

Abstract
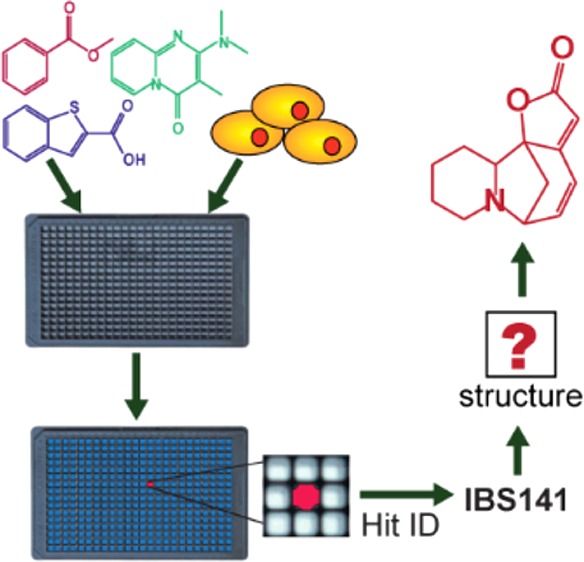
Compound libraries provide a starting point for multiple biological investigations, but the structural integrity of compounds is rarely assessed experimentally until a late stage in the research process. Here, we describe the discovery of a neuroprotective small molecule that was originally incorrectly annotated with a chemical structure. We elucidated the correct structure of the active compound using analytical chemistry, revealing it to be the natural product securinine. We show that securinine is protective in a cell model of Huntington disease and identify the binding site of securinine to its target, protein disulfide isomerase using NMR chemical shift perturbation studies. We show that securinine displays favorable pharmaceutical properties, making it a promising compound for in vivo studies in neurodegenerative disease models. In addition to finding this unexpected activity of securinine, this study provides a systematic roadmap to those who encounter compounds with incorrect structural annotation in the course of screening campaigns.
Keywords: Structure elucidation, high-throughput screen, natural product, PDI, neuroprotection
Small molecule screening libraries have been widely used in academia to understand biological pathways, to discover novel protein targets, and to discover protein inhibitors. Screening chemically diverse small molecules in phenotypic and biochemical assays has been a starting point to identify compounds with desired biological effects from large libraries of compounds. Alternatively, focused chemical libraries, assembled around an active compound, have been used to determine structure–activity relationships. Fragment libraries are emerging as a complementary strategy to high-throughput screening to identify lead compounds for drug design. These compound libraries can be synthesized or purchased, depending on the scope of the project. Regardless of the type of chemical library or its source, there is always likely to be some incorrect compound, due to errors in compound synthesis, characterization, formatting, and annotation, as well as unavoidable compound degradation.
If incorrect structure annotation involves inactive compounds, the errors are undetectable and less significant; although one must be wary of inferring structure–activity relationships from such negative results in high-throughput screening data. Irrespective, incorrect structure annotation becomes a significant problem when an active compound is mis-annotated. Such a situation occurred when we performed a phenotypic high-throughput screen for small molecules that prevent cell death in a cell model of Huntington’s disease (HD).1 Here, we describe how we were able to identify an unknown active compound that had been mischaracterized in our compound libraries and describe a systematic approach for revealing the identities of unknown small molecules encountered in high-throughput screens. Additionally, we identified the biophysical mechanism of action of the neuroprotective hit compound.
HD is a neurodegenerative disorder, characterized by a selective loss of medium spiny neurons in the striatum,2 due to the presence of mutant huntingtin gene (HTT). HTT normally has an average of 19 CAG repeats in its first exon, but pathogenesis develops when the CAG tract expands beyond 36 repeats. This produces an extensive polyglutamine repeat expansion near the N-terminus in the ubiquitously expressed huntingtin protein3 causing it to aggregate and induce neuronal toxicity and neuronal death. The mechanism underlying mutant-huntingtin-induced cell death in the striatum remains obscure; hence, developing therapeutic treatments for the disease has been challenging.
To address this challenge, we implemented a phenotypic high-throughput screen for small molecules that inhibit cell death in a PC12 cell culture model of HD.1 In this model, the PC12 cells were stably transfected with an inducible mutant-HTT plasmid.4 Induction of mutant HTT protein expression (mHTT-Q103) leads to cell death within 48 h, which was monitored using a fluorescent viability dye, Alamar blue.
We assembled a diverse small molecule library totaling 68,887 compounds from numerous commercial vendors. These compounds included natural products and their derivatives, synthetic drug-like compounds prefiltered in silico to penetrate the blood–brain barrier, and compounds with published biological activity.1 We screened this library in PC12 mHTT-Q103 cells to identify neuroprotective small molecules that prevent mutant-HTT-induced apoptosis. The identified hits from the primary screen were retested in an array of serial dilutions to validate the desired biological activity. One such compound, IBS141, was a potent small molecule hit that showed reproducible activity in this PC12 mHTT-Q103 cell viability assay with EC50 of 3.3 μM (Figure 1a). We pursued this compound in follow-up assays to probe its biological activity.1
Figure 1.

Neuroprotective hit compound, IBS141, has an incorrect structure annotation. (a) Overview of high-throughput screen used to identify IBS141. Reported structure of IBS141 in the vendor database. Viability dose–response curve of IBS141 in PC12 mHTT-Q103 cells. (b) Viability dose–response curve and proton NMR spectra of IBS141 and CBG141. CBG141 is a compound with the same annotated structure as IBS141 but from a different chemical supplier. All viability data represent the average of triplicates ± SD (n = 3) and plotted as percent of DMSO treated uninduced cells.
To establish the chemical stability of IBS141, we measured the mass and biological activity of the compound after incubation in aqueous buffer. IBS141 was incubated at 37 °C for zero, one, two, or three days in PC12 growth medium before aliquots were analyzed by LC–MS (for mass determination) or added to induced PC12 mHTT-Q103 cells (for biological activity evaluation). After 48 h of treatment, Alamar blue was added to cells and viability assessed on a fluorescence plate reader by quantifying the fluorescence of the dye. IBS141 showed the same neuroprotection in PC12 mHTT-Q103 cells at all the time points (Figure S1a, Supporting Information), indicating that heat incubation did not degrade its biological activity. However, by LC–MS, IBS141 showed a single mass peak of a 218.1 m/z at all the time points (Figure S1b, Supporting Information). This was unexpected because the predicted mass of IBS141, based on its assigned structure in the database, was 269.38. We hypothesized that this 51 m/z difference between observed and predicated mass might be due to the degradation of the original IBS141 stock. To test this, we purchased a fresh batch of IBS141 from the same supplier (InterBioScreen, cat. # STOCK1N-05465). The new batch of IBS141 had the same biological activity and potency in PC12 mHTT-Q103 cells (Figure 1b), and the same mass of a 218.1 m/z as the original batch, but still differing from the mass assigned to its structure in the vendor database. Next, we purchased the compound with the assigned structure of IBS141 from a different vendor (ChemBridge, cat. # 5118173). This new compound, CBG141, had the correct expected mass by LC–MS of a 269.38 m/z, but no activity in the PC12 mHTT-Q103 cell viability assay (Figure 1b). Furthermore, the proton (1H) NMR of CBG141 and IBS141 showed two different NMR spectra (Figure 1b). The major difference is seen in the 6–7 ppm region of the proton spectra; IBS141 shows 1H resonance signals in this region, but CBG141 does not. Protons with chemical shifts observed at 6–7 ppm are due to the presence of vinylic or aromatic groups. As there were no protons in the putative IBS141 structure that could generate such peaks, this was an indication that the structure of this hit compound was not consistent with its assigned structure in our database.
It is not uncommon to encounter mischaracterized compounds in the course of HTS campaigns. Such mischaracterization can occur due to compound degradation, robotic formatting errors, sample contamination, or supplier errors. Frequently, when researchers encounter this situation, they simply abandon the active compound, as structure elucidation can be challenging. However, in this case, we attempted to elucidate the structure of IBS141 due to its rare and striking activity in the assay and because we sought a systematic approach to solve such problems in the future.
To elucidate the structure of IBS141, thorough spectroscopic and analytical characterization were performed (Figure 2a). High-resolution mass spectrometry (HRMS) yielded a molecular composition of C13H15O2N, a molecular weight of 217.267, and a monoisotopic mass of 217.110279 m/z for the unknown active compound. Additionally, elemental analysis was performed to identify traces of C, H, N, B, P, S, Cl, and F elements. Results from elemental analysis confirmed the same molecular composition as was determined by HRMS, C13H15O2N. From the molecular formula, an index of hydrogen deficiency (also known as degree of unsaturation) was calculated. The unknown IBS141 had seven degrees of unsaturation, indicating that there were a total of seven rings, double bonds, and/or triple bonds in the structure.
Figure 2.

Systematic approach that helped determine the structure of unknown active IBS141 is securinine. (a) Analytical chemistry experiments and the conclusions from the data. (b) SciFinder database search of the acquired data on the unknown IBS141 identifies securinine as the single structure to match all the criteria.
Next, we used TLC stains to tests for the presence of specific functional groups that would account for one nitrogen and two oxygen elements in the compound’s molecular formula. The ninhydrin stain, which tests for the presence of primary and secondary amines, was negative, indicating that there might be a tertiary amine, a nitro group, a nitrile, or an amide in the structure. The bromocresol green stain, which tests for presence of functional groups with a pKa of less than five, was negative, indicating that no carboxylic acids were present in the structure. Schiff’s reagent that tests for the presence of aldehydes and the 2,4-dinitrophenylhydrazine (DNPH) reagent that stains for ketones and aldehydes were both negative. This indicated that there was an alcohol, an ether, an epoxide, or an ester as an oxygen-containing functional group in the compound.
To further elucidate the presence of functional groups, an IR spectrum was obtained (Figure 2a). The unknown active substance had a characteristic strong carbonyl peak at wavenumber 1738.21 cm–1 and a C–O bond vibration band in the 1300–1000 cm–1 region (with a major peak at 1250.87 cm–1). This suggested the presence of an ester functional group. Furthermore, the absence of peaks at 2300–2100 cm–1 indicated no alkynes or nitriles. There were also no absorptions beyond 3000 cm–1, which ruled out the presence of aromatic, alcohol, or amide groups. From molecular formula we knew there was a nitrogen in the structure. The absence of strong N–H stretch at 3500–3300 cm–1 indicated that it must be a tertiary amine, which does not have an N–H bond vibration. We also observed a strong absorption at 1626.49 cm–1. Since we ruled out the presence of primary amines, a peak in this position is indicative of an alkene C=C bond stretch.
Next, we performed 1H NMR and 13C NMR analyses (Figure 2a). As mentioned earlier, the proton spectrum showed resonance signals in the 7.0–5.5 ppm region, with a singlet at 5.5 ppm, a doublet at 6.60 ppm, and a doublet of doublets at 6.41 ppm. Since IR ruled out the presence of aromatic groups, this was indicative of vinylic conjugated protons.
The standard 1D 13C NMR produced 13 peaks, each corresponding to a carbon atom in the unknown active IBS141. To determine the carbon multiplicity, DEPT-135 13C NMR was performed. The DEPT-135 pulse sequence shows primary and tertiary carbon peaks with a positive phase, while secondary carbon peaks show signals with a negative phase. To distinguish between primary and tertiary carbons, we also performed DEPT-90 13C NMR; this spectrum is enhanced for signals from tertiary carbons only. With this information, we were able to identify the multiplicity of each carbon in the standard 1D 13C NMR spectrum (Figure 2a). After assigning each carbon, we searched 13C NMR databases for molecules that could generate the unique peak pattern of quaternary and tertiary carbons seen from 180 to 85 ppm. Our search resulted in three possible substructures that matched the observed pattern (Figure 2a).
Figure 2a summarizes our findings at this stage. We knew the unknown active compound had the molecular formula of C13H15NO2, with no other atoms present. It was a multiring compound with at least one double bond and a sum of seven rings and/or double bonds. An amine and an ester were the only functional groups present within a conjugated π, but nonaromatic, system. The final compound had to contain one of the three possible substructures observed in 13C NMR spectrum. We searched the SciFinder database, which allows for further filtering. First, we searched for all entries with the molecular formula of C13H15NO2, which yielded 3952 compounds. Then, we limited these 3952 structures to those containing one of the three substructures identified from NMR studies. Lastly, we eliminated any compounds with aromatic functional groups. These criteria resulted in only one compound, the natural product securinine (Figure 2b).
We purchased genuine securinine from TimTec (cat. # ST057165) and tested it alongside IBS141 in the PC12 mHTT-Q103 cell viability assay (Figure 3a). Securinine gave an identical activity and potency profile as unknown active IBS141 in the cell viability assay. Furthermore, NMR analysis of securinine showed that both 1H NMR and 13C NMR spectra of securinine were identical to those of IBS141 (Figure 3b,c). From these data we concluded that IBS141 was securinine.
Figure 3.
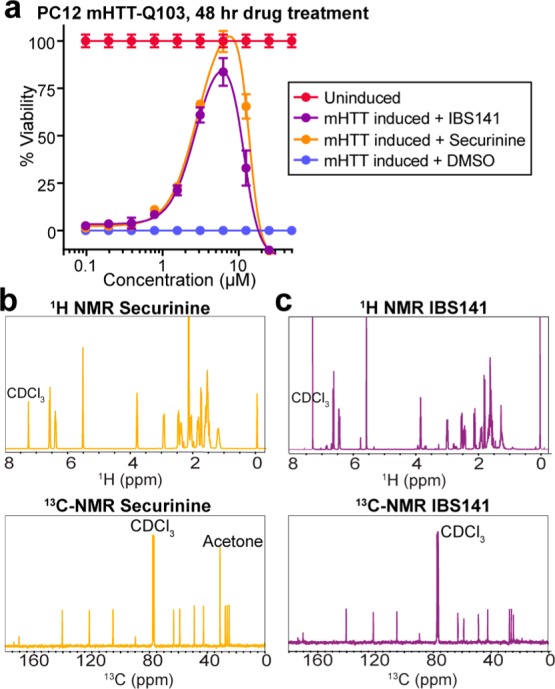
Validating the biological and chemical properties of securinine as the correct annotated structure for IBS141. (a) Viability dose–response curve in PC12 mHTT-Q103 cells of securinine and IBS141. Data are plotted as mean percent of DMSO treated uninduced cells ± SD. Experiments were performed in triplicate. Proton and carbon NMR spectra of (b) securinine and (c) IBS141 are identical.
After determining the correct structure of the neuroprotective compound, we investigated the biophysical mechanism of action of securinine. We previously showed that securinine can competitively inhibit compound 16F16 binding to protein disulfide isomerase (PDI).1 16F16 is an irreversible PDI inhibitor that covalently modifies two cysteines in the active site of PDI.5 We hypothesized that securinine may also be interacting at the same site on PDI as 16F16. To better understand this interaction, we used 1H–15N heteronuclear single quantum correlation (HSQC) NMR studies to identify the binding site of securinine on PDI. For these NMR studies we expressed and purified 15N-labled catalytic a domain of PDI (referred to as PDIa). The a domain contains the CGHC active site, can perform the same reduction and oxidation reactions as full length PDI,6 and is small enough (15 kDa) for NMR binding studies. Upon securinine binding to PDIa numerous chemical shift perturbations were observed in the protein resonance peaks (Figure 4a). The most notable changes were seen in peaks that substantially broadened (disappeared) with compound titration (i.e., R80, E98, W111, and A43); majority of these were from the residues around the CGHC active site of PDI (Figure 4b). Another significant change was observed in the appearance of H38 peak upon compound titration. In the protein alone spectrum H38 is undergoing a rapid hydrogen exchange with the solvent that accounts for the absence of its HSQC peak.7 Presence of securinine prevents this exchange. Additional chemical shifts were in the region opposite the active site (i.e., L10, A23, H24, Y26, N90, and G91). These shifts are indicative of the conformational change in the protein upon securinine binding. Based on the mapped chemical shift perturbation data, securinine may be binding adjacent to the active site of PDI and inducing a conformation change in the protein. Securinine interaction with H38 and R103 of PDI may lead to destabilization of active site C36 residue, thus inhibiting PDI function. Both H38 and R103 residues are needed to stabilize the negatively charged cysteine thiolate transition state7 of PDI. An alternative mechanism of inhibition is that the α,β-unsaturated lactone pharmacophore in securinine may be acting as a Michael acceptor for the nucleophilic attack by H38.8 This would result in a formation of an irreversible complex between securinine and PDI, leading to PDI inhibition.
Figure 4.
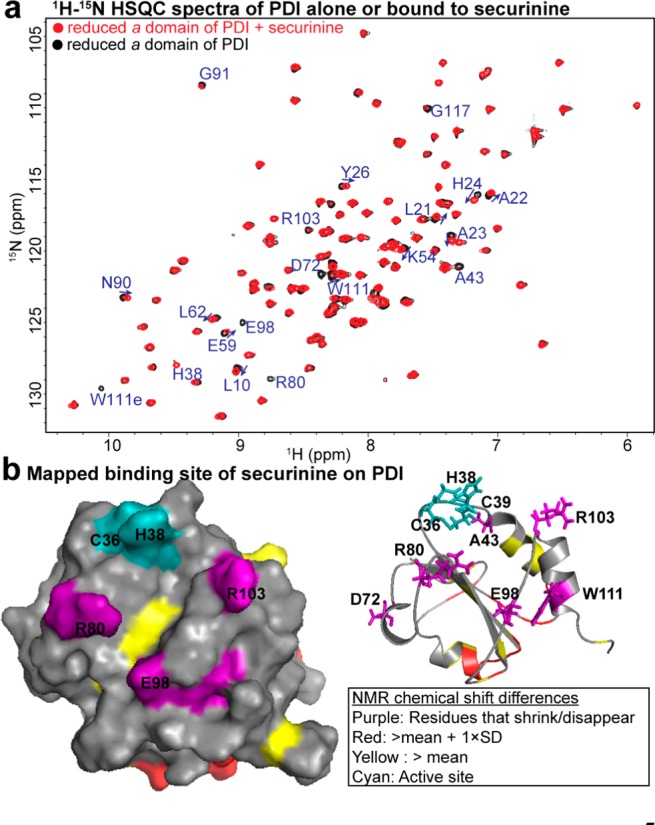
Chemical shift perturbations upon securinine binding to PDI. (a) 15N-HSQC spectra of PDIa alone or liganded with securinine. Most perturbed residues are in blue. (b) Chemical shift changes mapped onto the surface and ribbon depiction of PDIa (PDB: 1MEK, model #35). The residue numbering is based on the mature PDI protein sequence.
To investigate the binding mechanism further, we monitored the intrinsic tryptophan (Trp) fluorescence of PDIa in the presence of securinine. As shown in Figure 5a, Trp fluorescence was strongly quenched upon addition of securinine in a concentration-dependent manner with a dissociation constant (Kd) of 758 ± 129 μM (Figure 5b). Next, we monitored Trp fluorescence of PDIa after performing the jump dilution method to test for reversibility of binding. PDIa (1 mM) was incubated with high concentration of securinine (4.5 mM). The complex was then diluted 100-fold and Trp fluorescence recorded. As a control, the same procedure was performed on a sample containing phenylarsine oxide (PAO), a covalent irreversible modifier of vicinal thiol groups such as those found in the active site of PDI. After dialysis, the securinine–PDIa complex (now at 45 μM securinine and 10 μM PDIa) and control PAO–PDIa samples still showed complete quenching of intrinsic Trp fluorescence (Figure 5c). This result indicated that securinine, like PAO, binds irreversibly to PDI.
Figure 5.

Intrinsic tryptophan fluorescence assay shows irreversible binding of securinine to PDI. (a) Fluorescence emission spectra and (b) normalized intensity at 330 nm of PDIa in the presence of varying concentrations of securinine. (c) Emission spectra of PDIa after ligand–protein complex is formed at high concentration and then diluted 100-fold. The Trp fluorescence is still quenched in securinine treated samples and in PAO control after the dilution indicating irreversible binding to PDIa by the ligand. All spectra are normalized by the maximum fluorescence intensity of the PDIa-only sample (black). Data are plotted as mean ± SEM and fitted to a log-normal distribution.
Next, we wanted to investigate if securinine would be a good candidate for in vivo studies by evaluating its in vitro metabolic stability. Securinine had a low intrinsic clearance value of 8.3 mL/min/g in mouse liver microsomes compared to the control compound 7-ethoxycoumarin (intrinsic clearance of 24.58 mL/min/g) (Table S1, Supporting Information), and it was also relatively stable in mouse plasma with a half-life of 1.6 h (Table S2, Supporting Information). Furthermore, only 49% of securinine is bound to plasma proteins, compared to warfarin, an anticoagulant agent, with 94% plasma protein binding (Table S3, Supporting Information), indicating that in vivo more than 50% of securinine may be free to be distributed to tissues to exert pharmacological effects.
Securinine is a naturally occurring alkaloid that can be extracted from the plant Securinega suffruticosa. It has been shown to act as a central nervous system stimulant, at least in part by blocking the GABA binding site on the GABAA receptors.9,10 Securinine showed neuroprotection activities in rats with β-amyloid toxicity10 and in ameliorating the symptoms in patients with ALS.11 In our study, we found that securinine is neuroprotective in cell culture (Figure 3a) and in corticostriatal brain slice cultures of HD.1 Since PC12 cells do not have functional GABAA receptors,12 we found that neuroprotection mechanism of securinine is caused by inhibition of PDI.1 We show that securinine binds adjacent to the active site of PDI by forming an irreversible complex with the protein (Figure 5c).
Given our experience, we emphasize the importance of verifying the composition of active compounds at an early stage of a screening project. At a minimum, a mass spectrum should always be measured on all compounds of interest. If misidentification occurs, we have outlined steps that one can take to identify an unknown active small molecule. We believe this will be useful to both chemists and biologists as these steps can save time and resources for other researchers that are facing similar situations of misidentified compounds and can ultimately allow further studies and development of active compounds once their structures are identified.
Acknowledgments
We are grateful to Dr. Yasuhiro Itagaki for HRMS data, Dr. Luis Avila for assistance with FT-IR experiments, and Dr. John Decatur for assistance and the use of the Columbia Chemistry NMR core facility instruments provided by NIH 1S10RR25431-1A1 and NSF CHE-0840451 grant.
Glossary
ABBREVIATIONS
- HD
Huntington’s disease
- HTT
huntingtin gene
- 2,4-DNPH
2,4-dinitrophenylhydrazine
- HSQC
heteronuclear single quantum correlation
- PDIa
a domain of protein disulfide isomerase
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00014.
Supplementary figures and tables, compound characterization, and biological assays (PDF)
Author Contributions
A.K. performed the experiments and analyzed the data. A.K. and B.R.S. designed the experiments and wrote the manuscript. All authors have given approval to the final version of the manuscript.
This research was funded by the Howard Hughes Medical Institute, National Institute of Health (5R01CA097061, 5R01GM085081, R01CA161061), and New York Stem Cell Science (C026715) to B.R.S., and the Training Program in Molecular Biophysics (T32GM008281) to A.K.
The authors declare no competing financial interest.
Supplementary Material
References
- Hoffstrom B. G.; Kaplan A.; Letso R.; Schmid R. S.; Turmel G. J.; Lo D. C.; Stockwell B. R. Inhibitors of protein disulfide isomerase suppress apoptosis induced by misfolded proteins. Nat. Chem. Biol. 2010, 6, 900–6. 10.1038/nchembio.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mink J. W. The basal ganglia: focused selection and inhibition of competing motor programs. Prog. Neurobiol. 1996, 50, 381–425. 10.1016/S0301-0082(96)00042-1. [DOI] [PubMed] [Google Scholar]
- Kaplan A.; Stockwell B. R. Therapeutic approaches to preventing cell death in Huntington disease. Prog. Neurobiol. 2012, 99, 262–80. 10.1016/j.pneurobio.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiken C. T.; Tobin A. J.; Schweitzer E. S. A cell-based screen for drugs to treat Huntington’s disease. Neurobiol. Dis. 2004, 16, 546–55. 10.1016/j.nbd.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Kaplan A.; Gaschler M. M.; Dunn D. E.; Colligan R.; Brown L. M.; Palmer A. G.; Lo D. C.; Stockwell B. R. Small molecule-induced oxidation of protein disulfide isomerase is neuroprotective. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, E2245–52. 10.1073/pnas.1500439112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darby N. J.; Creighton T. E. Functional properties of the individual thioredoxin-like domains of protein disulfide isomerase. Biochemistry 1995, 34, 11725–35. 10.1021/bi00037a009. [DOI] [PubMed] [Google Scholar]
- Hernandez G.; Anderson J. S.; LeMaster D. M. Electrostatic stabilization and general base catalysis in the active site of the human protein disulfide isomerase a domain monitored by hydrogen exchange. ChemBioChem 2008, 9, 768–78. 10.1002/cbic.200700465. [DOI] [PubMed] [Google Scholar]
- Liu X. W.; Sok D. E. Inactivation of protein disulfide isomerase by alkylators including alpha, beta-unsaturated aldehydes at low physiological pHs. Biol. Chem. 2004, 385, 633–37. 10.1515/BC.2004.078. [DOI] [PubMed] [Google Scholar]
- Beutler J. A.; Karbon E. W.; Brubaker A. N.; Malik R.; Curtis D. R.; Enna S. J. Securinine alkaloids: a new class of GABA receptor antagonist. Brain Res. 1985, 330, 135–40. 10.1016/0006-8993(85)90014-9. [DOI] [PubMed] [Google Scholar]
- Lin X.; Jun-Tian Z. Neuroprotection by D-securinine against neurotoxicity induced by beta-amyloid (25–35). Neurol. Res. 2004, 26, 792–6. 10.1179/016164104225014148. [DOI] [PubMed] [Google Scholar]
- Copperman R.; Copperman G.; Marderosian A. D. Letter: Securinine. JAMA 1974, 228, 288. 10.1001/jama.1974.03230280016013. [DOI] [PubMed] [Google Scholar]
- Hales T. G.; Tyndale R. F. Few cell lines with GABAA mRNAs have functional receptors. J. Neurosci. 1994, 14, 5429–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.