

Abstract
Unlike traditional virus isolation and sequencing approaches, sequence-independent amplification based viral metagenomics technique allows one to discover unexpected or novel viruses efficiently while bypassing culturing step. Here we report the discovery of the first Sicinivirus isolate (designated as strain JSY) of picornaviruses from commercial layer chickens in mainland China by using a viral metagenomics technique. This Sicinivirus isolate, which contains a whole genome of 9,797 nucleotides (nt) excluding the poly(A) tail, possesses one of the largest picornavirus genome so far reported, but only shares 88.83% and 82.78% of amino acid sequence identity to that of ChPV1 100C (KF979332) and Sicinivirus 1 strain UCC001 (NC_023861), respectively. The complete 939 nt 5′UTR of the isolate strain contains at least twelve stem-loop domains (A–L), representing the highest set of loops reported within Sicinivirus genus. The conserved 'barbell-like' structure was also present in the 272 nt 3′UTR of the isolate as that in the 3′ UTR of Sicinivirus 1 strain UCC001. The 8,586 nt large open reading frame encodes a 2,862 amino acids polyprotein precursor. Moreover, Sicinivirus infection might be widely present in commercial chicken farms in Yancheng region of the Jiangsu Province as evidenced by all the tested stool samples from three different farms being positive (17/17) for Sicinivirus detection. This is the first report on identification of Sicinivirus in commercial layer chickens with a severe clinical disease in mainland China, however, further studies are needed to evaluate the pathogenic potential of this picornavirus in chickens.
Introduction
Birds are well known as an important reservoir for emerging pathogens [1], however, as the limitation of traditional virus isolation and sequencing approaches, it is difficult to find unexpected or unknown viruses. The whole metagenome sequencing of environmental viral communities [2], termed "viral metagenomics", has dramatically accelerated the viral discovery process. Thus, many new avian picornavirus species such as Melegrivirus A [3], Gallivirus A [4], Avisivirus A [5], and the recently accepted genera Sicinivirus [6] have been identified by metagenomic techniques.
The family Picornaviridae belongs to the order Picornavirales and is composed of 29 genera, including three new accepted genera Kunsagivirus, Sakobuvirus and Sicinivirus (http://www.ictvonline.org/). Among these genera, the first member of Sicinivirus was identified and genetically characterized from commercial broiler chickens in Ireland [6]. Picornaviruses are small, non-enveloped, positive-sense, single-stranded RNA viruses with a genome size of ~7.1–9.7 kb [6,7]. In most cases, the order of the encoded polyprotein precursor is L, VP0, VP3, VP1, 2A, 2B, 2C, 3A, 3B, 3C, and 3D, except canine picodicistrovirus (CPDV) of Dicipivirus genera that encode two polyprotein precursors, and possibly Megriviruses which contains a potential second open reading frame (ORF) at the 3′ part of the genome [8,9].
Here we report the discovery of the first Sicinivirus isolate (designated as strain JSY) of picornaviruses in commercial layer chickens with a severe clinical disease in mainland China by using a viral metagenomics technique. The Sicinivirus strain JSY was completely sequenced and the complete 5′ and 3′ untranslated region (UTR) were further characterized. At least twelve stem-loop domains (A–L) were observed in the 5′ UTR of Sicinivirus for the first time. In addition, comparative genome and phylogenetic analysis showed that the Sicinivirus strain JSY has certain sequence differences when compared to the previously reported Sicinivirus strains. The discovery of this novel strain facilitates further understanding of the molecular characteristics of the recently recognized genera Sicinivirus.
Materials and Methods
Ethics Statement
The present study was approved in accordance with the animal welfare guidelines (IACUC-2010) of the Animal Care and Use Committee of Institute of Animal Husbandry and Veterinary Medicine Beijing Academy of Agriculture and Forestry Sciences. Clinical samples were collected according to the Regulations for the Administration of Affairs Concerning Experimental Animals of the State Council of the People’s Republic of China. As our research is supported by China Agriculture Research System, we are responsible for diagnosis and control of poultry diseases. We declared that we had permissions from the farm owners to collect the samples and conduct this study, and further confirmed that the permissions here did not involve endangered or protected species.
Clinical samples and screening for viral pathogens
In May 2014, a high mortality was occurred in some commercial chicken farms in Yancheng region of the Jiangsu Province (China). The affected chickens ranged in age from 30 to >65 days. Clinical signs were lethargy, tendency to huddle, decreased feeding and drinking, and diarrhea with white green faeces. Illness rates were up to 50–80% in tested chicken flocks, and mortality rates were 30–50%. Gross lesions were characterized by severely swollen livers with distinct petechial and ecchymotic haemorrhages spots, translucent pericardial substance effusion, severe splenomegaly, kidney swelling, as well as thymus haemorrhages and bursa of Fabricius atrophy. 9 faecal samples from one of the above farms (designated as farm A) in Yancheng region were collected for detecting potential viral pathogens. These fecal samples were re-suspended 1:10 (w/v) in phosphate-buffered saline (PBS), and then centrifuged at 4,200 × g for 5 min. The supernatants were further filtered through 0.22 μm filters (Millipore, USA), and aliquoted and stored at -80°C until use. Viral DNA and RNA from the resultant supernatants were extracted using an AllPrep DNA/RNA Mini Kit or a QIAamp MinElute Virus Spin Kit (Qiagen, Germany), cDNAs were prepared using a SuperScript® First-Strand Synthesis System for reverse transcriptase (RT)-PCR (Invitrogen, USA) following the manufacturer’s instructions, then subjected to the regular PCR/RT-PCR detection of chicken pathogens including infectious bursal disease virus (IBDV), infectious bronchitis virus (IBV), avian encephalomyelitis virus (AEV) and chicken anaemia virus (CAV). The primers are listed in Table 1.
Table 1. Primers used in this study.
Primers based on:
| Primers | Sequences (5’-3’) | Position* | Fragment size (bp) |
|---|---|---|---|
| IBDVF a | CTGACTACCGGCATCGACA | 301 | 149 |
| IBDVR a | CCACTTGCCGACCATGA | 449 | |
| IBVF b | CCTAAGAACGGTTGGAAT | 24,408 | 741 |
| IBVR b | TACTCTCTACACACACAC | 25,148 | |
| AEVF c | GGGAAAGAGGATGAAGGAGGA | 1,956 | 811 |
| AEVR c | ACTCTTCTACCAACTCGTCATC | 2,766 | |
| CAVF d | GCAGTAGGTATACGCAAGG | 328 | 187 |
| CAVR d | CTGAACACCGTTGATGGTC | 514 | |
| KN | GACCATCTAGCGACCTCCACNNNNNNNN | - | - |
| RA01N | GCCGGAGCTCTGCAGATATCNNNNNNNNNN | - | - |
| K | GACCATCTAGCGACCTCCAC | - | - |
| RA01 | GCCGGAGCTCTGCAGATATC | - | - |
| S1F | GGACCCGTGACTATACCGTT | 676 | 1,212 |
| S1R | CCGCCGCCCAAGATAAGACA | 1,887 | |
| S2F | TGCTTTATCGCTCTTACCGAA | 1,669 | 1,247 |
| S2R | GATCGCCAAAGCATGAAGT | 2,915 | |
| S3F | GGAATACCACCACTGGCACT | 2,810 | 1,112 |
| S3R | AAACTCCGGTCCAGCTCGAA | 3,921 | |
| S4F | CAACCAGCGCCCTATACAAC | 3,797 | 1,492 |
| S4R | TCTTTAAGACCGTGTTTCTC | 5,288 | |
| S5F | CGCATAACTAATATTGAGCTTCC | 5,167 | 1,324 |
| S5R | CGTAGTACTCACAGGAGACGGAA | 6,490 | |
| S6F | CTCCCTCCTTGCCTCTCGT | 6,402 | 631 |
| S6R | ATCCAATGCCACCAACTCGT | 7,032 | |
| S7F | GCACCTACTTCCTAATACTCCTA | 6,966 | 1,473 |
| S7R | CCGGCTGACTGATTCATGT | 8,438 | |
| S8F | GCTCTATTTCTCGCGTTGCAAT | 8,331 | 652 |
| S8R | GCCTCCCTTCATGATGTACCAC | 8,982 | |
| S3RFO | CAGCTCCCACCATCTATCCC | 8,878 | - |
| S3RFI | TCAGCCATGCTCACTCACC | 9,046 | - |
| S5RRO | AGACAGAGCCAGAACAATAGGTG | 1,006 | - |
| S5RRI | ATTGCGCTCCATGCCGAAC | 951 | - |
| S5RRO2 | CCCCAAGGCAACTGTTACCA | 459 | - |
| S5RRI2 | GAGTGTCCATGCCATCTAACCTT | 313 | - |
Virus nucleic acid isolation, Metagenomic library construction, sequencing and genome walking
As previously described [10], one of the above filtered supernatants were treated with a mixture (160 μl of final volume) of 14 units of TURBO DNase (Ambion), 20 molecular biology units (MBU) of Baseline-ZERO DNase (Epicentre) and 20 units of RNase ONE (Promega) in 1 × TURBO DNase Buffer for 90 min at 37°C. Viral nucleic acids were then extracted using the QIAamp MinElute Virus Spin Kit (Qiagen, Germany) according to the manufacturer’s instructions.
Viral nucleic acid libraries were then constructed by sequence-independent RT-PCR amplification as previously described [10,11]. Briefly, 11 μl of extracted RNA was mixed with 1 μl of 10 mM dNTP Mixture and 1 μl of 100 μM primer KN or RA01N (Table 1), incubated at 65°C for 5 min, and chilled on ice. A reaction mix consisted of 4 μl of 5 × First-Strand buffer, 1 μl of 100 mM DTT, 1 μl of RNase inhibitor, and 200 units of SuperScript III reverse transcriptase (Invitrogen). The reaction was then incubated at 25°C for 5 min and 50°C for 60 min. Second strand cDNA was synthesized by incubating with 5 units of Klenow fragment polymerase (New England BioLabs) at 37°C for 60 min followed by inactivation at 75°C for 10 min. The PCR reaction mixture consisted of 10 μl of double-stranded cDNA, 0.25 μl of Ex Taq (5 units/μl, Takara), 5 μl of 10 × Ex Taq Buffer, 4 μl of 2.5 mM dNTP Mixture, and 1 μl of 100 μM primer K or RA01 (Table 1). For PCR procedure, 35 cycles of 98°C for 10 s, 56°C for 30 s, and 72°C for 2 min were used, followed by 10 min of final extension at 72°C. The PCR products ranging from 200 bp to 2,000 bp were separated and purified from a 1% agarose gel, the purified fragments were cloned into pEASY®-T5 Zero vector (TransGen Biotech, China), then six hundreds of single colonies were randomly selected and sequenced using commercial vector specific M13 forward primer.
A total of 511 reads were assembled using the SeqMan program, which is part of the Lasergene sequence analysis software package (DNASTAR Inc., USA). Single contigs were compared to GenBank using BLASTx. Subsequently, special PCR primers were designed based on the obtained sequences to walk the entire genome of the focused virus. Terminal sequences were obtained using a kit for rapid amplification of cDNA ends (RACE) (Clontech, Japan), both of the reported RACE primers and designed inner primers were listed (Table 1). For each fragment, at least three clones (if conflict occurs, up to eight clones) were sequenced to determine the consensus sequence of any given region.
The obtained genome sequence was submitted to NetPicoRNA 1.0 server [12] and also aligned with the available polyproteins of ChPV1 100C (KF979332) [1] and Sicinivirus 1 strain UCC001 (NC_023861) [6] to predict possible polyprotein cleavage sites. Conserved protein domains/families within the polyprotein were identified by BLASTP tools [12]. The sequence alignment results were edited using GeneDoc software. RNA secondary structure were modeled and refined by free energy minimization using Mfold (http://mfold.rna.albany.edu/?q=mfold/RNA-Folding-Form) [13] and RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) [14]. The generated secondary structure was drawn and analyzed by Rnaviz version 2.0.3 [15] and further edited using WPS Office 2013 (http://www.wps.cn/).
Phylogenetic analyses
Representative members of the 29 officially recognized genera and three other Sicinivirus sequences of family Picornaviridae were downloaded from NCBI, GenBank accession numbers of these sequences were listed in Table 2. Multiple sequence alignments of the Sicinivirus strain JSY and 32 sequences downloaded above were performed using Clustal Omega [16], and this alignment was determined online (http://www.ebi.ac.uk/Tools/msa/clustalo/). The phylogenetic tree based upon the results of multiple sequence alignment was constructed using the Molecular Evolutionary Genetics Analysis (MEGA) software version 6.0.6 [17] applying the maximum-likelihood method based on the JTT matrix-based model [18], the robustness of the phylogenetic constructions was evaluated by bootstrapping with 1,000 replicates, initial trees for the heuristic search were automatically obtained by applying neighbour-join and BioNJ algorithms.
Table 2. Pairwise amino acid sequence identities of Sicinivirus Strain JSY (KP779642) compared to other representative members.
| Genus | Species | GenBank accession no | Genome features | Pairwise amino acid identity(%) | ||||
|---|---|---|---|---|---|---|---|---|
| Size (nt) | G+C (%) | P1 | P2 | P3 | Polyprotein | |||
| Aphthovirus | Foot-and-mouth disease virus—type O | NC_004004 | 8,134 | 55.27 | 17 | 23.79 | 25.18 | 22.4 |
| Aquamavirus | Seal picornavirus type 1 | NC_009891 | 6,718 | 43.69 | 17.21 | 17.32 | 21.97 | 19.27 |
| Avihepatovirus | Duck hepatitis A virus 1 R85952 | NC_008250 | 7,711 | 43.51 | 17.72 | 17.53 | 21.4 | 19.37 |
| Avisivirus | turkey/M176-TuASV/2011/HUN | KC465954 | 7,532 | 44.97 | 17.29 | 17.43 | 21.83 | 19.79 |
| Cardiovirus | Encephalomyocarditis virus | NC_001479 | 7,835 | 49.47 | 19.6 | 19.77 | 26.63 | 22.59 |
| Cosavirus | Cosavirus A strain HCoSV-A1 | NC_012800 | 7,632 | 43.75 | 18.04 | 24.66 | 26.01 | 23.15 |
| Dicipivirus | Canine picodicistrovirus 209 | NC_021178 | 8,785 | 41.58 | 18.91 | 27.25 | 27.53 | NA* |
| Enterovirus | Poliovirus | NC_002058 | 7,440 | 46.34 | 19.97 | 17.21 | 27.62 | 23.58 |
| Erbovirus | Equine rhinitis B virus 1 | NC_003983 | 8,828 | 48.79 | 18.58 | 18.9 | 27.3 | 22.01 |
| Gallivirus | Turkey gallivirus | NC_018400 | 8,496 | 48.28 | 24.46 | 37.66 | 50.4 | 36.47 |
| Hepatovirus | Hepatitis A virus | NC_001489 | 7,478 | 37.86 | 14.18 | 22.87 | 25.28 | 21.14 |
| Hunnivirus | BHUV1/2008/HUN | NC_018668 | 7,583 | 45.58 | 17.55 | 22.63 | 25.14 | 21.18 |
| Kobuvirus | Aichi virus | NC_001918 | 8,251 | 58.9 | 28.46 | 36.59 | 45.36 | 34.56 |
| Kunsagivirus | roller/SZAL6-KuV/2011/HUN | KC935379 | 7,272 | 53.01 | 17.63 | 20.29 | 21.7 | 18.96 |
| Megrivirus | turkey/B407-THV/2011/HUN | NC_023858 | 9,739 | 45.64 | 18.54 | 26.32 | 34.42 | 26.15 |
| Mischivirus | Miniopterus schreibersii picornavirus 1 | JQ814851 | 8,468 | 47.44 | 17.6 | 21.18 | 25.82 | 22.92 |
| Mosavirus | Mosavirus A2 SZAL6-MoV/2011/HUN | NC_023987 | 8,398 | 45.48 | 19.79 | 24.07 | 28.83 | 23.17 |
| Oscivirus | Turdivirus 3 | NC_014413 | 7,678 | 46.6 | 21.89 | 29 | 40.65 | 31 |
| Parechovirus | Human parechovirus | NC_001897 | 7,348 | 39.47 | 17.15 | 19.72 | 22.05 | 19.17 |
| Pasivirus | Swine pasivirus 1 | NC_018226 | 6,916 | 43.07 | 14.14 | 19.07 | 19.97 | 18.06 |
| Passerivirus | Turdivirus 1 | NC_014411 | 8,035 | 57.92 | 35.17 | 39.52 | 52.49 | 41.19 |
| Rosavirus | Rosavirus 2 GA7403 | NC_024070 | 8,931 | 51.41 | 20.06 | 28.08 | 32.24 | 27.58 |
| Sakobuvirus | Feline sakobuvirus A isolate FFUP1 | NC_022802 | 7,807 | 55.51 | 27.47 | 36.6 | 48.24 | 36.11 |
| Salivirus | Salivirus A isolate 02394–01 | NC_012986 | 7,989 | 56.68 | 26.86 | 39.9 | 40.35 | 33.5 |
| Sapelovirus | Avian sapelovirus | NC_006553 | 8,289 | 42.7 | 19.97 | 23.9 | 28.23 | 23.07 |
| Senecavirus | Seneca valley virus | NC_011349 | 7,310 | 51.41 | 17.51 | 22.65 | 26.65 | 22.42 |
| Sicinivirus | Chicken picornavirus 1 100C | KF979332 | 8,331 | 55.2 | 77.89 | 92.6 | 95.96 | 88.83 |
| Chicken picornavirus 1 55C | NC_024765 | 8,287 | 55.56 | 75.27 | 92.89 | 96.44 | 88.13 | |
| Sicinivirus 1 strain UCC001 | NC_023861 | 9,243 | 54.29 | 75.3 | 83.79 | 87.29 | 82.78 | |
| Sicinivirus UCC1 | KF366619 | 8,329 | 54.13 | 78.66 | 83.7 | 70.54 | 79.03 | |
| Teschovirus | Porcine teschovirus 1 | NC_003985 | 7,117 | 44.79 | 19.48 | 22.66 | 26.48 | 22.81 |
| Tremovirus | Avian encephalomyelitis virus | NC_003990 | 7,055 | 44.73 | 14.02 | 19.27 | 25.99 | 21.19 |
*NA, the genome sequence of NC_021178 encodes two polyprotein precursors. P1, P2, P3 and Polyprotein of Sicinivirus strain JSY compared to the representative members of the 29 officially recognized genera. The highest amino acid identities were presented by boldface numbers.
Detection of Sicinivirus in clinical stool samples
A total of 17 chicken faecal samples from three commercial chicken farms (including 9, 5 and 3 stool samples from farms A [Nanyangzheng farm], B [Bufengzheng farm], and C [Dongzheng farm], respectively) in Yancheng region were detected for the presence of Sicinivirus. These three farms are located in different parts of Yancheng region, with the distances between each other being more than 20 kilometers. All of the faecal samples were collected one fresh dropping of chickens showing various degrees of clinical disease. A pair of primers S8F and S8R (Table 1) targeting the conserved 3D coding region of the Sicinivirus strain JSY were used to amplify a 652 bp fragment. RNA was extracted and reverse transcribed as mentioned above. For PCR procedure, 35 cycles of 98°C for 10 s, 58°C for 30 s, and 72°C for 1 min were used, followed by 10 min of final extension at 72°C. The PCR products were run on a 1% agarose gel for specific fragment detection.
Nucleotide sequence accession number
The full-length genomic nucleotide sequence of the Sicinivirus strain JSY was deposited in GenBank under accession number KP779642.
Results
Detection of clinical samples
Four pairs of primers were used to perform conventional PCR/RT-PCR detection, and the results showed that all the samples were negative for IBDV [19], IBV [20], AEV [21] and CAV [22]. In order to find other potential viral pathogens within these samples, cDNA from one of the fecal samples was generated as mentioned above to construct the metagenomic library for the further sequencing.
Overview of sequence data
The metagenomic sequencing data returned 511 useful reads. These reads were classified based on the best BLASTx expectation (E) scores. Summaries of the taxonomic classifications are shown in Fig 1. Among these sequences, only 8% of sequence reads (41 reads) matches to viruses, including Sicinivirus, avian leukosis virus (ALV), avian nephritis virus (ANV), chicken Gallivirus 1 and unclassified chicken Picornavirus. Interestingly, 82.9% of virus reads (34 reads) matches to different regions of the unexpected Sicinivirus genome, with about 75% nucleotide sequence identity. Besides, 7.3% of virus reads (3 reads) matches to ALV, 4.9% of virus reads (2 reads) matches to ANV, and the remaining 2 reads matches to chicken Gallivirus 1 and to another unclassified chicken Picornavirus (2.4% of virus reads each), respectively (Fig 1). The metagenomic sequencing data indicate that the predominant virus in the sample was the unexpected Sicinivirus.
Fig 1. Sequence classification of obtained reads based on BLASTx.
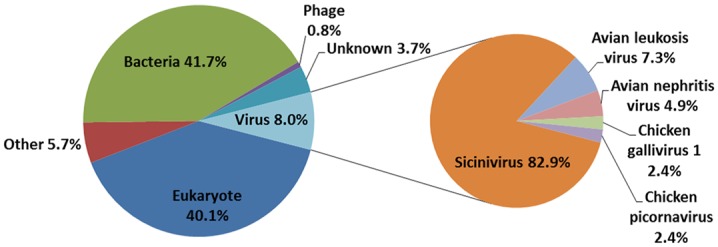
Percentages of reads with similarity to those of eukaryotes, bacteria, phages, viruses, unclassifiable sequences (other, including plasmid vector sequences) and to unknown sequences (Left). Percentages of viral sequence reads are classified by viral types (Right).
Genome organization and coding potential of Sicinivirus
Based on the sequence of specific PCR products and RACE fragments of both terminals, a Sicinivirus with a genome size of 9,797 nt was obtained. To our knowledge, the identified Sicinivirus isolate (designated as strain JSY) possess the largest picornavirus genome so far reported. The genome G+C content is 55.27%, with a typical picornaviruses genome organization as follows: 5′ UTR (1–939 nt)–L (940–2,319 nt)–VP0 (2,320–3,342 nt)–VP3 (3,343–3,960 nt)–VP1 (3,961–4,800 nt)– 2A (4,801–5,388 nt)– 2B (5,389–5,976 nt)– 2C (5,977–6,996 nt)– 3A (6,997–7,446 nt)– 3B (7,447–7,536 nt)– 3C (7,537–8,106 nt)– 3D (8,107–9,522 nt)– 3′ UTR (9,526–9,797 nt). The single 8,586 nt large open reading frame encodes the polyprotein precursor of 2,862 amino acids (Fig 2A).
Fig 2. Genome organization and the conserved picornaviral motifs.

(A) Predicted genome organization possesses conserved picornaviral motifs of Sicinivirus JSY (KP779642), ChPV1 100C (KF979332), ChPV1 55C (NC_024765), Sicinivirus 1 UCC001 (NC_023861) and Sicinivirus UCC1 (KF366619); the predicted cleavage sites of Sicinivirus JSY are indicated above the junction region. (B) Amino acid sequence alignment of Sicinivirus, Turdivirus 1 (NC_014411), Turkey gallivirus (NC_018400), Feline sakobuvirus A isolate FFUP1 (NC_022802), Aichi virus (NC_001918) and Salivirus A isolate 02394–01 (NC_012986), the identified motifs are indicated with jacinth boxes.
Analysis of the 5′UTR and 3′UTR
The predicted 939 nt 5′ UTR of the identified Sicinivirus was significantly larger than those of the published Sicinivirus (KF979332, and NC_024765, NC_023861) [1,6]. The results of GenBank BLASTx search showed that the partial 5′ UTR of the Sicinivirus strain JSY shared 94%, 77%, and 75% nucleotide sequence homology with the other Sicinivirus 1 strain UCC001 (position 600–737) (NC_023861), turkey 'TuASV' (position 421–479 and 666–715) (KC465954) [5] and turkey 'Gallivirus' (position 440–511 and 633–637) (NC_018400) [4], respectively, which could correspond to the apical part of domains I, J and K of a type II internal ribosomal entry site (IRES) of Sicinivirus as reported previously [5] (Fig 3). In addition, RNA secondary structure analysis of the 939 nt 5′ UTR sequence showed that the Sicinivirus strain JSY contains at least twelve stem-loop domains (A–L) [23], including the top/largest stem loop I and the eIF4G binding domains J and K [24,25] (Fig 3). This is the first characterization of the complete 5′ UTR structure for genus Sicinivirus. A 27 nt (position 736–762) pyrimidine-rich tract p(Y) and two tetraloop GNRA motifs (position 442–445 and 463–466) are also present (Fig 3). Furthermore, the Sicinivirus strain JSY contains a conserved Y7–X47–ATG motif, where the Y7 defines a 7 nt pyrimidine tract and X47 is 47 nt space from the ATG, this motif is similar to that of the published Sicinivirus sequence [6].
Fig 3. Predicted RNA secondary structure of the Sicinivirus JSY 5' UTR as determined by Mfold and RNAfold.

The complete structure of the 5'UTR (A to L, indicates the type II IRES) has been annotated (inset). Two GNRA motifs and the pyrimidine-rich region are illustrated with gray background boxes.
The 272 nt 3′ UTR sequence of the Sicinivirus strain shared 77% and 79% nucleotide sequence homology with Sicinivirus 1 strain UCC001 (position 1–271) [6] and turkey 'gallivirus' (position 179–230) (NC_018400) [4], respectively. The predicted secondary RNA structure of the 3′ UTR of the Sicinivirus strain possesses a 48 nt ‘barbell-like’ structure (Fig 4) as reported previously [4,6].
Fig 4. Predicted RNA secondary structure and the conserved 3' UTR motifs.

(A) Complete structure of the 3'UTR of Sicinivirus JSY (KP779642), the Poly(Y) tract and the 'barbell-like' structure are illustrated with gray background boxes. (B) Nucleotide sequence alignment of Sicinivirus, Turdivirus 1 (NC_014411), Turkey gallivirus (NC_018400), Feline sakobuvirus A isolate FFUP1 (NC_022802), Aichi virus (NC_001918) and Salivirus A isolate 02394–01 (NC_012986), the conserved regions are indicated with jacinth boxes.
Analysis of coding regions
The myristylation site GSISST was recognized in the VP0 protein of the Sicinivirus strain JSY as reported by Bullman S et al (2014) [6]. The 2C protein of the Sicinivirus strain contains a conserved NTP-binding site GxxGXGKS (X, uncharged; x, variable) motif as GPPGCGKS [5,26] and the DDLxQ motif as DDVGQ, which is associated with the putative helicase activity [5,27]. As the published Sicinivirus 1 strain UCC001 [6], the active-site cysteine in motif GxCG (x, variable) as GLCG [24] and the RNA binding domain (KFRDI) as QFKDL were present in the 3C protein of the Sicinivirus strain JSY. In addition, the RNA-dependent RNA polymerase (3D protein) of the strain also possesses the highly conserved motifs KDE[LI]R as KDELR, GG[LMN]PSG as GGNPSG, YGDD and FLKR as observed for the published Sicinivirus [6]. All the corresponding sites were labeled in Fig 2B.
For viral capsid protein VP1, the identified Sicinivirus strain JSY shows only 66% and 60.5% of amino acid sequence identity to that of ChPV1 100C and Sicinivirus 1 strain UCC001, respectively. Furthermore, residues I118 and L120 of the seven drug-binding pocket [28,29] sites in rhv_like capsid domain (cd00205) [9] were different among these five studied Sicinivirus sequences (Fig 5).
Fig 5. Amino acid sequence alignment of VP1 polypeptide of the five Sicinivirus.
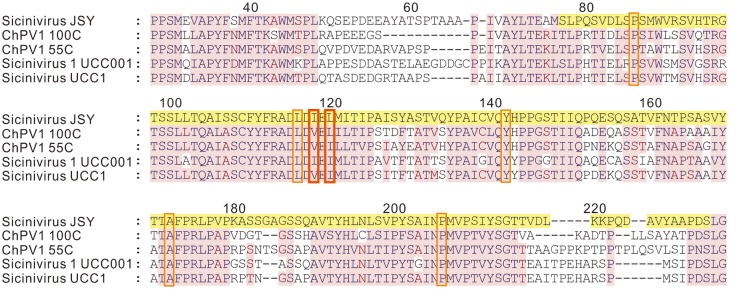
Drug-binding pocket sites of Sicinivirus JSY (KP779642), ChPV1 100C (KF979332), ChPV1 55C (NC_024765), Sicinivirus 1 UCC001 (NC_023861) and Sicinivirus UCC1 (KF366619) are illustrated with jacinth frames, I118 and L120 positions are labeled with jacinth frames in bold. rhv_like capsid domain (cd00205) in Sicinivirus JSY (KP779642) is indicated with yellow background.
Comparative genomic and phylogenetic analysis
Pairwise comparisons showed that the viral polyprotein sequences of the Sicinivirus strain JSY shared only 18.06% (swine Pasivirus 1, NC_018226) [30] to 41.19% (Turdivirus 1, NC_014411) [31] amino acid sequence identity with the downloaded representative members except Sicinivirus genera. Meanwhile, the viral polyprotein shared 88.83%, 88.13%, 82.78%, and 79.03% amino acid sequence identity with chicken Picornavirus 1 100C, chicken Picornavirus 1 55C [1], Sicinivirus 1 strain UCC001 [6], and Sicinivirus UCC1 (KF366619) (Unpublished), respectively (Table 2). These viruses with high degree of amino acid sequence similarity grouped together and formed a distinct cluster among picornaviruses based on phylogenetic tree of P1, 2C, and 3D proteins (Fig 6).
Fig 6. Phylogenetic analyses of the Sicinivirus strain JSY.
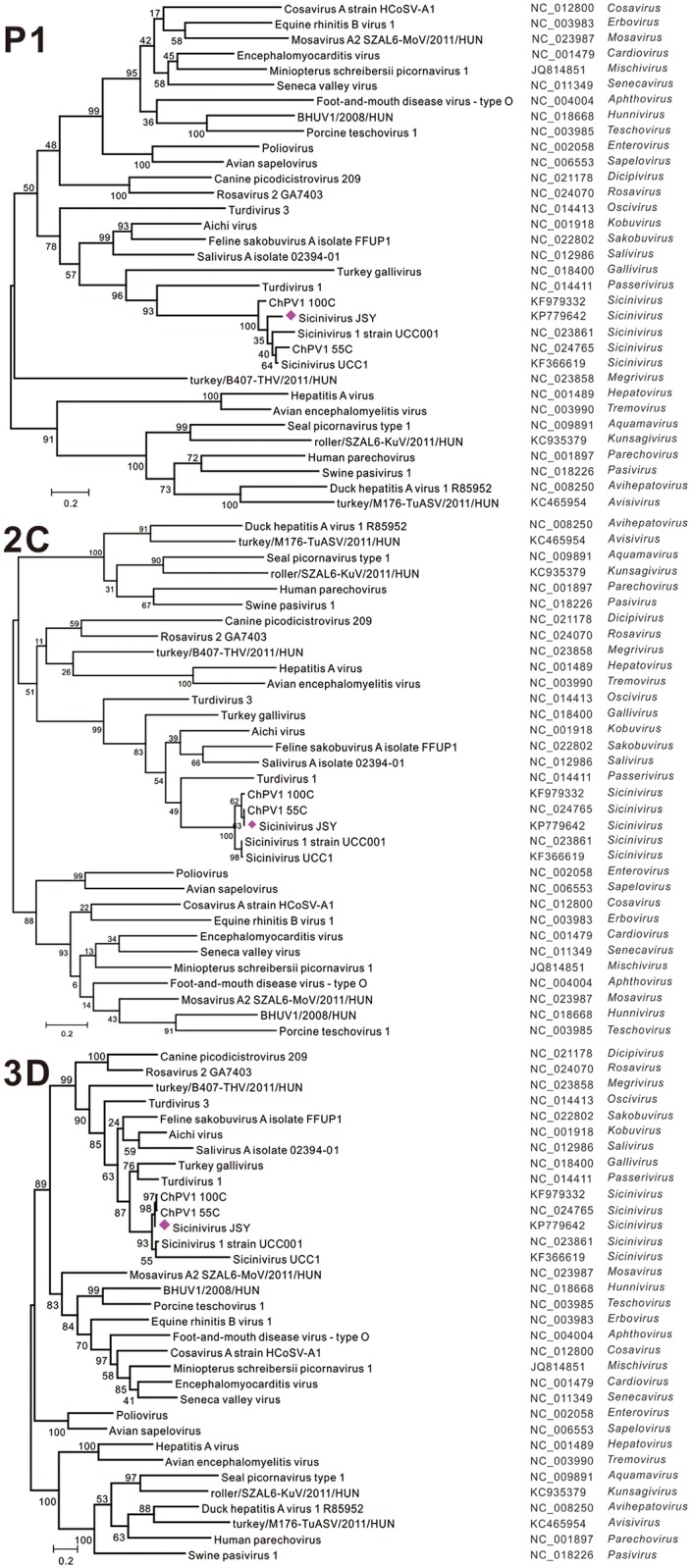
Phylogenetic relationship between Sicinivirus strain JSY and the representative members of the 29 officially recognized genera based upon the complete amino acid sequences of picornavirus P1, 2C, and 3D coding regions. The phylogenetic tree was constructed using the Molecular Evolutionary Genetics Analysis (MEGA) [17] applying the maximum-likelihood method based on the JTT matrix-based model [18], the robustness of the phylogenetic constructions was evaluated by bootstrapping with 1,000 replicates, initial trees for the heuristic search were obtained automatically by applying neighbour-join and BioNJ algorithms.
Confirmation of Sicinivirus in clinical faecal samples
Sicinivirus distribution in Mainland China is not restricted to one sample analysed by metagenomics. A RT-PCR survey targeted to a 652 bp fragment of the 3Dpol gene of Sicinivirus demonstrated the presence of this virus in stool samples of other 8 animals from the same farm. Furthermore, Sicinivirus was also detected in 5 and 3 animals of two other commercial chicken farms of the same region (Fig 7). The generated 17 PCR products in total were further confirmed by sequencing, and these PCR products shared almost 100% nucleotide sequence identity with the Sicinivirus strain JSY.
Fig 7. Detection of Sicinivirus in clinical stool samples by RT-PCR.

M, Molecular marker D2000; Lanes 1–9, stool samples from farm A; Lanes 10–14, stool samples from farm B; Lanes 15–17, stool samples from farm C; Lanes 18, negative control. The fecal sample that was used to perform the metagenomic analysis is indicated with an orangey-red asterisk (Lane 4).
Discussion
A range of random amplification methods coupled with Next Generation Sequencing platforms, such as Roche-454 [32], Illumina HiSeq [33], and Ion Torrent PGM [34], have been recently used to discover unexpected and unknown viruses. However, plasmid cloning and Sanger sequencing remains still as a practical alternative for the rapid identification of viruses in clinical samples. In the present study, we chose to amplify the constructed library, and clone the purified fragments into traditional T-vector as previously described by Victoria et al (2008) [35]. BLAST analysis showed that 34 of the obtained 511 viral metagenomic sequences (6.65%) shared nucleic acid similarities to Sicinivirus. The results revealed that the described approach might be a relatively convenient and cost-effective method for quick screening unexpected or unknown viruses from clinical samples.
By comparing with other Sicinivirus strains, the Sicinivirus strain JSY identified in the present study shares 73.99%, 78.81%, 79.61%, and 73% nucleotide sequence identities with Sicinivirus 1 strain UCC001 [6], chicken picornavirus 1 100C, chicken picornavirus 1 55C, and Sicinivirus UCC1 [1] at the genome level, respectively. Bullman et al (2014) reported that the A-I domains of type II IRES were missed in the Sicinivirus 1 strain UCC001, thus indicating that the 373nt 5' UTR sequence of the strain was incomplete. Lau et al (2014) reported the sequences of chicken picornavirus 1 100C and chicken picornavirus 1 55C, these two strains share similar genome structure, including a short Leader (L) protein (21 aa) but not the predicted type II-like IRES [1] (Fig 2A). In addition, the polyprotein of the identified Sicinivirus strain JSY shows high amino acid identities (88.13% and 88.13%) to ChPV1_55C and ChPV1_100C, respectively (Table 2), which indicates the close genetic relationship of Sicinivirus strain JSY with the two Siciniviruses that were found in Hong Kong [1]. However, when the upstream sequences (about 880 bp upstream of VP0 gene) of ChPV1_55C and ChPV1_100C were compared with the corresponding region of the Sicinivirus strain JSY reported in this study, the nucleotide sequence identity was 74.9% and 66.6%, respectively, suggesting that the 5' region of these two strains identified in Hong Kong probably have not been sequenced completely. The other Sicinivirus UCC1 was also identified in Ireland by Bullman et al (Unpublished), it only contains a partial L sequence (Fig 2A) and the upstream sequence has not been fully sequenced as well.
Considerable variation was observed in the P1 region, the identified Sicinivirus strain JSY shows 75.27% to 78.66% amino acid sequence identity to four other Sicinivirus sequences (Table 2). The P1 region comprises virus capsid proteins VP0, VP3, and VP1. As viral polypeptide VP1 is the most surface-exposed capsid protein [36], and harbors important immunogenic sites [37,38], also contributes to virus attachment and entry [39,40]. Therefore, the VP1 region of picornaviruses, such as foot-and-mouth disease virus (FMDV), has been conventionally used to investigate the genetic relatedness of different isolates [41] and infer evolutionary dynamics including tracing the origin and movement of the outbreak strains [42]. Among these Sicinivirus strains, two pocket sites I118 and L120 (Fig 5) were seen in the VP1 polypeptide, which are different from the rhv_like capsid domain (cd00205). However, the effect of the differences on structure, pathogenicity and evolution still needs further evaluation.
For picornaviruses, five types of IRES have been reported, classified as type I [43], type II [44], type III [45], type IV [46] and type V [47], each type of IRES has a different characteristic structure and initiation occurs via a distinct mechanism [47]. Type II IRES requires eIF4G/eIF4A to form a 48S complex, and can function without eIF4E and factors associated with ribosomal scanning. Unlike type I IRES, which requires bind various IRES trans-acting factors (ITAFs), type II IRES requires fewer ITAFs [44,47]. Further analysis of the 5' UTR region showed that the Sicinivirus strain JSY contains a type II IRES. In the present study, we determined the full-length sequence of the 5' UTR region of Sicinivirus and found that the 5' UTR of Sicinivirus contains at least twelve stem-loop domains (A-L) for the first time.
Besides the predominant Sicinivirus, we also obtained other virus-related reads, including ALV (3 reads), ANV (2 reads), two other genera (also of Picornaviridae family) from the present study. Three ALV-related reads shared 99.5%, 100% and 94.1% nucleotide sequence homology with endogenous ALV [48], natural recombinant ALV-E/A virus PDRC-1039 [49] and ALV strain BR170E, respectively. The two ANV reads shared 76.1% and 89.4% nucleotide sequence homology with ANV1 and ANV3, respectively [50,51]. However, the whole genomic sequencing of these viruses was not successful due mainly to the low homology of these obtained reads, and the low abundance of associated viruses in the clinical samples.
Conclusions
In the present study, the viral metagenomics technique has been used to test a clinical sample. Unexpectedly, among 511 reads, 34 reads (82.9% of total virus reads) showed sequence similarities to viruses from the recently discovered genus Sicinivirus, suggesting that this virus was the predominant type in the tested sample. The viral polyprotein of the Sicinivirus isolate only had 88.83% and 82.78% of amino acid sequence identity to that of ChPV1 100C and Sicinivirus 1 strain UCC001, respectively. Moreover, we determined for the first time the full-length sequence of the 5' UTR region of Sicinivirus and found that it contains at least twelve stem-loop domains (A-L). Sicinivirus infection might be widely present in commercial chicken farms in Yancheng region of the Jiangsu Province as evidenced by all the tested stool samples from three different chicken farms being positive (17/17) for Sicinivirus by RT-PCR detection. This is the first report on identification and genome characterization of Sicinivirus from chickens in mainland China, however, further studies are needed to evaluate the pathogenic potential of this picornavirus in chickens.
Data Availability
All relevant data are within the paper.
Funding Statement
This work was supported by a grant from China Agriculture Research System (CARS-42). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Lau SK, Woo PC, Yip CC, Li KS, Fan RY, Bai R, et al. (2014) Chickens host diverse picornaviruses originated from potential interspecies transmission with recombination. J Gen Virol 95(Pt 9):1929–1944. 10.1099/vir.0.066597-0 [DOI] [PubMed] [Google Scholar]
- 2. Willner D, Furlan M, Haynes M, Schmieder R, Angly FE, Silva J, et al. (2009) Metagenomic analysis of respiratory tract DNA viral communities in cystic fibrosis and non-cystic fibrosis individuals. PLoS One 4(10):e7370 10.1371/journal.pone.0007370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Honkavuori KS, Shivaprasad HL, Briese T, Street C, Hirschberg DL, Hutchison SK, et al. (2011) Novel picornavirus in Turkey poults with hepatitis, California, USA. Emerg Infect Dis 17(3):480–487. 10.3201/eid1703.101410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boros A, Nemes C, Pankovics P, Kapusinszky B, Delwart E, Reuter G (2012) Identification and complete genome characterization of a novel picornavirus in turkey (Meleagris gallopavo). J Gen Virol 93(Pt 10):2171–2182. 10.1099/vir.0.043224-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boros A, Nemes C, Pankovics P, Kapusinszky B, Delwart E, Reuter G (2013) Genetic characterization of a novel picornavirus in turkeys (Meleagris gallopavo) distinct from turkey galliviruses and megriviruses and distantly related to the members of the genus Avihepatovirus. J Gen Virol 94(Pt 7):1496–1509. 10.1099/vir.0.051797-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bullman S, Kearney K, O'Mahony M, Kelly L, Whyte P, Fanning S, et al. (2014) Identification and genetic characterization of a novel picornavirus from chickens. J Gen Virol 95(Pt 5):1094–1103. 10.1099/vir.0.061085-0 [DOI] [PubMed] [Google Scholar]
- 7. Lau SK, Yip CC, Tsoi HW, Lee RA, So LY, Lau YL, et al. (2012) Clinical features and complete genome characterization of a distinct human rhinovirus (HRV) genetic cluster, probably representing a previously undetected HRV species, HRV-C, associated with acute respiratory illness in children. J Clin Microbiol 45(11):3655–3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Woo PC, Lau SK, Choi GK, Huang Y, Teng JL, Tsoi HW, et al. (2012) Natural occurrence and characterization of two internal ribosome entry site elements in a novel virus, canine picodicistrovirus, in the picornavirus-like superfamily. J Virol 86(5):2797–2808. 10.1128/JVI.05481-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boros A, Pankovics P, Knowles NJ, Nemes C, Delwart E, Reuter G (2014) Comparative complete genome analysis of chicken and Turkey megriviruses (family picornaviridae): long 3' untranslated regions with a potential second open reading frame and evidence for possible recombination. J Virol 88(11):6434–6443. 10.1128/JVI.03807-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Victoria JG, Kapoor A, Li L, Blinkova O, Slikas B, Wang C, et al. (2009) Metagenomic analyses of viruses in stool samples from children with acute flaccid paralysis. J Virol 83(9):4642–4651. 10.1128/JVI.02301-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Allander T, Tammi MT, Eriksson M, Bjerkner A, Tiveljung-Lindell A, Andersson B (2005) Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc Natl Acad Sci U S A 102(36):12891–12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Blom N, Hansen J, Blaas D, Brunak S (1996) Cleavage site analysis in picornaviral polyproteins: discovering cellular targets by neural networks. Protein Sci 5(11):2203–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Research 31(13):3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gruber AR, Lorenz R, Bernhart SH, Neubock R, Hofacker IL (2008) The Vienna RNA websuite. Nucleic Acids Research 36(Web Server issue):W70–4. 10.1093/nar/gkn188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. De Rijk P, Wuyts J, De Wachter R (2003) RnaViz 2: an improved representation of RNA secondary structure. Bioinformatics 9(2):299–300. [DOI] [PubMed] [Google Scholar]
- 16. Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30(12):2725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jones DT, Taylor WR, Thornton JM (1992) The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 8(3):275–282. [DOI] [PubMed] [Google Scholar]
- 19. Liu J, Wei L, Jiang T, Shi L, Wang J (2007) Reduction of infectious bursal disease virus replication in cultured cells by proteasome inhibitors. Virus Genes 35(3):719–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ji J, Xie J, Chen F, Shu D, Zuo K, Xue C, et al. (2011) Phylogenetic distribution and predominant genotype of the avian infectious bronchitis virus in China during 2008–2009. Virol J 8:184 10.1186/1743-422X-8-184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wei L, Chee LL, Wei T, Kwang J, Zhou J, Wang J, et al. (2008) The VP1 protein of avian encephalomyelitis virus is a major host-protective immunogen that serves as diagnostic potential. J Virol Methods 49(1):56–62. [DOI] [PubMed] [Google Scholar]
- 22. Noteborn MH, Verschueren CA, Van Roozelaar DJ, Veldkamp S, Van Der Eb AJ, de Boer GF (1992) Detection of chicken anaemia virus by DNA hybridization and polymerase chain reaction. Avi Pathol 21(1):107–118. [DOI] [PubMed] [Google Scholar]
- 23. Stewart SR, Semler BL (1997) RNA determinants of picornavirus cap-independent translation initiation. Seminars Virol 8(3):242–255. [Google Scholar]
- 24. Ghazi F, Hughes PJ, Hyypia T, Stanway G (1998) Molecular analysis of human parechovirus type 2 (formerly echovirus 23). J Gen Virol 79 (Pt 11):2641–2650. [DOI] [PubMed] [Google Scholar]
- 25. Belsham GJ (2009) Divergent picornavirus IRES elements. Virus Res 139(2):183–192. 10.1016/j.virusres.2008.07.001 [DOI] [PubMed] [Google Scholar]
- 26. Tseng CH, Knowles NJ, Tsai HJ (2007) Molecular analysis of duck hepatitis virus type 1 indicates that it should be assigned to a new genus. Virus Res 123(2):190–203. [DOI] [PubMed] [Google Scholar]
- 27. Gorbalenya AE, Koonin EV, Wolf YI (1990) A new superfamily of putative NTP-binding domains encoded by genomes of small DNA and RNA viruses. FEBS Lett 262(1):145–148. [DOI] [PubMed] [Google Scholar]
- 28. Grant RA, Hiremath CN, Filman DJ, Syed R, Andries K, Hogle JM (1994) Structures of poliovirus complexes with anti-viral drugs: implications for viral stability and drug design. Curr Biol 4(9):784–797. [DOI] [PubMed] [Google Scholar]
- 29. Phelps DK, Speelman B, Post CB (2000) Theoretical studies of viral capsid proteins. Curr Opin Structural Biol 10(2):170–173. [DOI] [PubMed] [Google Scholar]
- 30. Sauvage V, Ar Gouilh M, Cheval J, Muth E, Pariente K, Burguiere A, et al. (2012) A member of a new picornaviridae genus is shed in pig feces. J Virol 86(18):10036–10046. 10.1128/JVI.00046-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Woo PC, Lau SK, Huang Y, Lam CS, Poon RW, Tsoi HW, et al. (2010) Comparative analysis of six genome sequences of three novel picornaviruses, turdiviruses 1, 2 and 3, in dead wild birds, and proposal of two novel genera, orthoturdivirus and paraturdivirus, in the family picornaviridae. J Gen Virol 91(Pt 10):2433–2448. 10.1099/vir.0.021717-0 [DOI] [PubMed] [Google Scholar]
- 32. Reuter G, Boros A, Kiss T, Delwart E, Pankovics P (2014) Complete genome characterization of mosavirus (family picornaviridae) identified in droppings of a European roller (Coracias garrulus) in Hungary. Arch Virol 159(10):2723–2729. 10.1007/s00705-014-2113-4 [DOI] [PubMed] [Google Scholar]
- 33. Boros Á, Pankovics P, Adonyi Á, Phan TG, Delwart E, Reuter G (2014) Genome characterization of a novel chicken picornavirus distantly related to the members of genus avihepatovirus with a single 2A protein and a megrivirus-like 3′ UTR. Infection, Genetic Evol 28(0):333–338. [DOI] [PubMed] [Google Scholar]
- 34. Jakhesara SJ, Prasad VV, Pal JK, Jhala MK, Prajapati KS, Joshi CG (2014) Pathotypic and sequence characterization of Newcastle disease viruses from vaccinated chickens reveals circulation of genotype II, IV and XIII and in India. Transbound Emerg Dis 10.1111/tbed.12294 [DOI] [PubMed] [Google Scholar]
- 35. Victoria JG, Kapoor A, Dupuis K, Schnurr DP, Delwart EL (2008) Rapid identification of known and new RNA viruses from animal tissues. PloS Pathogens 4(9):e1000163 10.1371/journal.ppat.1000163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Acharya R, Fry E, Stuart D, Fox G, Rowlands D, Brown F (1989) The three-dimensional structure of foot-and-mouth disease virus at 2.9 A resolution. Nature 337(6209):709–716. [DOI] [PubMed] [Google Scholar]
- 37. Logan D, Abu-Ghazaleh R, Blakemore W, Curry S, Jackson T, King A, et al. (1993) Structure of a major immunogenic site on foot-and-mouth disease virus. Nature. 362(6420):566–568. [DOI] [PubMed] [Google Scholar]
- 38. Beck E, Feil G, Strohmaier K (1983) The molecular basis of the antigenic variation of foot-and-mouth disease virus. EMBO J 2(4):555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jackson T, King AM, Stuart DI, Fry E (2003) Structure and receptor binding. Virus Res 91(1):33–46. [DOI] [PubMed] [Google Scholar]
- 40. Burman A, Clark S, Abrescia NG, Fry EE, Stuart DI, Jackson T (2006) Specificity of the VP1 GH loop of foot-and-mouth disease virus for alphav integrins. J Virol 80(19):9798–9810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Beck E, Strohmaier K (1987) Subtyping of European foot-and-mouth disease virus strains by nucleotide sequence determination. J Virol 61(5):1621–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Samuel AR, Knowles NJ (2001) Foot-and-mouth disease type O viruses exhibit genetically and geographically distinct evolutionary lineages (topotypes). J Gen Virol 82(Pt 3):609–621. [DOI] [PubMed] [Google Scholar]
- 43. de Breyne S, Yu Y, Unbehaun A, Pestova TV, Hellen CU (2009) Direct functional interaction of initiation factor eIF4G with type 1 internal ribosomal entry sites. Proc Natl Acad Sci U S A 106(23):9197–9202. 10.1073/pnas.0900153106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Niepmann M (2009) Internal translation initiation of picornaviruses and hepatitis C virus. Biochim Biophys Acta 1789(9–10):529–541. 10.1016/j.bbagrm.2009.05.002 [DOI] [PubMed] [Google Scholar]
- 45. Ali IK, McKendrick L, Morley SJ, Jackson RJ (2001) Activity of the hepatitis A virus IRES requires association between the cap-binding translation initiation factor (eIF4E) and eIF4G. J Virol 75(17):7854–7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hellen CU, de Breyne SA (2007) distinct group of hepacivirus/pestivirus-like internal ribosomal entry sites in members of diverse picornavirus genera: evidence for modular exchange of functional noncoding RNA elements by recombination. J Virol 81(11):5850–5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sweeney TR, Dhote V, Yu Y, Hellen CU (2012) A distinct class of internal ribosomal entry site in members of the kobuvirus and proposed salivirus and paraturdivirus genera of the picornaviridae. J Virol 86(3):1468–1486. 10.1128/JVI.05862-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Smith A, Benkel BF (2009) Novel avian leukosis virus-related endogenous proviruses from layer chickens: characterization and development of locus-specific assays. Poultry Sci 88(8):1580–1585. [DOI] [PubMed] [Google Scholar]
- 49. Barbosa T, Zavala G, Cheng S (2008) Molecular characterization of three recombinant isolates of avian leukosis virus obtained from contaminated Marek's disease vaccines. Avi Dis 52(2):245–252. [DOI] [PubMed] [Google Scholar]
- 50. Zhao W, Hua XG, Yuan L, Cui L, Shan TL, Dai XQ, et al. (2011) Sequence analyses of the representative Chinese-prevalent strain of avian nephritis virus in healthy chicken flocks. Avi Dis 55(1):65–69. [DOI] [PubMed] [Google Scholar]
- 51. de Wit JJ, Dam GB, de Laar JM, Biermann Y, Verstegen I, Edens F, et al. (2011) Detection and characterization of a new astrovirus in chicken and turkeys with enteric and locomotion disorders. Avi Pathol 40(5):453–461. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.