms, PLP1 (contains exon 3a and b) and DM-20 (contains only exon 3a). The smaller DM-20 protein lacks a 35 amino acid hydrophilic peptide (encoded by exon 3b) which is near the center of the protein and is exposed to the cytoplasm (Gow et al., 1997). Remaining exons are present in both mRNAs. Wild-type oligodendrocytes extend processes from the cell body and incorporate DM-20/PLP1 into the plasmalemma (green, Figure 1Da). These processes elaborate membrane sheets that are also DM-20/PLP1 positive. The arrowhead points to the nucleus, which is not labeled by anti-CHOP antibodies. However, nuclei of DM-20/PLP1-positive oligodendrocytes from msd mice are robustly CHOP positive, which appears yellow (arrowhead, Figure 1Db), and demonstrates activation of the UPR. In these cells, DM-20/PLP1 staining is largely confined to the perinuclear region, which is a common feature of oligodendrocytes from Plp1 mutant animals (Williams and Gard, 1997). Debris surrounding the cell body marks the locations of cell processes likely resorbed as accumulating mutant proteins perturbed cell function (arrows).
UPR Induction Correlates with Plp1 Expression in Mutant Mice
In view of our in vitro data, we examined expression of the Chop gene in msd mice with age to determine if induction parallels transcription from the Plp1 gene. Expression of Plp1 in spinal cord is low in the perinatal period, and we detect message on Northern blots beginning at P4 in wild-type mice, with a rapid increase thereafter (Figure 2A). Message levels in msd mice are detectable by P7 but remain roughly constant beyond this age until death. Chop expression in msd mice increases dramatically during the postnatal period, beginning at P7 and corresponding with the age that we detect Plp1 expression. Chop expression is maximal between P16–18 and decreases by P21, near the age of death. This decline may be related to depletion of the oligodendrocyte lineage similar to that reported for the allelic jimpy (jp) mouse (Knapp et al., 1986), which harbors a splice site mutation causing exon 5 skipping and a frameshift in the remaining coding region (Hudson et al., 1987). Although Chop is weakly expressed in controls, the level is constant with age and indicates that induction of this gene is not a feature of normal CNS development. Similar results for Plp1 and Chop are observed in msd brains.
Figure 2. Temporal Induction of Chop and Atf3 in Spinal Cord from Mutant Mice Correlates with Plp1 Expression.

(A) Northern blots from wild-type (wt) and msd mice between P4–P21 express Plp1, Chop, and Atf3. Ethidium bromide-stained 18S rRNA serves as loading controls.
(B) Northern blots from wild-type (wt), jp, and rsh mice at P18 and P21 show that Chop and Atf3 are elevated in the mutants.
(C) Northern blots from msd, jp, and rsh mice show induction of molecular chaperones.
(D) Northern blots from optic nerve of msd mice show induction of UPR effector genes
(E) Northern blots from tunicamycin-treated 3T3 cells (2 μg/mL, 4 hr), wild-type, and msd mice show that Doc genes are not induced by mutant Plp1 expression.
(F) Doc1 expression cannot be detected by nonquantitative RT-PCR from wild-type or msd mice after 35 cycles. Expression of Docs4 and 6 is detected.
(G) Ribonuclease protection assays show that Doc4 expression is decreased in msd mice to 57% of controls.
(H) Northern blots show that Doc4 transcripts are processed differently in wild-type and msd mice.
In Figure 1C, we observe that mRNA encoding a second bZip transcription factor, ATF3, is induced in 293T cells expressing mutant PLP1. Atf3 is also strongly induced in spinal cords of msd mice, although the time course of induction lags behind that of Chop by as much as 6 days (Figure 2A). Furthermore, Atf3 expression levels decrease earlier than Chop, from approximately P15.
Previously, we demonstrated accumulation of mutant DM-20/PLP1 in the perinuclear region of msd and rsh mouse oligodendrocytes (Gow et al., 1998). We interpret these data in terms of protein misfolding, which suggests that a common molecular mechanism accounts for the phenotypes of these allelic mutants. In support of this notion, we find that both Chop and Atf3 are induced in spinal cords of rsh mice compared to littermates at P21 (Figure 2B) as well as in jp mice. Together, these data reveal important similarities in pathogenesis for all three mouse mutants despite distinct genetic lesions and disease severities in these animals.
Steady-state mRNA levels of three molecular chaperones in spinal cords of msd, rsh, and jp mice are shown in Figure 2C. In msd mice, mRNAs for Bip, Erp59, and Erp72 are increased, particularly at P13, when the behavioral phenotype becomes apparent. In similar fashion, jp and rsh mice exhibit roughly 2-fold increases in the levels of molecular chaperones, which again points to activation of the UPR. Previously, we reported expression of six molecular chaperones induced to varying extents in msd and jp mice, which implies broad induction of this class of stress-induced genes (Southwood and Gow, 2001). It is likely that relatively modest increases in expression of molecular chaperones in Figure 2C reflect the low abundance of oligodendrocytes in spinal cord (~10% of total cells). Indeed, 5- to 10-fold increases in expression are apparent in optic nerve (Figure 2D), in which oligodendrocytes comprise over 50% of cells.
Chop Target Genes in the CNS
CHOP is widely believed to play a central role in the UPR, although this role has not been precisely determined. Target genes, or Downstream-Of-CHOP (Doc) genes, have been identified using representational difference analysis from tunicamycin-treated fibroblasts (Wang et al., 1998), which prompted us to examine expression of these genes in Plp1 mutants. Four Doc genes induced by CHOP are Chop itself (Doc2); Carbonic Anhydrase VI (Doc1); a gene encoding the cytoskeleton binding protein, advillin (Doc6); and Doc4, encoding the evolutionarily conserved teneurin-4 protein. Although expressed by tunicamycin-treated 3T3 cells (Figure 2E), Northern blots demonstrate that these genes are not induced in msd spinal cord compared with littermates. Indeed, Docs1 and 6 are not detected, and Doc4 mRNA levels actually decrease in the mutants. By nonquantitative RT-PCR, we detect Doc4 and Doc6 but not Doc1 mRNAs, indicating that this latter gene, at least, is not a target of CHOP in the CNS (Figure 2F).
Quantification of Doc4 levels by ribonuclease protection assays indicate that expression is decreased to 57% of wild-type levels, using β-actin as an internal control (Figure 2G). In addition, Northern blots show that Doc4 transcripts are differentially processed in msd compared to littermates (Figure 2H). Thus, two forms of Doc4 mRNA, differing in size by 1–2 kilobases, are apparent in wild-type tissue, while only the larger message is present in mutants. The significance of this observation is currently unclear, and we do not know if alternative splicing and/or the use of alternate polyadenylation signals account for the difference. In either case, Doc4 mRNA processing in response to ER stress was not observed in earlier studies (Wang et al., 1998).
Oligodendrocytes Express CHOP and ATF3 In Vivo
Induction of the Chop and Atf3 genes in myelin mutant mice prompted us to identify cells expressing these transcription factors and to determine if the proteins are nuclear localized. Anti-DM-20/PLP1 antibodies strongly label white matter tracts in the cerebellum of P18–21 wild-type mice (Figure 3A). However, CHOP is not detected in any cells in this field (Figure 3B), commensurate with a low level of steady-state mRNA during normal postnatal development. The DNA stain DAPI reveals glial cell nuclei in the white matter (Figure 3C).
Figure 3. CHOP Is Localized to Nuclei of DM-20/PLP1+ Oligodendrocytes in P18 Cerebellum from msd and rsh Mice.

(A) Immunofluorescence labeling in wild-type mice using anti-DM-20/PLP1 antibodies.
(B) CHOP is not detected in this field.
(C) DAPI stain shows locations of nuclei.
(D) DM-20/PLP1 staining in msd mice is largely perinuclear (arrowheads).
(E and F) Nuclei are stained with anti-CHOP antibodies (arrowheads).
(G) Anti-DM-20/PLP1 antibodies label rsh myelin sheaths in the pons.
(H and I) Oligodendrocyte nuclei are labeled with anti-CHOP antibodies (arrowheads). Inset shows an oligodendrocyte at high magnification with CHOP in the nucleus (arrowhead). Scale bars, 20 μm in (B); inset, 3 μm.
Disease in msd mice is accompanied by profuse and widespread oligodendrocyte apoptosis which, by P18, is at an advanced stage in many CNS regions (Gow et al., 1998). Few oligodendrocytes remain in cerebellar white matter, and DM-20/PLP1 staining is principally localized to perinuclear regions of these cells (inset, Figures 3D and 3F). Many oligodendrocytes in the cerebellum express CHOP at high levels, and, most importantly, this protein is nuclear localized (inset, Figures 3E and 3F). Expression and nuclear localization of CHOP in rsh oligodendrocytes is similar to that in msd mice (inset, Figures 3H and 3I). Thus, together with our previous findings that DM-20/PLP1 trafficking is disrupted in msd and rsh oligodendrocytes (Gow et al., 1998), Figures 2 and 3 demonstrate similarities in the pathogenesis of these allelic mutants.
Brainstem is another heavily myelinated region of CNS, and DM-20/PLP1 staining in wild-type tissue is shown in Figure 4A. ATF3 is not detected in this field (Figure 4B). In contrast, ATF3-positive oligodendrocytes are observed in msd and rsh mice (white arrowheads, Figures 4D and 4E, 4G and 4H), although some oligodendrocytes are ATF3 negative (black arrowheads). In addition, PLP1/DM-20 expression is not detected in some ATF3-positive cells (arrows, Figures 4G–4I).
Figure 4. ATF3 Is Localized to Nuclei of DM-20/PLP1+ Oligodendrocytes in Brainstem from msd and rsh.

(A) Immunofluorescence labeling of white matter from wild-type mice using anti-DM-20/PLP1 antibodies.
(B) ATF3 is not detected in this field. Scale bar, 20 μm.
(C) DAPI stain shows locations of nuclei.
(D) DM-20/PLP1 staining in msd mice is largely perinuclear (arrowheads).
(E and F) Nuclei in some oligodendrocytes are stained with anti-ATF3 antibodies (white arrowheads); others are unstained (black arrowheads).
(G) Anti-DM-20/PLP1 antibodies label oligodendrocytes (arrowheads) and myelin sheaths in rsh mice.
(H and I) Some cells are labeled with anti-ATF3 antibodies (white arrowheads), while others are ATF3− (black arrowheads). Arrows show ATF3+, DM-20/PLP1− cells.
Oligodendrocytes and Microglia Express CHOP and ATF3 in Brains of Mutant Mice
Morphometric analyses throughout the brains of three P18 msd mice indicate that 100% of CHOP-positive nuclei (n = 300 cells) are from DM-20/PLP1-positive oligodendrocytes (Figure 5). Similarly, counts from two rsh mice reveal that >99% of CHOP-positive oligodendrocytes express DM-20/PLP1 (n = 113). However, ATF3-positive, DM-20/PLP1-negative cells account for ~17% (n = 109) in msd and 31% (n = 143) in rsh brains. Colocalization analyses (averages from two mice) using cell-specific markers reveal that Mac-3-positive microglia account for 19% ± 3.7% and 35% ± 2.2% of ATF3-positive nuclei in msd (n = 321) and rsh (n = 293), respectively. On the other hand, we observe <2% colocalization for ATF3 and S100β (astrocytes) and <1% for ATF3 and Mac-1 (macrophages) or ATF3 and CD3ε (T cells). Thus, the major cell populations that express ATF3 in CNS of rsh and msd mice are oligodendrocytes and microglia.
Figure 5. Oligodendrocytes and Microglia Comprise the CHOP+ and ATF3+ Cell Populations in Brains of msd and rsh Mice.
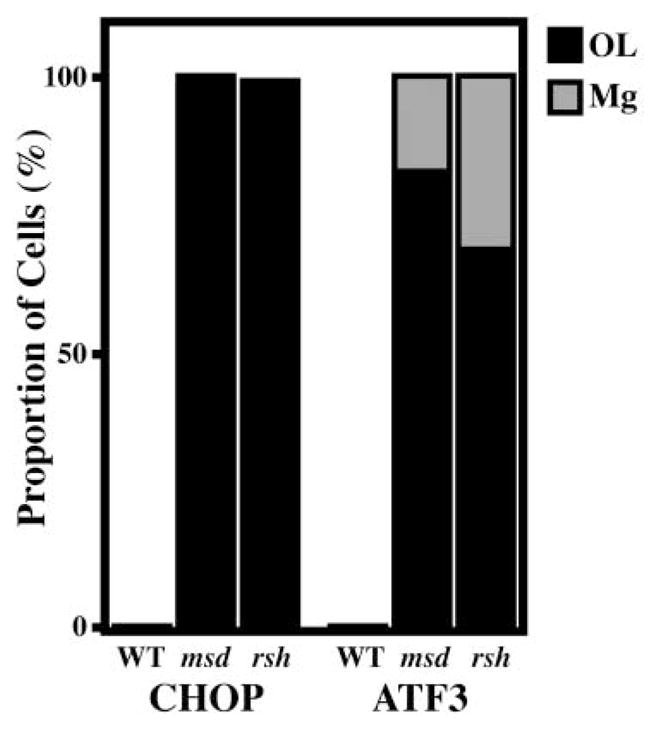
Essentially all CHOP+ cells in the CNS of msd and rsh mice are oligodendrocytes. Oligodendrocytes and microglia are the major cell populations comprising ATF3+ cells.
The UPR Is Induced Specifically in White Matter of a PMD Patient
To determine the relevance to PMD of our conclusions from previous (Gow et al., 1994b, 1998; Gow and Lazzarini, 1996; Southwood and Gow, 2001) and current analyses, we examined expression of UPR effector genes in autopsy specimens from a patient with multiple sclerosis (MS) and a PMD patient with severe disease caused by a splice site mutation in intron 6 of the PLP1 gene that causes an in-frame deletion of the 66 base pair exon 6 (Δexon6) in mRNAs from cultured skin fibroblasts (Hobson et al., 2000). We confirm these data using white matter mRNA by reverse transcriptase-PCR and sequencing of the amplified products; open reading frames from the dominant cDNA species encode putative proteins of 27.8 kDa (PLP1Δexon6) and 24 kDa (DM-20Δexon6). Furthermore, expression of cloned cDNAs in our COS-7 cell paradigm (Gow, 2002; Gow and Lazzarini, 1996) causes accumulation of mutant PLP1 and DM-20 in the ER of transfected cells (Figure 6A).
Figure 6. CHOP and ATF3 Are Induced in White Matter from a PMD Patient Harboring the Δexon6 Mutation.

(A) Immunofluorescence labeling of mutant PLP1 (Aa) and DM-20 (Ab) encoded by cDNAs from a Δexon6 PMD patient show protein accumulating in the ER of transfected COS-7 cells. Scale bar, 5 μm.
(B) Northern blot of white matter (WM) and gray matter (GM) from a multiple sclerosis patient (MS) and a Δexon6 PMD patient. 18S serves as loading controls.
(C) An oligodendrocyte (arrow) labeled with anti-MBP antibodies (Ca) expresses CHOP (Cb) in the nucleus (Cc). Three additional oligodendrocytes (arrowheads and inset) express CHOP in the cytoplasm. Scale bar, 5μm.
Oligodendrocytes are widely known to be localized to white matter tracts (WM); however, these cells also reside in gray matter (GM), as reflected in Northern blots of patient tissue probed with human PLP1 cDNA (Figure 6B). In an MS patient, CHOP is expressed at comparable steady-state levels in gray and nonplaque white matter regions. Gray matter is largely comprised of neuronal cell bodies, while white matter is rich with glial cells; thus, different cell types in apparently normal tissue express CHOP at low but comparable levels. In contrast, we observe a 5-fold increase in CHOP between white and gray matter from the Δexon6 patient, indicating that this gene is selectively induced in the glial cell population. Furthermore, ATF3, BIP, ERp59, and ERp72 are selectively induced in white matter from this patient. Thus, these data are consistent with our findings in vitro and in animal models demonstrating activation of the UPR in response to coding region mutations in the PLP1 gene. Comparable CHOP expression in gray matter of the MS and Δexon6 patients suggests that metabolic stress in neurons is not a significant pathogenic feature of PMD. Interestingly, BIP expression in gray matter from the MS patient is elevated above white matter; however, its significance is currently unknown.
MBP-Positive Oligodendrocytes in a PMD Patient Localize CHOP to the Nucleus
To determine the identity of CHOP-expressing cells in white matter from the Δexon6 patient, we labeled paraffin sections from neocortex with antibodies against myelin basic protein (MBP) and CHOP (Figure 6C). MBP strongly labels myelin membrane around axons (double arrow, Figure 6Ca) as well as the plasma membrane of an oligodendrocyte cell body (arrow). This cell also expresses CHOP in the nucleus (Figure 6Cb), which is evident from its colocalization with the nuclear stain DAPI (Figure 6Cc). Three additional cells are weakly MBP positive and express CHOP in the cytoplasm (arrowheads, inset). Presumably, these cells are at the initial stage of synthesizing myelin proteins such as MBP and DM-20/PLP1 and have induced the UPR, although it appears that nuclear translocation of CHOP has not yet occurred. We are unable to detect DM-20/PLP1 or ATF3 in brain sections from this patient using several antibodies.
CHOP Modulates Disease Severity in rsh Mice
The data presented in Figures 1–5 suggest a role for CHOP in pathogenesis associated with coding region mutations in the PLP1 gene. Early in vivo experiments demonstrate that epithelial cells in the kidneys of mice undergo CHOP-dependent apoptosis with a predictable time course after acute tunicamycin toxicity (Wang et al., 1998).
To determine if CHOP modulates pathogenesis associated with Plp1 mutations, we crossed Chop null mice (Zinszner et al., 1998) with rsh mice to generate Chop null/rsh double mutants. A Kaplan-Meier analysis of these mice is shown in Figure 7, where the ordinate indicates the number of mice for each genotype (see Experimental Procedures for husbandry details). Wild-type and Chop null mice, which have no known phenotype, live beyond 20 weeks of age. Similarly, rsh mice exhibit normal longevity, as shown previously (Schneider et al., 1992). In dramatic contrast, disease in Chop null/rsh mice is substantially more severe than that conferred by the rsh mutation alone, and double mutants exhibit frequent seizures induced by sudden noise, handling, or startle responses. These mice die as early as 5 weeks of age and have an average life span of less than 10 weeks. Despite apparent longevity, rsh mice that harbor a single functional Chop gene exhibit occasional seizures, consistent with an intermediate phenotype stemming from haplo-insufficiency at the Chop locus. Together, these data provide direct evidence for a pivotal role of CHOP in pathogenesis and demonstrate that the activity of this transcription factor serves to ameliorate the toxic effects of mutant DM-20/PLP1 in oligodendrocytes.
Figure 7. Kaplan-Meier Analysis and Oligodendrocyte Apoptosis in Chop Null/rsh Mice.

(A) Survival over 20 weeks in sib cohorts of wild-type, rsh, Chop null, Chop null/rsh, and Chop-het/rsh mice. The ordinate indicates the number of mice in each cohort.
(B) Levels of apoptosis are 5-fold higher in cervical spinal cord from P16 msd mice compared to controls.
(C) Electron micrographs show (Ca) a morphologically normal oligodendrocyte from a Chop null mouse surrounded by myelinated fibers and (Cb) an apoptotic oligodendrocyte from a Chop null/rsh mouse with condensed chromatin (arrowhead) surrounded by large unmyelinated axons (asterisks). Scale bar, 1μm.
Oligodendrocyte Apoptosis in Chop Null/rsh Mice
In a hunt for cellular mechanisms that account for high mortality in Chop null/rsh mice, we examined levels of apoptosis in CNS, which are elevated for Plp1 mutants with severe forms of disease (Schneider et al., 1992). At early postnatal ages, the number of TUNEL-positive cells in cervical spinal cord from Chop null/rsh mice is elevated ~5-fold over age-matched controls (Figure 7B), and by 10 weeks, apoptotic oligodendrocytes are widespread in optic nerve and spinal cord from double mutants. A morphologically normal oligodendrocyte surrounded by myelinated axons (arrows) in optic nerve from a Chop null mouse is shown in Figure 7Ca. On the other hand, many oligodendrocytes in Chop null/rsh mice have abnormally shaped nuclei and exhibit regions of condensed chromatin around the nuclear membrane (arrowhead, Figure 7Cb). Moreover, oligodendrocyte loss and dysfunction is evident from the large number of unmyelinated axons in these mutants (asterisks). Thus, these data demonstrate that CHOP plays a critical role in protecting oligodendrocytes from the detrimental consequences of unfolded proteins in the ER and are consistent with the correlation between oligodendrocyte death and shortened life span of PLP1 mutants with severe forms of disease, such as msd and jp mice.
Discussion
Although several features of the UPR are well-characterized, a rapid expansion in our understanding of this signaling cascade has occurred only since identification of the upstream initiating elements of the pathway—such as ATF6, Ire1, and PERK—as well as molecular chaperones and transcription factors, which effect changes in gene expression upon UPR activation. In this study, we examine expression of two effector proteins, CHOP and ATF3, in PMD brain as well as animal models, and find them to be nuclear localized in oligodendrocytes expressing mutant PLP1 gene products. Furthermore, we show induction of several molecular chaperones. Finally, we demonstrate direct involvement of CHOP in the pathogenesis of PMD and reveal novel and important features of the UPR in oligodendrocytes, as indicated below.
First, CHOP does not serve identical functions in oligodendrocytes to those demonstrated in fibroblasts and kidney proximal tubule epithelial cells. For example, DNA condensation and fragmentation in kidney cells from tunicamycin-treated Chop null mice are not prominent features of pathology, in stark contrast to control animals. Thus, CHOP is required for apoptosis of these cells (Zinszner et al., 1998). However, oligodendrocytes in optic nerve and spinal cord from Chop null/rsh mice undergo apoptosis with greater frequency than in controls (Figure 7). These data reveal a major divergence in importance of CHOP as a central mediator of cell death for different cell types which, conceivably, could be related to the cell type-specific target genes of this transcription factor. Nevertheless, available data convincingly indicates that CHOP is a key regulatory component of the UPR in kidney cells and oligodendrocytes.
Additional diversity in cell type-specific functions of Chop is revealed in a recent examination of diabetic Akita mice. This mouse harbors a C96Y missense mutation in the Insulin2 gene, and a single mutant allele leads to hyperglycemia in the first month of life (Oyadomari et al., 2002). Interestingly, hyperglycemia develops at a slightly later age in the absence of the Chop gene (i.e., Ins2WT/C96Y, Chop null mice), and pancreatic β cells undergo cell death with reduced frequency. On the other hand, apoptosis of β cells in Akita mice with two mutant Insulin2 alleles is equally widespread in the presence or absence of CHOP. These data indicate that Chop is not required for pancreatic β cell death in the context of the UPR, similar to oligodendrocytes. Nonetheless, CHOP in β cells exhibits weak proapoptotic properties which contrast with its strong prosurvival role in oligodendrocytes from rsh mice.
In support of cell type-specific functions for CHOP, we find that target genes in oligodendrocytes overlap with but are distinct from those in other cell types. Thus, Chop induction in msd mice does not induce expression of Docs 1, 4, and 6, in contrast to mouse embryonic fibroblasts treated with tunicamycin (Wang et al., 1998). The molecular basis underlying this cell-specific target gene selection is currently unknown, but a likely possibility is the partnering of CHOP with oligodendrocyte-specific bZip transcription factors. Nevertheless, we cannot exclude the prospect that target gene selection arises from unique aspects of UPR induction, such as acute tunicamycin exposure versus persistent PLP1 gene expression.
A second feature of the UPR to emerge from this study is that CHOP expression (and ATF3) is not a prelude to programmed cell death under all conditions, an important divergence that dramatically alters the life span of and disease severity in Plp1 mutant mice. In msd and jp mice, oligodendrocyte apoptosis is widespread in the CNS (Gow et al., 1998) and depletes this cell lineage shortly before the animals die (Knapp et al., 1986). On the other hand, oligodendrocyte populations in white matter tracts of rsh mice are slightly expanded compared to controls (Fanarraga et al., 1992), which may reflect compensation to overcome diminished myelin synthesis and/or reduced numbers of axons ensheathed per cell. These data suggest common features of pathogenesis in rsh, msd, and jp mice, despite substantial differences in life span. Thus, activation of the UPR per se does not govern oligodendrocyte survival or disease severity, which must be regulated by additional factors (Gow and Lazzarini, 1996).
Third, UPR activation appears to be limited to genetic forms of PMD causing coding region mutations in PLP1. For example, immunocytochemical analyses of brain and spinal cord from 4e-Plp mice, which overexpress a wild-type Plp1 transgene in oligodendrocytes (Kagawa et al., 1994), indicate that the UPR is not induced; indeed, we are unable to detect expression of either CHOP or ATF3 in oligodendrocytes from these mutants. Furthermore, expression of these transcription factors is not induced in white matter from a PMD patient harboring a PLP1 duplication (data not shown). Morphological evidence also distinguishes disease caused by coding region mutations versus overexpression. In transgenic mice overexpressing Plp1, the major ultrastructural defect observed in oligodendrocytes is distention of Golgi cisternae (Kagawa et al., 1994; Readhead et al., 1994), while Plp1 coding region mutations distend the ER (Duncan, 1990). In accord with these ultrastructural changes, we anticipate a UPR associated with the latter but not the former genetic form of PMD. However, perhaps the strongest evidence of a distinct pathogenic mechanism for Plp1 overexpression is revealed by a recent study showing inappropriate targeting to lysosomes of cholesterol and lipid raft components, which are normally sorted to the Golgi (Simons et al., 2002).
Consistent with variable disease severity observed for different PLP1 coding region mutations, we speculate that upstream elements of the UPR confer a graded response to metabolic stress: the greater the accumulation, the more intense the UPR and the higher the likelihood of apoptosis. Several studies support this model. For example, we previously described a cellular mechanism that accounts for disease severity in PMD on the basis of the amount of PLP1 gene products accumulating in the ER (Gow and Lazzarini, 1996). The interpretation from these data and additional mutations (A.G., unpublished data) is illustrated in Figure 8A. Thus, eight missense mutations causing ER accumulation of both DM-20 and PLP1 (including msd) give rise to severe disease (Figure 8Aa). In contrast, nine mutations that disrupt trafficking to the cell surface of PLP1 but not DM-20 (including rsh) cause mild disease (Figure 8Ab). Furthermore, wild-type or mutant forms of DM-20 that traverse the secretory pathway can transport mutant PLP1 out of the ER, which, presumably, reduces levels of misfolded protein in this organelle (Gow and Lazzarini, 1996). Finally, we show in Figure 1C that, on a per cell basis, the glycoprotein synthesis inhibitor tunicamycin induces Chop expression far more effectively than does PLPmsd, although Atf3 and BiP are induced with similar efficiencies.
Figure 8. Possible Models to Account for the Correlation between Disease Severity in PMD and DM-20/PLP1 Trafficking Defects.

(A) UPR portrayed as a graded response that is proportional to the level of misfolded protein in the ER; the higher the level, the stronger the UPR. (Aa) Mutations causing ER accumulation of both PLP1 and DM-20 induce a strong UPR. (Ab) Mutations causing accumulation of PLP1 but not DM-20 induce a weak response.
(B) UPR portrayed as an off/on switch upon misfolded protein accumulation. (Ba) PLP1 and DM-20 accumulation induces the UPR. DM-20 is toxic and disrupts cell survival pathways (?). (Bb) Accumulation of only PLP1 in the ER induces the UPR but does not affect cell survival pathways.
Other studies provide evidence of variable disease severity that is consistent with a graded UPR. When expressed in oligodendrocytes of transgenic mice, the H-2Kb MHC protein accumulates in the ER and causes hypomyelination above a threshold of transgene expression (Baerwald et al., 2000; Power et al., 1996; Turnley et al., 1991). This threshold can be breached in hemizygotes by high-level transgene expression or by mating two asymptomatic lines. Moreover, expression significantly above this threshold also decreases life span to 3 weeks and causes widespread, nonimmune-mediated oligodendrocyte apoptosis. In analogous experiments, high-level expression of H-2Kb in pancreas causes widespread apoptosis of β cells and diabetes, again by a nonimmune mechanism (Allison et al., 1988). Presumably, these cells are particularly sensitive to overwhelming the ER or disrupting the UPR, because knockout mice that lack the gene encoding the UPR initiator receptor kinase PERK also exhibit high levels of β cell death and diabetes (Harding et al., 2001).
Despite considerable precedence for graded UPR signaling, we cannot formally exclude the possibility that severe forms of PMD result specifically from disrupting DM-20 trafficking (Figure 8B). For example, rsh and msd mutations may activate the UPR in oligodendrocytes to similar extents; however, accumulation of DM-20 in the ER of msd oligodendrocytes may activate additional pathways (“?” in Figure 8Ba) that block survival mechanisms normally regulated by the UPR. Alternatively, accumulation of mutant DM-20 in the ER but not mutant PLP1 may be particularly toxic to cells.
Recent appreciation of the UPR has led to speculation that disrupting or overwhelming this signaling cascade might play a role in pathogenesis for a number of degenerative diseases (Aridor and Balch, 1999). Herein, we show that PLP1 mutations induce the UPR and that disruption of this pathway dramatically increases disease severity. Thus, PMD is the first example of a degenerative disease where the UPR modulates pathogenesis. In the search for additional UPR-linked diseases, it is important to develop criteria that exclude diseases arising from other pathogenic mechanisms. In this vein, null alleles should confer a phenotype that is significantly more mild than or clinically distinct from at least some coding region mutations. This criterion rules out mutations that simply abolish protein function. This is the case for PLP1 and for α1-ANTITRYPSIN, where null mutations cause emphysema and missense mutations accumulating in the ER cause hepatitis (Dycaico et al., 1988).
A second distinguishing feature of UPR-linked diseases is dominant or semidominant inheritance, at least at the level of individual cells. PMD is technically a recessive disease, because PLP1 is located on the X chromosome, and carrier females rarely develop symptoms. However, mutant PLP1 gene products act dominantly because the jp phenotype cannot be rescued with a wild-type Plp1 transgene (Schneider et al., 1995). Additional features for this class of diseases require in vivo and in vitro expression studies to determine if mutant genes encode secretory pathway proteins that accumulate in the ER. Thus, protein aggregation is a prominent feature of trinucleotide repeat diseases such as Huntington disease, but this aggregation often occurs in the nucleus and would not be expected to induce the UPR.
Experimental Procedures
Primary Oligodendrocyte Cultures and Transfected Cells
Brains from P2 male pups were dissociated by trituration through 18G, 21G, and 25G needles in HEPES-buffered MEM (Life Technologies), pelleted, and plated onto polyornithine-coated T25 flasks in high-glucose DMEM containing 15% FCS (Hyclone), 1 mM pyruvate, 1.5 g/l glucose, and Pen-Strep (Life Technologies). Cultures were grown at 37°C for 2 weeks, with media changes every 2–3 days. Oligodendrocyte progenitors were recovered by light trypsin digestion (ATV trypsin, Irvine Scientific, CA) and differentiated at low density on coated plastic 35 mm dishes (Nunclon) in high-glucose DMEM containing 2% FCS, pyruvate, glucose, and antibiotics. cDNAs encoding wild-type and mutant DM-20/PLP1 were expressed for 1–2 days using pCMV5 (Lorence et al., 1990) in FUGENE-6-transfected COS-7 or 293T cells (Gow, 2002).
Autopsy Specimens from PMD and MS Patients
Gray and white matter regions were dissected from the cortex of the Δexon6 PMD patient at autopsy (9 hr postmortem) and either frozen on dry ice or immersion fixed in 10% formalin for several weeks at 4°C before paraffin processing. Brain hemispheres from the MS patient (13.5 hr postmortem) were frozen on dry ice at autopsy and stored at −80°C.
Northern Blotting, RT-PCR, and Ribonuclease-Protection Assays
Total RNA was purified from homogenates of frozen tissue by CsCl centrifugation using a Beckman SW50.1 rotor (Chirgwin et al., 1979). Several spinal cords from young mice were pooled, and human tissue samples weighed 0.5–1 gm. Ten microgram RNA samples were run on 1%–1.5% formaldehyde-agarose gels, blotted, probed (Gow et al., 1999), and washed at 58°C (Chop, Atf3 probes) or 55°C (other probes) in 0.2 × SSPE and 0.1% SDS. Exposures of autoradiograms were adjusted to yield nonsaturated hybridization signals with similar intensities for each sample. The signals were digitized, quantified by integration of the scanned peaks, and adjusted for exposure time. Oligo-dT priming from 10 μg of total mRNA was performed as recommended (Invitrogen), and 20% of each reaction was used for 35 cycle-PCR with primers defined previously (Wang et al., 1998). Product identities were confirmed by size and restriction enzyme mapping. Probes for ribonuclease protection assays were 32P-UTP labeled using T7 polymerase (Stratagene). Standard hybridization conditions (Ambion) were used prior to RNase A treatment and nondenaturing PAGE in 5% gels. Probes: Doc4, 230 bp of 3′ coding region; β-actin, 72 bp of 5′ coding region.
Immunocytochemistry and TUNEL Labeling
Mice were perfused intracardially for 30 min with 4% paraformaldehyde in 0.1 M sodium phosphate buffer (pH 7.2). Brains were dissected, cut along the midline, and embedded in O.C.T. at −80°C. Ten μm parasagittal sections were immunolabeled (Gow et al., 1998). Seven μm sections of cortex from the Δexon6 PMD patient were de-waxed, hydrated, treated with 1 × TUF fluid as recommended (Zymed, CA), and immunolabeled. Antibodies used were rat anti-DM-20/PLP1 (1:5, hybridoma AA3, Yamamura et al., 1991); mouse anti-MBP (1:1000, mix of hybridomas SMI94 and SMI99, Sternberger Inc.); rabbit anti-CHOP (1:300, a kind gift from Dr. David Ron, Skirball Institute, New York); rabbit anti-ATF3 (1:300, C-19, Santa Cruz); 2 × mouse anti-S100β antibodies (1:100, SH-B1, Sigma); rat anti-Mac-1 (1:50, Pharmingen); rat anti-Mac-3 (1:50, Pharmingen); hamster anti-CD3ε (1:50, Pharmingen). Pharmingen antibodies were used to label 6 μm fresh frozen brain sections fixed/ permeabilized with methanol for 10 min, then subsequently fixed with 2% paraformaldehyde and labeled with anti-ATF3 antibodies. Secondary antibodies were from Vector Labs (Burlingame, CA) and Southern Biotech (Birmingham, AL). TUNEL labeling was carried out on 10 μm transverse spinal cord cryostat sections (Gow et al., 1998).
Kaplan-Meier Analysis
This analysis was carried out in three concurrent phases. First, Chop null male mice (Zinszner et al., 1998) with a wild-type Plp1 gene (i.e., Chop−/−, X/Y) on a mixed B6C3H F1 (Taconic Farms, NY) / 129 Sv/SW background were mated to rsh carrier female mice (i.e., Chop−/−, rsh/X) on a B6C3H F1 (Taconic Farms) background (gift from Dr. S. Billings-Gagliardi, University of Massachusetts, MA), then to the Chop−/−, rsh/X progeny to generate Chop−/−, rsh/X carrier females. These double mutants were sib mated to Chop−/−, X/Y mice, and male progeny with genotypes of either Chop−/−, X/Y (Chop null) or Chop−/−, rsh/Y (Chop null/rsh) were generated with equal frequency from ten litters for analysis. Second, Chop−/−, rsh/X carrier females were mated with wild-type hybrid mice to generate Chop−/−, rsh/Y mice from one litter for analysis (Chop-het/rsh). Third, Chop−/−, rsh/X females were sib mated with Chop−/−, X/Y males to recover the rsh allele on a Chop−/− background. Female Chop−/−, rsh/X carriers were sib mated with Chop−/−, X/Y males to generate male wild-type and rsh sib cohorts from six litters for analysis. Cohorts of Chop null and Chop null/rsh or Wild-Type and rsh mice comprise multiple sets of sibs housed in open cages. A nonparametric ANOVA (Kruskal-Wallis test) of Chop null/rsh mice revealed no statistical differences in average life span between individual litters (p = 0.36).
Acknowledgments
We dedicate this research to the memory of Dr. M.E. Hodes at the University of Minneapolis for his eminent contributions to the molecular characterization of PLP1 gene mutations for so many families living with Pelizaeus-Merzbacher disease. We acknowledge excellent technical support from Jingsong Li, Milena Pariali, Brunella Bertucci, Anita Chalasani, and Cunli Xiang; patient referrals by Drs. Grace Hobson (DuPont, Delaware) and Franca Cambi (Thomas Jefferson University, Philadelphia); as well as critical discussions and reagents from Drs. David Ron (Skirball Institute of Biomolecular Medicine, New York), John Kamholz, and Gerard Tromp (Wayne State University School of Medicine). This work was supported by grants to A.G. from the National Multiple Sclerosis Society (RG2891 B-2), National Institutes of Health (RO1 NS43783), and by grants to A.G. and J.G. from the Children’s Research Center of Michigan.
References
- Allison J, Campbell IL, Morahan G, Mandel TE, Harrison LC, Miller JFAP. Diabetes in transgenic mice resulting from over-expression of class I histocompatibility molecules in pancreatic β cells. Nature. 1988;333:529–533. doi: 10.1038/333529a0. [DOI] [PubMed] [Google Scholar]
- Aridor M, Balch WE. Integration of endoplasmic reticulum signaling in health and disease. Nat Med. 1999;5:745–751. doi: 10.1038/10466. [DOI] [PubMed] [Google Scholar]
- Baerwald KD, Corbin JG, Popko B. Major histocompatibility complex heavy chain accumulation in the endoplasmic reticulum of oligodendrocytes results in myelin abnormalities. J Neurosci Res. 2000;59:160–169. [PubMed] [Google Scholar]
- Chirgwin J, Przybyla A, MacDonald R, Rutter W. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979;18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- Duncan ID. Dissection of the phenotype and genotype of the X-linked myelin mutants. In: Duncan ID, Skoff RP, Colman DR, editors. Myelination and Dysmyelination. New York: New York Academy of Sciences; 1990. pp. 110–121. [DOI] [PubMed] [Google Scholar]
- Dycaico MJ, Grant SG, Felts K, Nichols WS, Geller SA, Hager JH, Pollard AJ, Kohler SW, Short HP, Jirik FR, et al. Neonatal hepatitis induced by alpha 1-antitrypsin: a transgenic mouse model. Science. 1988;242:1409–1412. doi: 10.1126/science.3264419. [DOI] [PubMed] [Google Scholar]
- Fanarraga ML, Griffiths IR, McCulloch MC, Barrie JA, Kennedy PG, Brophy PJ. Rumpshaker: an X-linked mutation causing hypomyelination: developmental differences in myelination and glial cells between the optic nerve and spinal cord. Glia. 1992;5:161–170. doi: 10.1002/glia.440050302. [DOI] [PubMed] [Google Scholar]
- Garbern J, Cambi F, Shy M, Kamholz J. The molecular pathogenesis of Pelizaeus-Merzbacher disease. Arch Neurol. 1999;56:1210–1214. doi: 10.1001/archneur.56.10.1210. [DOI] [PubMed] [Google Scholar]
- Gencic S, Hudson LD. Conservative amino acid substitution in the myelin proteolipid protein of jimpymsd mice. J Neurosci. 1990;10:117–124. doi: 10.1523/JNEUROSCI.10-01-00117.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow A. The COS-7 cell in vitro paradigm to study myelin proteolipid protein gene mutations. In: Potter N, editor. Neurogenetics: Methods And Protocols. Totowa, NJ: Humana Press; 2002. pp. 263–275. [DOI] [PubMed] [Google Scholar]
- Gow A, Lazzarini RA. A cellular mechanism governing the severity of Pelizaeus-Merzbacher disease. Nat Genet. 1996;13:422–428. doi: 10.1038/ng0896-422. [DOI] [PubMed] [Google Scholar]
- Gow A, Friedrich VL, Lazzarini RA. Intracellular transport and sorting of the oligodendrocyte transmembrane proteolipid protein. J Neurosci Res. 1994a;37:563–573. doi: 10.1002/jnr.490370503. [DOI] [PubMed] [Google Scholar]
- Gow A, Friedrich VL, Lazzarini RA. Many naturally occurring mutations of myelin proteolipid protein impair its intracellular transport. J Neurosci Res. 1994b;37:574–583. doi: 10.1002/jnr.490370504. [DOI] [PubMed] [Google Scholar]
- Gow A, Gragerov A, Gard A, Colman DR, Lazzarini RA. Conservation of topology, but not conformation, of the proteolipid proteins of the myelin sheath. J Neurosci. 1997;17:181–189. doi: 10.1523/JNEUROSCI.17-01-00181.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow A, Southwood CM, Lazzarini RA. Disrupted proteolipid protein trafficking results in oligodendrocyte apoptosis in an animal model of Pelizaeus-Merzbacher disease. J Cell Biol. 1998;140:925–934. doi: 10.1083/jcb.140.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow A, Southwood CM, Li JS, Pariali M, Riordan GP, Brodie SE, Danias J, Bronstein JM, Kachar B, Lazzarini RA. CNS myelin and sertoli cell tight junction strands are absent in Osp/Claudin 11-null mice. Cell. 1999;99:649–659. doi: 10.1016/s0092-8674(00)81553-6. [DOI] [PubMed] [Google Scholar]
- Griffiths IR, Scott I, McCulloch MC, Barrie JA, McPhilemy K, Cattanach BM. Rumpshaker mouse: a new X-linked mutation affecting myelination: evidence for a defect in PLP expression. J Neurocytol. 1990;19:273–283. doi: 10.1007/BF01217305. [DOI] [PubMed] [Google Scholar]
- Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, Schneider A, Zimmermann F, McCulloch M, Nadon N, Nave KA. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science. 1998;280:1610–1613. doi: 10.1126/science.280.5369.1610. [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD, Ron D. Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001;7:1153–1163. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- Hobson GM, Davis AP, Stowell NC, Kolodny EH, Sistermans EA, de Coo IF, Funanage VL, Marks HG. Mutations in noncoding regions of the proteolipid protein gene in Pelizaeus-Merzbacher disease. Neurology. 2000;55:1089–1096. doi: 10.1212/wnl.55.8.1089. [DOI] [PubMed] [Google Scholar]
- Hudson HD, Berndt JA, Puckett C, Kozak CA, Lazzarini RA. Aberrant splicing of proteolipid protein mRNA in the dysmyelinating jimpy mutant mouse. Proc Natl Acad Sci USA. 1987;84:1454–1458. doi: 10.1073/pnas.84.5.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikenaka K, Kagawa T. Transgenic systems in studying myelin gene expression. Dev Neurosci. 1995;17:127–136. doi: 10.1159/000111281. [DOI] [PubMed] [Google Scholar]
- Kagawa T, Ikenaka K, Inoue Y, Kuriyama S, Tsujii T, Nakao J, Nakajima K, Aruga J, Okano H, Mikoshiba K. Glial cell degeneration and hypomyelination caused by overexpression of myelin proteolipid protein gene. Neuron. 1994;13:427–442. doi: 10.1016/0896-6273(94)90358-1. [DOI] [PubMed] [Google Scholar]
- Katayama T, Imaizumi K, Sato N, Miyoshi K, Kudo T, Hitomi J, Morihara T, Yoneda T, Gomi F, Mori Y, et al. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat Cell Biol. 1999;1:479–485. doi: 10.1038/70265. [DOI] [PubMed] [Google Scholar]
- Klugmann M, Schwab MH, Puhlhofer A, Schneider A, Zimmermann F, Griffiths IR, Nave KA. Assembly of CNS myelin in the absence of proteolipid protein. Neuron. 1997;18:59–70. doi: 10.1016/s0896-6273(01)80046-5. [DOI] [PubMed] [Google Scholar]
- Knapp P, Skoff R, Redstone D. Oligodendroglial cell death in jimpy mice: an explanation for the myelin deficit. J Neurosci. 1986;6:2813–2822. doi: 10.1523/JNEUROSCI.06-10-02813.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Hoffman EP, Marks HG. The rump-shaker mutation in spastic paraplegia. Nat Genet. 1994;7:351–352. doi: 10.1038/ng0794-351. [DOI] [PubMed] [Google Scholar]
- Lorence MC, Murry BA, Trant JM, Mason JI. Human 3β-hydroxysteroid dehydrogenase/Δ5-4 isomerase from placenta: expression in nonsteroidogenic cells of a protein that catalyzes the dehydrogenation/isomerization of C21 and C19 steroids. Endocrinology. 1990;126:2493–2498. doi: 10.1210/endo-126-5-2493. [DOI] [PubMed] [Google Scholar]
- Ma Y, Hendershot LM. The unfolding tale of the unfolded protein response. Cell. 2001;107:827–830. doi: 10.1016/s0092-8674(01)00623-7. [DOI] [PubMed] [Google Scholar]
- Ma Y, Brewer JW, Diehl JA, Hendershot LM. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol. 2002;318:1351–1365. doi: 10.1016/s0022-2836(02)00234-6. [DOI] [PubMed] [Google Scholar]
- Niwa M, Sidrauski C, Kaufman RJ, Walter P. A role for presenilin-1 in nuclear accumulation of Ire1 fragments and induction of the mammalian unfolded protein response. Cell. 1999;99:691–702. doi: 10.1016/s0092-8674(00)81667-0. [DOI] [PubMed] [Google Scholar]
- Okada T, Yoshida H, Akazawa R, Negishi M, Mori K. Distinct roles of ATF6 and PERK in transcription during the mammalian unfolded protein response. Biochem J. 2002;16 doi: 10.1042/BJ20020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, Mori M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest. 2002;109:525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power C, Kong P, Trapp B. Major histocompatibility class I expression in oligodendrocytes induces hypomyelination in transgenic mice. J Neurosci Res. 1996;44:165–173. doi: 10.1002/(SICI)1097-4547(19960415)44:2<165::AID-JNR9>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Readhead C, Schneider A, Griffiths IR, Nave KA. Premature arrest of myelin formation in transgenic mice with increased proteolipid protein gene dosage. Neuron. 1994;12:583–595. doi: 10.1016/0896-6273(94)90214-3. [DOI] [PubMed] [Google Scholar]
- Rosenbluth J, Stoffel W, Schiff R. Myelin structure in proteolipid protein (PLP)-null mouse spinal cord. J Comp Neurol. 1996;371:336–344. doi: 10.1002/(SICI)1096-9861(19960722)371:2<336::AID-CNE12>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Sato N, Urano F, Yoon Leem J, Kim SH, Li M, Donoviel D, Bernstein A, Lee AS, Ron D, Veselits ML, et al. Upregulation of BiP and CHOP by the unfolded-protein response is independent of presenilin expression. Nat Cell Biol. 2000;2:863–870. doi: 10.1038/35046500. [DOI] [PubMed] [Google Scholar]
- Schneider A, Montague P, Griffiths I, Fanarraga M, Kennedy P, Brophy P, Nave KA. Uncoupling of hypomyelination and glial cell death by a mutation in the proteolipid protein gene. Nature. 1992;358:758–761. doi: 10.1038/358758a0. [DOI] [PubMed] [Google Scholar]
- Schneider A, Griffiths I, Readhead C, Nave KA. Dominant-negative action of the jimpy mutation in mice complemented with an autosomal transgene for myelin proteolipid protein. Proc Natl Acad Sci USA. 1995;92:4447–4451. doi: 10.1073/pnas.92.10.4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M, Kramer EM, Macchi P, Rathke-Hartlieb S, Trotter J, Nave KA, Schulz JB. Overexpression of the myelin proteolipid protein leads to accumulation of cholesterol and proteolipid protein in endosomes/lysosomes: implications for Pelizaeus-Merzbacher disease. J Cell Biol. 2002;157:327–336. doi: 10.1083/jcb.200110138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwood CM, Gow A. Molecular mechanisms of disease stemming from mutations in the proteolipid protein gene. Microsc Res Tech. 2001;52:700–708. doi: 10.1002/jemt.1054. [DOI] [PubMed] [Google Scholar]
- Stecca B, Southwood CM, Gragerov A, Kelley KA, Friedrich VLJ, Gow A. The evolution of lipophilin genes from invertebrates to tetrapods: DM-20 cannot replace PLP in CNS myelin. J Neurosci. 2000;20:4002–4010. doi: 10.1523/JNEUROSCI.20-11-04002.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- Turnley AM, Morahan G, Okano H, Bernard O, Mikoshiba K, Allison J, Bartlett PF, Miller JAFP. Dysmyelination in transgenic mice resulting from expression of class I histocompatibility molecules in oligodendrocytes. Nature. 1991;353:566–569. doi: 10.1038/353566a0. [DOI] [PubMed] [Google Scholar]
- Wang XZ, Kuroda M, Sok J, Batchvarova N, Kimmel R, Chung P, Zinszner H, Ron D. Identification of novel stress-induced genes downstream of chop. EMBO J. 1998;17:3619–3630. doi: 10.1093/emboj/17.13.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams WC, 2nd, Gard AL. In vitro death of jimpy oligodendrocytes: correlation with onset of DM-20/PLP expression and resistance to oligodendrogliotrophic factors. J Neurosci Res. 1997;50:177–189. doi: 10.1002/(SICI)1097-4547(19971015)50:2<177::AID-JNR7>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Nanba E, Zhang H, Sasaki M, Komaki H, Takeshita K. Jimpy(msd) mouse mutation and connatal Pelizaeus-Merzbacher disease. Am J Med Genet. 1998;75:439–440. [PubMed] [Google Scholar]
- Yamamura T, Konola JT, Wekerle H, Lees MB. Monoclonal antibodies against myelin proteolipid protein: identification and characterization of two major determinants. J Neurochem. 1991;57:1671–1680. doi: 10.1111/j.1471-4159.1991.tb06367.x. [DOI] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]