

Abstract
Background
Burkholderia cenocepacia is a soil-dwelling Gram-negative Betaproteobacterium with an important role as opportunistic pathogen in humans. Infections with B. cenocepacia are very difficult to treat due to their high intrinsic resistance to most antibiotics. Biofilm formation further adds to their antibiotic resistance. B. cenocepacia harbours a large, multi-replicon genome with a high GC-content, the reference genome of strain J2315 includes 7374 annotated genes. This study aims to annotate transcription start sites and identify novel transcripts on a whole genome scale.
Methods
RNA extracted from B. cenocepacia J2315 biofilms was analysed by differential RNA-sequencing and the resulting dataset compared to data derived from conventional, global RNA-sequencing. Transcription start sites were annotated and further analysed according to their position relative to annotated genes.
Results
Four thousand ten transcription start sites were mapped over the whole B. cenocepacia genome and the primary transcription start site of 2089 genes expressed in B. cenocepacia biofilms were defined. For 64 genes a start codon alternative to the annotated one was proposed. Substantial antisense transcription for 105 genes and two novel protein coding sequences were identified. The distribution of internal transcription start sites can be used to identify genomic islands in B. cenocepacia. A potassium pump strongly induced only under biofilm conditions was found and 15 non-coding small RNAs highly expressed in biofilms were discovered.
Conclusions
Mapping transcription start sites across the B. cenocepacia genome added relevant information to the J2315 annotation. Genes and novel regulatory RNAs putatively involved in B. cenocepacia biofilm formation were identified. These findings will help in understanding regulation of B. cenocepacia biofilm formation.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1993-3) contains supplementary material, which is available to authorized users.
Keywords: Burkholderia cenocepacia, Biofilms, dRNA-Seq, Transcription start site, Small RNAs, Antisense RNA, Genomic islands
Background
Burkholderia cenocepacia J2315 is a member of the Burkholderia cepacia complex (Bcc), a group of 18 species of closely related Gram-negative Betaproteobacteria [1] which occur in the soil rhizosphere and also play an important role as opportunistic pathogens in humans [2–4]. Bcc bacteria are intrinsically resistant to most antibiotics, and infections with Bcc bacteria are therefore difficult to treat. Bcc bacteria are also able to form biofilms, further adding to their recalcitrance to antibiotic treatment [4].
B. cenocepacia J2315 harbours a large 8.06 Mb multi-replicon genome with a high average GC-content of 66.9 %. The genome consists of two large replicons of 3.87 and 3.22 Mb, a smaller replicon 0.88 Mb and a plasmid 0.09 Mb in size, with 7261 annotated protein coding and 113 annotated RNA genes [5], including 74 tRNAs and 10 riboswitches. However, transcription start sites (TSS), 5′ untranslated regions (5′UTRs) of annotated genes and regulatory non-coding small RNAs have not yet been comprehensively analysed and annotated. Emerging new RNA sequencing techniques, notably differential RNA sequencing (dRNA-Seq, [6]), make it now possible to precisely map the transcription start sites over a whole genome, and at the same time discover novel genome features.
Primary transcripts of prokaryotes carry a triphosphate at their 5′-end, whereas 5′-ends derived from processing and degradation carry a monophosphate. The dRNA-Seq approach uses the properties of a 5′-monophosphate-dependent exonuclease (Terminator™ 5′-Phosphate-Dependent Exonuclease, TEX) to selectively degrade processed transcripts, thereby enriching for un-processed RNA species carrying a native 5′-triphosphate. TSS can then be identified by comparing TEX-treated with untreated RNA-seq libraries, as they appear as localised maxima in coverage enriched by TEX-treatment [6].
dRNA-Seq enables precise mapping of 5′ ends of transcripts, whereas coverage over the whole transcript length is usually poor and 3′ end of transcripts are only represented for short transcripts. For this reason the dRNA-Seq datasets were compared to conventional global RNA-seq data (gRNA-Seq) which provide more even coverage and a more comprehensive representation of full length transcription units. This approach aids in evaluating the function of an identified TSS, particularly for TSS internal to genes.
The aim of the present study is to identify genes expressed in B. cenocepacia biofilms and detect the regulatory elements that might be involved in biofilm formation and survival, as a prerequisite to develop new strategies in treatment of B. cenocepacia infections.
Results and discussion
TSS annotation
dRNA-Seq of duplicate biofilm-derived RNA samples resulted in datasets with 2.4–4.1 million mapped reads, gRNA-Seq of triplicate biofilm-derived RNA samples resulted in datasets with 23–33 million mapped reads (Additional file 1: Table S1). A total of 10843 TSS were automatically annotated based on the dRNA-Seq data (Additional file 2: Table S2), evenly distributed on forward and reverse strands. 3908 TSS remained after noise filtering on a minimum of 10 read starts (Table 1, Additional file 3: Table S3). These were then categorised according to their position in relation to annotated genes (Fig. 1a): TSS in intergenic regions, located ≤ 300 nt upstream of the start of and in sense with an annotated gene, were assigned primary TSS (pTSS) for the respective gene. TSS internal to annotated genes were assigned internal sense (isTSS) or antisense (asTSS). TSS in intergenic regions and not associated with any gene were assigned “orphan” (oTSS). Where TSS were positioned within 100 nt of and same sense to a primary or orphan TSS, they were designated secondary (sTSS).
Table 1.
Number of transcription start sites by category
| Replicon 1 | Replicon 2 | Relicon 3 | Plasmid | Total | |
|---|---|---|---|---|---|
| Genes | 3622 | 2859 | 781 | 100 | 7374 |
| TSS | 6815 | 3010 | 914 | 104 | 10843 |
| Total categorised TSS | 2595 | 1142 | 316 | 57 | 4010 |
| pTSS | 1271 | 671 | 136 | 11 | 2089 |
| depleted pTSS | 64 | 24 | 8 | 6 | 102 |
| pTSS internal same gene | 42 | 27 | 2 | 1 | 72 |
| pTSS internal upstream gene | 78 | 22 | 5 | 0 | 105 |
| oTSS | 237 | 126 | 57 | 8 | 428 |
| sTSS | 181 | 61 | 19 | 0 | 261 |
| isTSS | 502 | 140 | 55 | 15 | 712 |
| asTSS | 304 | 144 | 49 | 23 | 520 |
Fig. 1.
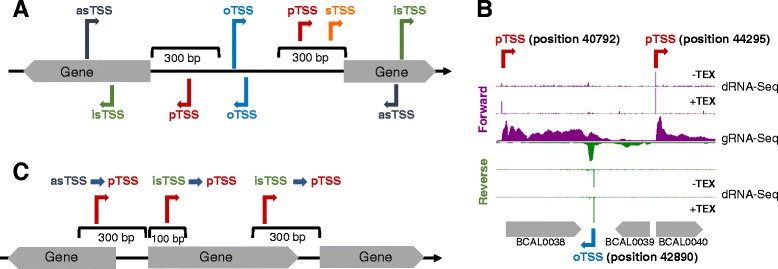
Categorisation of transcription start sites. Panel a: Initial TSS categorisation into primary (pTSS), secondary (sTSS), internal sense (isTSS) and internal antisense (asTSS) based on differential RNA sequencing. Panel b: Comparison of dRNA-Seq and gRNA-Seq data for genes BCAL0038-0040, visualised with the Integrated Genome Browser [51]. dRNA-Seq data are represented as read starts per base, with a vertical scale of 100 for forward reads and 7000 for reverse reads. gRNA-Seq data are represented as coverage, with a vertical scale of 500 for forward reads and 1000 for reverse reads. Panel c: Re-categorisation of internal TSS guided by leading edges of transcription identified by global RNA sequencing
TSS loci were also compared to the global transcriptome datasets by manual inspection. More than 90 % of intergenic pTSS were accompanied by an abrupt increase in coverage in the gRNA-Seq dataset (Fig. 1b), substantiating that they are bona fide loci for transcription initiation. For the purpose of differentiation from TSS based on dRNA-Seq data, we designate these abrupt increases in gRNA-Seq data coverage “leading edges of transcription” (LEs, [7]) for the rest of the manuscript.
LEs found internal to genes were used to assign function to internal TSS: where internal TSS were associated with LEs and positioned ≤300 nt upstream or ≤100 nt downstream and in sense with a gene lacking an intergenic pTSS, they were re-assigned pTSS for the respective gene (Fig. 1c).
Transcription can be primed by molecules other than NTPs, e.g. nano rRNAs [8]. The resulting primary transcripts do not carry a triphosphate at their 5′-end and are depleted by TEX-treatment. Where depleted local read start maxima coincided with a distinct LE, they were reported as depleted pTSS (Table 1, Additional file 3: Table S3), adding another 102 pTSS to the dataset.
In total 2089 pTSS were annotated over the whole B. cenocepacia genome for genes transcribed under biofilm conditions, representing 28 % of all annotated genes. This proportion appears realistic when comparing it to values found in similar studies (24 % for Salmonella enterica [9] and 51 % for Helicobacter pylori [10]), and considering that the large B. cenocepacia genome consists of a high number of non-essential genes [11] that are not all transcribed in the one growth condition analysed in the present study. Most pTSS were located on the large replicon (Table 1), which is to be expected since the large replicon harbours most essential genes [11]. Intergenic pTSS for three genes (BCAL3153, BCAL0301 and BCAL0672) were confirmed by 5′RACE (Additional file 4: Figure S1, panels A, B and C).
105 pTSS were located internal to an upstream gene (Table 1), in some cases the upstream gene was part of the same operon (Fig. 1). Two internal pTSS located in an upstream same sense gene, one with and one without LE, were analysed by 5′RACE (Additional file 4: Figure S1, panels E and F). Where a distinct LE was present, the pTSS was unambiguously confirmed, indicating that these adjacent genes do not constitute an operon. Where a LE was not apparent, transcription initiation as well as read-through from further upstream in the operon occurred. This confirms previous observations that transcription can be initiated or modulated at several loci within an operon, resulting in full length transcripts and alternative transcripts [12].
Promoters
As the present dataset is derived from analysis of only one condition, biofilm growth, promoter search focussed on the core promoter region with its −10 and −35 elements.
Sequences 60 nt upstream of pTSS, excluding TSS located in genomic islands, were submitted to Improbizer [13], a motif finding algorithm that considers location of sequence patterns within the input sequences and favours motifs that occur at the same place. Improbizer found 3 motifs (Additional file 5: Table S4), the first two of which were plausible candidates for a −10 and −35 box based on their sequence and their position relative to TSS. Of the 1733 analysed upstream sequences, more than 95 % possessed a 9 nt long AT-rich motif, on average at position −8 to −16 relative to the TSS (Fig. 2a, Additional file 5: Table S4). The more conserved part of this motif, with consensus sequence TAnAAT, is very similar to the conserved −10 hexamer of E. coli with consensus sequence TATAAT, regarding sequence and position relative to TSS [14]. A second, less conserved motif was found in 93 % of submitted sequences, it centred at position −34 (Fig. 2b). Its consensus sequence is TTGCC, making it similar to the conserved −35 box of E. coli [14] with consensus sequence TTGACA.
Fig. 2.

Sequence logos showing conserved motifs upstream of pTSS. Motifs are based on 1733 sequences upstream of primary TSS located in intergenic regions, analysed with Improbizer [13] (panels a and b), MEME [15] (panel c) and DMINDA [16] panel (d and e). X-axis: Average position relative to TSS. Y-axis: Sequence conservation
The same 1733 upstream sequences were also analysed with MEME [15], and DMINDA [16], confirming the more conserved first motif, with the same conserved part of the core promoter region containing the −10 consensus sequence TAnAAT and the same positioning relative to TSS (Fig. 2c, d, e). The second less conserved motif could not be confirmed by either MEME or DMINDA, presumably because it is too weak to be detected by algorithms which do not take the position of the motif into account.
The sequences up to 60 nt upstream of internal pTSS, asTSS, isTSS and oTSS were screened for occurrence of the conserved and AT-rich motif (Fig. 2a) to assess whether these TSS were derived from genuine transcription initiation or from sequencing artefacts. We used Motif Finder [13], a program which considers the location of the motif in query sequences when searching for matches. More than 95 % of internal pTSS were associated with a sequence match to the AT-rich motif, on average at the same position as the input motif (Additional file 5: Table S4). Furthermore, 94 % of asTSS, 92 % of oTSS and 81 % of isTSS were associated with a matching motif in the same position.
Overall, the occurrence and position of promoters for transcription initiation further corroborates that most TSS found by dRNA-Seq are bona fide. The lower incidence of promoters for isTSS indicates that isTSS can be also caused by TEX inhibition at strong secondary structures, as has been observed for Streptomyces coelicolor, another organism with high genomic GC content [17, 18].
MEME detected only 5 motifs with an e-value <0.001, only one of which, containing the proposed −10-box (Fig. 2c), was plausible as a promoter motif based on its conservation and convergence towards a specific position (Additional file 6: Table S5). DMINDA detected 16 motifs, only two of which, both AT-rich, were converging towards a specific position (Fig. 2d and e, Additional file 7: Table S6). Variations of motifs which could represent different sigma factor binding sites were not found in this analysis. Repeating the analysis with adjusted input paratmeters did not improve results. This is probably due to the relatively large number of input sequences (upstream sequences from all genes expressed under biofilm condition). Analysing subsets of these sequences, generated based on similar expression patterns in a microarray dataset [19] or on related functions, also did not result in plausible specific and conserved sigma factor binding site motifs (data not shown). A reason for this might be the large number of sigma factors encoded in the B. cenocepacia J2315 genome [5]. This bacterium possesses 20 sigma factors the target genes of which have not yet been characterised and which probably have overlapping target gene populations. A more in depth analysis of promoter sequences might therefore require experimental evidence regarding sigma factor target genes, generated e.g. by ChIP sequencing.
Length of 5′ UTRs and leaderless transcripts
The average length of 5′UTRs is 72 nt (Fig. 3), with a distribution peak between 21 and 30 nt; 75 % of 5′UTRs were between 17 and 126 nt long. This is in good agreement with values for other bacteria such as Salmonella enterica [9], Helicobacter pylori [10] and Streptomyces coelicolor [18].
Fig. 3.

5′UTR length distribution. Data represents 5′UTRs from primary TSS located in intergenic regions, excluding internal primary TSS
The length of a 5′UTR can be related to expression regulation of the corresponding gene. Long 5′UTRs may contain riboswitches or provide binding sites for small regulatory RNAs [20, 21]. Leaderless genes are translated by a different mechanism than genes with a leader sequence, and have been shown to be differentially regulated under stress conditions compared to leader-lead genes [22]. To investigate a possible link between length of 5′UTRs and gene function in B. cenocepacia J2315, we performed functional enrichment analysis on sub-sets of genes, genes without (≤10 nt) 5′UTR or with a long (>150 nt) 5′UTR.
The pTSS of 72 genes were located exactly at the annotated start codon and the pTSS of a further 42 genes was located ≤10 nt upstream of the annotated start codons. These transcripts were considered to be leaderless. 24 leaderless transcripts were tRNAs, consistent with the length of tRNA leader sequences in B. cenocepacia J2315, which ranges from 5 to 127 nt (see below). Functional enrichment analysis of the remaining leaderless transcripts revealed that, with 28 coding sequences (CDS), transcriptional regulators of various families are particularly over-represented (Additional file 8: Table S7). The TA-rich promoter motif was found directly upstream of genes with leaderless transcripts (Fig. 3), like reported for other bacteria [23], showing that leaderless genes possess a transcription initiation signal instead of a Shine-Dalgarno sequence.
187 CDS featured a long 5′UTR of >150 nt. Functional enrichment analysis performed on these genes revealed transcriptional regulators, nucleotide binding and membrane proteins as over-represented (Additional file 8: Table S7). Comparison of the respective sequences with the Rfam database revealed the yet unannotated S-adenosyl-L-homocysteine (SAH) riboswitch [20] in the 5′UTR of an adenosylhomocysteinase (BCAL0145, see below).
Transcription initiation in genomic islands
The genome of B. cenocepacia J2315 contains 14 genomic islands with a GC-content lower than genome average of 66.9 % or with CDS similar to prophages; these GI encompass 9.3 % of the total genome [5]. Internal TSS appear to occur at a higher density in genomic islands (Fig. 4), in agreement with observations made in E. coli and Salmonella sp. [24]. 18 % of all annotated isTSS and 25 % of asTSS are located in genomic islands, which is higher than expected given the proportion of genomic islands on the genome.
Fig. 4.
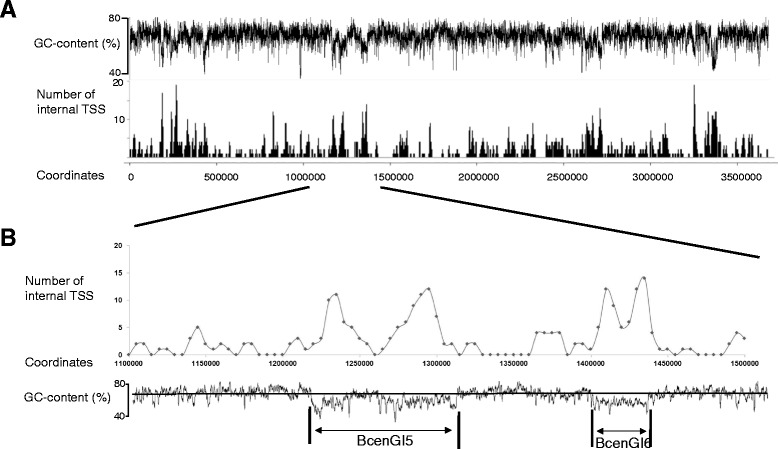
Distribution of internal TSS across replicon 1 of the B. cenocepacia J2315 genome. Data represents numbers of isTSS and asTSS within 10000 bp sliding windows and 5000 bp steps. Panel a: Number of internal TSS across the whole replicon 1. Panel b: Detail for genome positions 1,100,000–1,500,000 of replicon 1, including two genomic islands with a GC-content lower than the genome average. Position of genomic islands: BcenGI5 1222566..1315385, BcenGI6 1402882..1437088. The window size for displaying the GC-content is 500, the horizontal line depicts the genome average of 66.9 %
This indicates that, while genes in genomic islands can be transcribed, these loci are not completely compatible with the B. cenocepacia J2315 transcriptional machinery, and transcription initiation often does not result in a functional product [25].
Interestingly, the genomic region encoding exopolysaccharide synthesis genes implicated in capsule formation (BCAL3217-3246) also shows a higher density of internal TSS, thus confirming that this region was acquired by recent gene transfer and constitutes a genomic island [26]. Moreover, most TSS annotated on the plasmid were categorised as internal TSS, indicating that plasmid genes are also not entirely compatible with B. cenocepacia transcription mechanisms. We propose that the distribution of internal TSS across the B. cenocepacia genome can be used as a further criterion to identify genomic islands in B. cenocepacia, as genomic islands can display a higher density of internal TSS than genome background.
Re-annotation of start codons and discovery of un-annotated proteins
Gene calling for genome annotation usually predicts up to 99 % of all protein coding genes correctly, but the rate of gene calling errors can rise to 14 % in the GC-rich Burkholderia sp. genomes [27]. We used TSS mapping to screen for mis-annotated and un-annotated genes in the B. cenocepacia 2315 genome.
For 72 genes, 66 CDS and 6 RNAs, TSS mapping predicted the primary TSS internal and downstream of the annotated gene start, suggesting that an incorrect start codon might have been predicted and the gene is shorter than annotated. The internal TSS position for BCAL0063 was confirmed by 5′RACE (Additional file 4: Figure S1, panel D) as an example. All genes with internal and downstream pTSS, as well as leaderless genes, were screened for alternative start codons downstream of their annotated gene start. Unusually long 5′UTRs on the other hand could indicate that the corresponding gene is longer than annotated. 5′UTRs longer than 150 nt, as well as orphan TSS consistent with a 5′UTR according to gRNA-Seq data, were screened for open reading frames and upstream alternative start codons. 64 CDS for which an alternative start codon could be predicted, making the gene either longer or shorter, are listed in Table 2.
Table 2.
Alternative start codons for CDS with internal TSS or long 5′ UTRs ≥ 150 nt, as predicted by Prodigal
| TSS position | Strand | Gene | Annotated gene position | Alternative gene start | Annotated start codon | Alternative start codon | Alternative gene is |
|---|---|---|---|---|---|---|---|
| Replicon 1 | |||||||
| 108753 | + | BCAL0088 | 108758..109138 | 108842 | GTG | ATG | shorter |
| 175808 | + | BCAL0151 | 175965..177107 | 175872 | ATG | ATG | longer |
| 196936 | + | BCAL0175 | 196857..197147 | 196965 | ATG | ATG | shorter |
| 317647 | + | BCAL0289 | 317651..322474 | 317771 | GTG | ATG | shorter |
| 566857 | - | BCAL0515 | Complement (565664..566551) | 566617 | ATG | ATG | longer |
| 700318 | + | BCAL0646 | 700302..701309 | 700341 | GTG | ATG | shorter |
| 787134 | + | BCAL0722 | 787317..788582 | 787293 | GTG | GTG | longer |
| 937522 | + | BCAL0865 | 937677..938504 | 937566 | TTG | ATG | longer |
| 999283 | - | BCAL0916 | Complement (998559..999290) | 999245 | ATG | ATG | shorter |
| 1041114 | - | BCAL0952 | Complement (1040227..1040955) | 1041075 | ATG | ATG | longer |
| 1147555 | + | BCAL1059 | 1147734..1148927 | 1147695 | ATG | ATG | longer |
| 1161150 | - | BCAL1069 | Complement (1159161..1160903) | 1160996 | ATG | ATG | longer |
| 1207883 | - | BCAL1102 | Complement (1207594..1207914) | 1207848 | GTG | ATG | shorter |
| 1390938 | - | BCAL1277 | Complement (1388805..1390997) | 1390868 | ATG | ATG | shorter |
| 1463359 | - | BCAL1335 | Complement (1462753 ..1463376) | 1463334 | ATG | ATG | shorter |
| 1892553 | + | BCAL1715 | 1892545..1893150 | 1892578 | GTG | GTG | shorter |
| 1933621 | + | BCAL1753 | 1933609..1934565 | 1933672 | ATG | ATG | shorter |
| 2041280 | - | BCAL1849 | Complement (2040670..2041326) | 2041221 | ATG | ATG | shorter |
| 2067405 | - | BCAL1871 | Complement (2065350..2067203) | 2067380 | ATG | ATG | longer |
| 2120545 | + | BCAL1921 | 2120767..2121222 | 2120662 | GTG | ATG | longer |
| 2136329 | + | BCAL1937 | 2136797..2138446 | 2136644 | ATG | ATG | longer |
| 2656999 | - | BCAL2401 | Complement (2656576..2657001) | 2656953 | GTG | ATG | shorter |
| 2822661 | - | BCAL2559 | Complement (2822212..2822745) | 2822661 | TTG | ATG | shorter |
| 3011653 | + | BCAL2740 | 3011646..3012689 | 3011679 | ATG | ATG | shorter |
| 3037852 | - | BCAL2766 | Complement (3037324..3037656) | 3037734 | TTG | ATG | longer |
| 3093624 | - | BCAL2818 | Complement (3092132..3093673) | 3093598 | GTG | ATG | shorter |
| 3094006 | + | BCAL2819 | 3093965..3095413 | 3094037 | GTG | ATG | shorter |
| 3122838 | - | BCAL2841 | Complement (3121514..3122860) | 3122710 | GTG | ATG | shorter |
| 3257864 | - | BCAL2974 | Complement (3257551..3257931) | 3257820 | ATG | ATG | shorter |
| 3266225 | + | BCAL2981 | 3266173..3268080 | 3266245 | ATG | ATG | shorter |
| 3461898 | + | BCAL3168 | 3461889..3462575 | 3461937 | ATG | ATG | shorter |
| 3533713 | - | BCAL3229 | Complement (3531274..3533205) | 3533499 | GTG | ATG | longer |
| 3584399 | - | BCAL3275 | Complement (3583374..3584396) | ATG | TTG | shorter | |
| 3619212 | - | BCAL3302 | Complement (3616817..3619048) | 3619129 | GTG | ATG | longer |
| 3666930 | + | BCAL3349 | 3666923..3667387 | 3666965 | ATG | ATG | shorter |
| 3721637 | - | BCAL3395 | Complement (3719319..3721676) | 3721619 | ATG | ATG | shorter |
| Replicon 2 | |||||||
| 14979 | + | BCAM0014 | 15152..15757 | 15110 | ATG | ATG | longer |
| 184425 | - | BCAM0158 | Complement (182630..184141) | 184234 | ATG | ATG | longer |
| 570490 | + | BCAM0516 | 570437..570919 | 570545 | TTG | ATG | shorter |
| 713692 | + | BCAM0645 | 713658..715211 | 713733 | ATG | ATG | shorter |
| 878428 | + | BCAM0795 | 878392..878856 | 878428 | TTG | ATG | shorter |
| 904562 | + | BCAM0820 | 904770..905861 | 904728 | ATG | ATG | longer |
| 1009355 | + | BCAM0918 | 1009947..1011812 | 1009395 | GTG | ATG | longer |
| 1203868 | + | BCAM1112 | 1204104..1206368 | 1204068 | ATG | ATG | longer |
| 1405396 | + | BCAM1280 | 1405387..1406880 | 1405435 | ATG | ATG | shorter |
| 1419414 | + | BCAM1290 | 1419396..1420382 | 1419438 | TTG | ATG | shorter |
| 1967577 | + | BCAM1756 | 1967535..1969922 | 1967598 | GTG | ATG | shorter |
| 2032438 | + | BCAM1814 | 2032438..2034045 | 2032462 | ATG | ATG | shorter |
| 2291422 | - | BCAM2058 | Complement (2290470..2291126) | 2291303 | TTG | ATG | longer |
| 2291441 | + | BCAM2059 | 2291632..2292495 | 2291587 | ATG | ATG | longer |
| 2300468 | + | BCAM2066 | 2300462..2301877 | 2300489 | ATG | ATG | shorter |
| 2471730 | - | BCAM2210 | Complement (2471467..2471733) | 2471658 | ATG | ATG | shorter |
| 2613643 | + | BCAM2327 | 2613607..2614722 | 2613697 | ATG | ATG | shorter |
| 2703044 | + | BCAM2401 | 2703049..2703798 | 2703109 | ATG | ATG | shorter |
| 2703867 | + | BCAM2402 | 2703876..2704169 | 2703900 | GTG | ATG | shorter |
| 3026129 | + | BCAM2679 | 3026111..3026374 | 3026153 | TTG | ATG | shorter |
| 3058373 | - | BCAM2703 | Complement (3057460..3058437) | 3058353 | GTG | ATG | shorter |
| 3077665 | - | BCAM2719 | Complement (3077106..3077669) | 3077606 | ATG | ATG | shorter |
| 3133147 | + | BCAM2769 | 3133305..3134042 | 3133281 | GTG | ATG | longer |
| Replicon 3 | |||||||
| 69880 | - | BCAS0060 | Complement (68555..69688) | 69709 | ATG | ATG | longer |
| 80842 | - | BCAS0070 | Complement (79501..80850) | 80742 | TTG | ATG | shorter |
| 769919 | + | BCAS0706 | 770234..771562 | 770120 | TTG | ATG | longer |
| Plasmid | |||||||
| 2363 | + | pBCA003 | 2363..3220 | 2546 | ATG | ATG | shorter |
| 79981 | - | pBCA080 | Complement (79541..79750) | 79891 | ATG | ATG | longer |
To search the genome for un-annotated protein-coding genes, all oTSS and asTSS possessing a LE were screened for an open reading frame with an ATG start codon which could produce a protein ≥50 amino acid residues. These amino acid sequences were compared to the NCBI protein sequence database and hits with both >75 % query coverage and >40 % amino acid identity were retained (Additional file 9: Table S8). Most hits were annotated as hypothetical or conserved hypothetical proteins with no predicted function. In one case, a type II toxin-antitoxin module was discovered on the opposite strand of a gene currently annotated as BCAL1704, a conserved hypothetical protein. We propose to re-annotate this loci, to BCAL1704A, a ParD-type antitoxin with 81 amino acid residues, and BCAL1704B, a ParE-type toxin with 99 amino acid residues (Fig. 5).
Fig. 5.
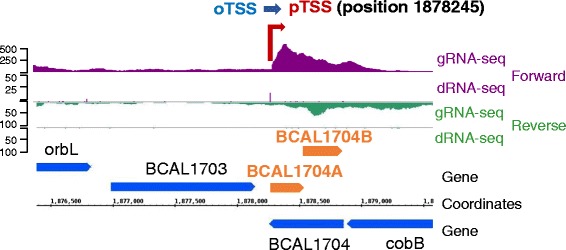
Re-annotation of a conserved hypothetical protein as a type II toxin-antitoxin system. New annotation: BCAL1704A, ParD, antitoxin component of, and BCAL1704B, ParE, toxin component of a type II toxin-antitoxin system. Position of ParD: 1878263..1878505. Position of ParE: 1878498..1878794. The start of ParE overlaps the 3′end of ParD by 8 nt. dRNA-Seq data are represented as read starts per base, gRNA-Seq data are represented as coverage
Antisense transcription
Most reads mapping to annotated genome features map in sense direction (>93 %), only 5–7 % map antisense, based on the gRNA-Seq dataset. Nonetheless, antisense transcription is pervasive in B. cenocepacia J2315, and reads map antisense to nearly all genome features (Additional file 10: Table S9). However, it is safe to assume that not all of these antisense transcripts have a function [28]. Genes with strong sense transcription tend to have a high number of antisense reads, likely a result of technical RNA sequencing artefacts. Moreover, antisense transcription can be a result of read-through from the 3′ end of a downstream opposite-sense gene or transcription initiation for an upstream opposite-sense gene.
We attempted to annotate antisense transcripts that might genuinely be involved in gene regulation, i.e. transcripts complementary to at least part of a gene and not belonging to any category mentioned above. For this purpose we filtered for genes with a minimum antisense-RPKM of 10 and a ratio of antisense-to-sense RPKM of >0.1, leaving 11 % of all genes. 105 of these featured an asTSS, or an oTSS located ≤ 300 bp downstream of the 3′end of a gene, with a LE in gRNA-Seq data (Additional file 11: Table S10). Among the genes with antisense transcription were 12 transposases, 12 transcriptional regulators and two toxin-antitoxin systems, the same categories for which functional antisense transcripts have been found in other bacteria [29]. These antisense RNAs might therefore also have a function in B. cenocepacia.
Genes induced under biofilm condition
To screen the RNA-seq data for genes potentially essential for biofilm growth of B. cenocepacia J2315, we compared RNA-seq data with published microarray datasets obtained from cells grown in a biofilm [30], from planktonic cells harvested in stationary phase, and from cells grown under reduced oxygen levels [19] as well as under various stress conditions [19, 30]. The aim was to find genes with high expression in biofilms (gRNA-Seq RPKM >100) and induced in biofilms while not induced under any other condition.
Only the first two genes of a multi-subunit K+-transport system (kdpA-kdpE, BCAL2379-2383) met these criteria. These five genes are organised in two operons with two annotated TSS, the first of which is 100-fold stronger expressed than the second (Fig. 6a). The first operon contains the structural K+-transport and ATPase genes, the second contains the two-component regulatory system required for induction of kdpABC. qPCR analysis of kdpA confirmed its induction in biofilms compared to planktonic cultures in logarithmic and stationary phase (Fig. 6b). In E. coli, the Kdp-system is an inducible high affinity K+-pump essential for intracellular K+-homoeostasis under salt stress, [31]. The two-component system kdpDE has also been implicated in virulence in various pathogenic bacteria [31]. In Bacillus sp., the Kdp-system was found to be up-regulated in swarming cells [32] and necessary for biofilm formation [33], and the Kdp-system was up-regulated in Staphylococcus aureus biofilms [34]. These observations indicate that, apart from its role in osmoadaptation, the Kdp-system also plays an important role in biofilm formation, presumably also in B. cenocepacia. Work to characterise the relevance of these genes for B. cenocepacia biofilm formation and persistence, using deletion and conditional mutants, is ongoing.
Fig. 6.

Expression of the kdp-system in B. cenocepacia J2315 biofilms. Panel a: dRNA-Seq and gRNA-Seq data for the kdp-operon. dRNA-Seq data are represented as read starts per base, gRNA-Seq data are represented as coverage. Red arrows: pTSS, at position 2640727 (kdpA) and 2635837 (kdpD). Panel b: Quantitative RT-PCR of kdpA in biofilms and planktonic cultures. Bars represent expression relative to planktonic logarithmic cultures and are based on three biological replicates. Orange: Planktonic culture in logarithmic phase, harvested at 5 × 108 CFU/ml. Light blue: Planktonic culture in late logarithmic phase, harvested at 1 × 109 CFU/ml. Brown: Planktonic culture in stationary phase, harvested at 2.2 × 109 CFU/ml. Green: Biofilm grown in microtiter plates
Transcription of annotated non-coding RNAs
The published genome annotation [5] includes rRNAs and small non-coding RNAs such as tRNAs, several riboswitches and essential RNAs.
For 47 of the 74 annotated tRNA genes a TSS was found, positioned 5–127 nucleotides upstream of the annotated gene start. Most tRNA transcripts are, as expected, processed at position +1 of the annotated gene, marked by a local maximum in read starts in the -TEX library and depletion in the + TEX library.
Exceptions are the tRNAs for histidine and selenocysteine, which are processed at position −1 and −10, respectively. 16S rRNA genes have a TSS at position −237, the co-transcribed 23S and 5S rRNA are processed at +3 and +2, respectively. TSS for annotated riboswitches are positioned at annotated gene start (thiamine pyrophosphate) and 5 nt (glycine) or 8–14 nt upstream (cobalamin).
Of the annotated essential RNAs, transfer messenger RNA has an 11 bp 5′-leader element. RNase P appears longer than annotated, its TSS is located at position −51 and the transcript shows no obvious processing site. The signal recognition particle appears shorter than annotated, with a TSS at position +2.
Overall, dRNA-Seq data are in good agreement with annotated non-coding RNAs, showing that small RNAs are detected with our experimental approach.
Candidate regulatory small RNAs
Apart from annotated tRNAs, riboswitches and conserved essential RNAs, bacterial genomes also contain other small non-coding RNAs that are involved in post transcriptional gene expression regulation [21, 35]. For a preliminary screening of TSS for highly expressed novel small RNAs, sequences following oTSS, isTSS and asTSS, as well as 5′UTRs longer than the average of 72 nt, were compared to the Rfam database.
In this manner nine small RNAs were found in the B. cenocepacia J2315 genome (Table 3). One of these constitutes the 6S RNA, which was already predicted and its expression confirmed by other studies on B. cenocepacia [30, 36]. Two small RNAs are phage-related regulatory RNAs located on genomic island BcenGI9. Two Rfam hits constitute conserved regulatory motifs: the SAH riboswitch located in the 5′UTRs of BCAL0145, an adenosylhomocysteinase, and the Burkholderiales-specific sucA RNA motif located in the 5′UTR of BCAL1515, sucA, an enzyme of the citric acid cycle. The sucA RNA motif probably constitutes a riboswitch [37]. The remaining four novel small RNAs all are from the same family, “toxic small RNAs”, which were found to be toxic if introduced into E. coli on a cloning vector [38]. Expression of these toxic small RNAs has been confirmed by Northern blotting in four strains of B. cenocepacia, including strain J2315 [38]. However, their function in B. cenocepacia is unknown.
Table 3.
Novel non-coding small RNAs in B. cenocepacia J2315 with hits in Rfam database
| Preliminary name | Strand | Length (nt)b | Terminator sequence | Adjacent genes | Relative orientation | Genome position | Rfam ID | Name | |
|---|---|---|---|---|---|---|---|---|---|
| ncS03 | + | 58 | yes | BCAL0197 | BCAL0198 | → → ← | 221314..221371 | RF02278 | Toxic small RNA |
| ncS05 | - | 67 | yes | BCAL0436 | BCAL0437 | ← ← → | Complement (479440..479506) | RF02278 | Toxic small RNA |
| ncS17 | + | 200 | yes | BCAL2667 | BCAL2668 | → → → | 2935785..2935984 | RF00013 | 6S RNA |
| ncS23 | + | 81 | yes | BCAL2965 | BCAL2965a | → → → | 3246834..3246914 | RF01394 | isrK |
| ncS24 | + | 90 | no | BCAL2965 | BCAL2965a | → → → | 3246937..3247026 | RF01695 | C4a |
| ncS27 | - | 92 | yes | BCAL3348a | BCAL3349 | → ← → | Complement (3666557..3666648) | RF02278 | Toxic small RNA |
| ncS62 | + | 57 | yes | BCAM1871 | BCAM1872 | → → ← | 2089713..2089769 | RF02278 | Toxic small RNA |
| ncR1 | + | 119 | no | BCAL0144 | BCAL0145 | → → → | 168973..169091 | RF01057 | SAH riboswitch |
| ncR2 | + | 118 | no | BCAL1514 | BCAL1515 | → → → | 1676458..1676575 | RF01070 | SucA RNA motif |
aC4 forms one transcriptional unit with isrK
bLength of non-coding RNAs is inferred from dRNA-Seq data and terminator structures (when present) and is not yet experimentally confirmed
The sequence of small RNAs is generally only conserved between closely related bacterial species and can vary dramatically in primary sequence and secondary structure between bacterial genera [39]. On the other hand, most small RNAs in the Rfam database are derived from well-studied species such as E. coli, which are not closely related to Burkholderia. Since the Rfam algorithm first performs a BLAST search, functional homologues from distantly related species are unlikely to produce a hit and novel small RNAs specific for B. cenocepacia are likely to be overlooked by this approach.
To identify putative regulatory small non-coding RNAs not yet represented in the Rfam database, we compared sequences derived from oTSS to non-coding RNAs experimentally confirmed in B. cenocepacia strains in other studies using RNA-sequencing and Northern blotting [36], co-purification with Hfq-protein [40] or microarrays [30, 41–43], if they showed the following properties: strong transcription initiation with a coverage >300 reads in dRNA-Seq data, a defined 3′ end in dRNA-Seq data or a transcript appearing short (<500 nt), truncated or missing in gRNA-Seq data.
Homologues of six short transcripts with strong transcription initiation from oTSS were also expressed in B. cenocepacia strains AU1054 and HI2424 under conditions mimicking the human lung and the soil environment (Table 4) [36]. One of these was confirmed to be a small RNA by Northern blotting in the same study. One short transcript was present in the RNA fraction co-purified with the Hfq-protein of B. cenocepacia J2315 [40]. Hfq is an RNA chaperone which mediates base pairing of small regulatory RNAs with their target mRNA [44] and B. cenocepacia J2315 harbours the Hfq gene as two non-identical homologues [5], making this non-coding RNA a plausible candidate for a regulatory small RNA. These findings show that with the approach used in this study we could identify transcripts which could encode non-coding regulatory small RNAs. Their strong expression in biofilms suggests that these small RNAs might have a role in adaptation of B. cenocepacia to biofilm conditions.
Table 4.
Putative novel non-coding RNAs expressed in B. cenocepacia and experimentally confirmed by previous studies
| Preliminary name | Strand | Length (nt)b | Terminator sequence | Adjacent genes | Relative orientation | Genome position | Replicon | Type of experiment | |
|---|---|---|---|---|---|---|---|---|---|
| ncS04 | + | 105 | yes | BCAL0264 | BCAL0265 | ← → ← | 292949..293053 | 1 | Co-purification with Hfq [40] |
| ncS06 | + | 117 | no | BCAL0549 | BCAL0550 | → → ← | 603652..603828 | 1 | RNA-Seq and Northern blot [36] |
| ncS11 | - | 208 | yes | BCAL2293 | BCAL2294 | → ← ← | Complement (2545296..2545503) | 1 | RNA-Seq [36] |
| ncS18 | + | 178 | no | BCAL2713 | BCAL2714 | → → → | 2979006..2979183 | 1 | RNA-Seq [36] |
| ncS21a | + | 361 | yes | BCAL2737 | BCAL2738 | → → → | 3008232..3008591 | 1 | RNA-Seq [36] |
| ncS33 | + | 93 | no | BCAM1725 | BCAM1726 | → → → | 1926664..1926756 | 2 | RNA-Seq [36] |
| ncS36 | + | 60 | no | BCAM2207 | BCAM2208 | ← → → | 2468880..2468939 | 2 | RNA-Seq [36] |
ancS21 is associated with an open reading frame and potentially constitutes a protein, see Additional file 6: Table S5
bLength of non-coding RNAs is inferred from dRNA-Seq data and terminator structures (when present) and is not yet experimentally confirmed
A detailed analysis of putative novel small non-coding RNAs expressed in B. cenocepacia J2315 biofilms and their involvement in biofilm formation is ongoing.
Conclusions
This study is the first genome-wide analysis of TSS in B. cenocepacia. Through differential RNA-Sequencing, bioinformatics methods and 5′RACE we annotated the primary TSS for 2089 genes expressed in biofilms, defined alternative start codons for 64 genes, identified novel protein sequences and characterised antisense transcription. 15 non-coding RNAs highly expressed in biofilms and a potassium uptake system strongly induced under biofilm conditions were identified that could be involved in biofilm formation and survival. Comparison of dRNA-Seq data with gRNA-Seq data proved to be invaluable for TSS categorisation and interpretation.
The data presented in this study will provide the starting point for evaluation of the regulatory processes involved in B. cenocepacia biofilm formation and could reveal novel targets for antibiotic therapy.
Methods
Bacterial strain and culture conditions
B. cenocepacia strain J2315 (LMG 16656) was grown in Luria-Bertani broth (LBB, Oxoid, Hampshire, UK) at 37 °C. Biofilms were grown in 96-well microtiter plates and cells were harvested as described previously [45]. Planktonic cultures were grown in 250 ml glass flasks, incubated at 37 °C in a shaking incubator at 150 rpm and harvested as described previously [46].
RNA extraction and sequencing
For differential RNA-sequencing (dRNA-Seq, [6]), total RNA was extracted from cell pellets of two biological biofilm replicates using the RiboPure Bacteria kit (Life Technologies, Renfrewshire, UK). RNA samples were then split and one aliquot treated with Terminator™ 5′ monophosphate-dependent exonuclease (TEX). A separate library was constructed from each aliquot, TEX-treated and untreated. rRNA depletion and RNA fragmentation steps were omitted. 5 'triphosphates were removed using tobacco acid pyrophosphatase and an RNA adapter was ligated to the 5′-monophosphate of the RNA. RNA was then polyadenylated and first-strand cDNA synthesis performed using an oligo(dT)-adapter primer. The resulting cDNA was PCR-amplified to about 20-30 ng/μl. A library-specific barcode for multiplex sequencing was part of a 3′-sequencing adapter. The following adapter sequences flank the cDNA inserts: TrueSeq Sense primer 5′AATGATAC GGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′ and TrueSeq Antisense NNNNNN primer (NNNNNN = 6n barcode for multiplexing) 5′-CAAGCAGAAGACGGCATACGAGAT-NNNNNN-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATC(dT25)-3′. The resulting cDNA libraries were sequenced using a HiSeq 2000 machine (Illumina) in single-read mode and running 100 cycles.
For global transcriptome sequencing (gRNA-Seq), RNA of three biological biofilm replicates was extracted as described previously [45]. Total RNA was depleted of ribosomal RNA using the Ribo-zero Magnetic Gram-Negative Bacteria kit (Epicentre, Madison, WI, USA) and used for Illumina paired-end sequencing generating 100 bp reads [45].
Mapping of RNA seq reads
In order to assure a high sequence quality, the Illumina reads in FASTQ format were trimmed with a cut-off phred score of 20 by the program fastq_quality_trimmer from FASTX toolkit version 0.0.13 (http://hannonlab.cshl.edu/fastx_toolkit/). The following steps were performed using the subcommand “create”, “align” and “coverage” of the tool READemption [47] version 0.3.5. The poly(A)-tail sequences were removed and a size filtering step was applied in which sequences shorter than 12 nt were eliminated. The collections of remaining reads were mapped to the reference genome sequences (accessions AM747720, AM747721, AM747722, and AM747723) using segemehl software version 0.2.0 [48]. Coverage plots in wiggle format representing the number of aligned reads per nucleotide were generated based on the aligned reads just considering the first base of each read.
TSS annotation and classification
Mapping output from dRNA-Seq was split by replicon and coverted to SAM format (using samtools 1.2). Those SAM files were used as input for TSSAR, a tool for automated de novo TSS annotation [49], to map all loci with coverage maxima which are enriched in the TEX-treated library. Default parameters (p-value threshold 0.001, noise threshold 2, merge range 5) were used, the two biological replicates were pooled and TSS within 5 nt of each other were clustered into one. Genome regions with read start distributions that do not conform to a Poisson distribution are omitted from TSSAR analysis [49]. Such regions were then manually annotated by scanning the respective wiggle files for nucleotides with an abrupt increase in coverage; except regions internal to rRNA genes where the abundance of mapped reads was too high to allow robust TSS annotation. A noise filter with a minimum coverage of 10 read starts was applied, based on the normalised number of read starts per base, to ensure reported TSS are robust. TSS were then classified according to their genomic context. All TSS positions were assigned relative to the start of the associated annotated gene, with the first base of the gene being position +1, upstream positions start with −1.
We used the B. cenocepacia strain J2315 annotation deposited in the EMBL database under accession numbers AM747720, AM747721, AM747722, and AM747723 [5] for assigning and classifying TSS. This newer annotation includes 21 non-coding RNAs other than rRNAs and tRNAs: riboswitches for e.g. thiamine and cobalamin and the essential RNAs tmRNA, ribonuclease P and bacterial signal recognition particle. In contrast to this, the older annotation deposited at NCBI under accession numbers NC_011000, NC_011001, NC_011002 and NC_011003 contains only rRNAs and tRNAs and lacks all genes annotated as pseudogenes in the newer annotation.
Rapid amplification of cDNA ends (RACE)
To confirm TSS, the 5′end of selected transcripts was determined by RACE. For 5′RACE, RNA was transcribed with gene specific primers and a homopolymeric tail added to the 3′ end of the resulting cDNA. The tailed cDNA was then amplified with nested gene specific primers and a primer complementary to the homopolymer tail. We used the 5′ RACE System for Rapid Amplification of cDNA Ends (Life Technologies, Paisley, UK) with the following changes to standard protocol: the reverse transcriptase provided with the kit was replaced by ThermoScript reverse transcriptase (Life Technologies, Paisley, UK), temperature for first strand synthesis was elevated to 60 °C and the additional protocol for transcripts with high GC-content was followed. The resulting amplicons were cloned into E. coli using the pGEM®-T Vector system (Promega, WI, USA) and JM109 high-efficiency competent cells (Promega, WI, USA). Vector inserts amplified from clones were analysed by Sanger-sequencing. RACE primer sequences are available as supplementary data (Additional file 12: Table S11).
Quantitative RT-PCR analysis
For quantitative RT-PCR (qPCR), planktonic and biofilm cultures were grown and harvested as described previously [45, 46]. RNA extraction from cell pellets was performed with a modified protocol, using the RiboPure Bacteria kit (Life Technologies, Renfrewshire, UK) with the following changes to manufacturer’s instructions: before transferring the RNA to the filter cartridge, 1.25 instead of 0.5 volumes of ethanol were added to retain a higher proportion of small RNAs. Before DNase treatment, RNA was denatured by heating to 65 °C for 5 min and DNase incubation time was increased from 30 min to 60 min. The RNA extract was then DNase-digested (NEB, Ipswich, MA, USA) for a second time for 60 min and extracted with phenol-chloroform (Roti-Aqua-P/C/I for RNA extraction, Carl Roth, Karlsruhe, Germany). Extracted RNA was precipitated with 2.5 volumes ethanol-sodium acetate mix (ethanol : 3 M Na-acetate 30:1, pH 6.5) over night at −20 °C, centrifuged and washed with 70 % ethanol. The RNA pellet was air dried and re-dissolved in water.
cDNA generation and quantitative RT-PCR was performed as described previously [50] using eight control genes with minimal expression changes across all tested conditions for data normalisation. All eight control genes were used for normalisation in every condition. Primer sequences of target and control genes are shown in Additional file 10: Table S9.
Further bioinformatical analysis
dRNA-Seq and gRNA-Seq data were visualised with the Integrated Genome Browser version 8.3.1. [51] for manual comparison. Novel proteins were searched for by comparing sequences to the NCBI non-redundant protein sequence database using pBLAST [52], novel non-coding small RNAs were searched for by comparing DNA sequences to the Rfam database [39]. The TransTerm algorithm [53] was used to screen for Rho-independent transcriptional terminator structures. Functional enrichment analysis was performed with the DAVID web tool [54], using a custom background gene list consisting of all genes with an assigned pTSS. Alternative start codons were predicted using Prodigal [55]. DNA sequence motifs upstream of pTSS were identified with Improbizer [13] using default parameters and motifs were then searched for in sequences upstream TSS belonging to other categories with Motif Matcher [13]; both programs consider location of the motif. Improbizer and Motif Matcher are available as web tools at https://users.soe.ucsc.edu/~kent/improbizer/index.html. Sequences upstream of pTSS were also submitted to MEME [15] and DMINDA [16] for comparison and cross-validation. Input parameters were default, except for minmum and maximum motif length in MEME which were 8 and 50, respectively.
Supporting data and software
The dRNA-Seq raw sequencing data was submitted to ArrayExpress under accession number E-MTAB-3381. The gRNA-Seq raw reads are available in ArrayExpress under accession number E-MTAB-2079 [45]. A script that performs the READemption and TSSAR based analysis can be retrieved from https://zenodo.org/record/17358 (DOI: 10.5281/zenodo.17358).
Plots for read starts per nucleotide from dRNA-Seq and for coverage from gRNA-Seq as well as data for TSS, candidate regulatory RNAs and alternative annotations are available on the Burkholderia genome database [56] beta site (www.burkholderia.com).
Acknowledgements
This study was funded by the Belgian Science Policy Office within their Interuniversity Attraction Poles (IAP) program, (Phase VII/2012–2017, Project P7/28). The authors wish to thank Bert Remmerie for help with TSS categorisation.
Abbreviations
- Bcc
Burkholderia cepacia complex
- BcenGI
Burkholderia cenocepacia genomic island
- bp
Base pairs
- cDNA
Complementary DNA
- CDS
Coding sequence
- CFU
Colony forming units
- dRNA-Seq
Differential RNA sequencing
- gRNA-Seq
Global RNA sequencing
- Hfq
Host factor q
- kdp
Potassium transport pump
- LBB
Luria-Bertani broth
- LE
Leading edge of transcription
- Mb
Mega base pairs
- nt
Nucleotide
- NTP
nucleoside triphosphate
- TEX
Terminator exonuclease
- tRNA
Transfer RNA
- rRNA
Ribosomal RNA
- RACE
Rapid amplification of cDNA ends
- RPKM
Reads per kilo base per million mapped reads
- SAH
S-adenosyl-L-homocysteine
- UTR
Untranslated region
- TSS
Transcription start site (pTSS: Primary TSS, asTSS: Internal antisense TSS, isTSS: Internal sense TSS, oTSS: Orphan TSS, sTSS: Secondary TSS)
Additional files
Number of mapped reads per RNA sample. (XLSX 11 kb)
Transcription start sites identified by differential RNA sequencing and automated annotation. (XLSX 489 kb)
Annotated and categorised TSS in the B. cenocepacia J2315 genome. (XLSX 353 kb)
Transcription start sites confirmed by 5′RACE. dRNA-Seq data are represented as number of read starts per base and gRNA-Seq data as read coverage, both are visualised using the Integrated Genome Browser [51]. dRNA-Seq data are represented as read starts per base, gRNA-Seq data are represented as coverage. Blue arrows depict representative sequences derived from 5′RACE analysis. Panel A: pTSS of BCAL3153, confirmed by 2 out of 8 RACE sequences, 6 sequences were shorter than the putative 5′UTR. Panel B: pTSS of BCAL0672, confirmed by 5 out of 12 RACE sequences, 7 sequences were shorter than the putative 5′UTR. The isTSS internal to BCAL0672 was not confirmed by 5′RACE, transcripts originating from this locus are probably truncated. Panel C: pTSS of BCAL0301 confirmed by 2 out of 7 RACE sequences, 5 sequences were shorter than the putative 5′UTR. The isTSS located internal to BCAL0300 could not be confirmed, transcripts originating from this locus are probably truncated. Panel D: Internal pTSS of BCAL0063. 4 out of 5 RACE sequences confirmed the internal pTSS, one read was shorter. Panel E: isTSS within BCAL3201: 3 out of 9 RACE sequences confirmed the isTSS, 3 were shorter and 3 longer than the transcript originating at this TSS. Sequences were therefore derived from transcripts originating from this TSS as well as from TSS further upstream in the operon. Panel F: pTSS for BCAL3391, internal to BCAL3390: confirmed by 6 out of 6 RACE sequences. (PDF 114 kb)
Promoter motifs in sequences upstream of TSS based on Improbizer [13] analysis. The last nt of all sequences is position +1 of the transcript, the TSS. Motifs with scores >2 were regarded as positive matches. Motif sequences are indicated in capital letters. (XLSX 436 kb)
Promoter motifs based on MEME [15] analysis. (XLSX 124 kb)
Promoter motifs based on DMINDA [16] analysis. Motif sequences are shown in context with the expression pattern of the respective genes, based on microarray data [19]. (XLSX 319 kb)
Genes belonging to enriched functionally related gene groups among leaderless transcripts and among transcripts with a 5′UTR longer than 150 nt. (XLSX 12 kb)
Amino acid sequences associated with orphan TSS or internal antisense TSS and with homologues in the non-redundant protein sequence database. (XLSX 18 kb)
Number of mapped reads and RPKM values sense and antisense for all genes of the B. cenocepacia J2315 genome. (XLSX 2108 kb)
Genes with antisense transcripts. (XLSX 20 kb)
Oligonucleotides used in this study. (DOCX 17 kb)
Footnotes
Competing interests
The authors declare no competing financial and non-financial interests.
Authors’ contributions
AS analysed the RNA-seq data, performed quantitative RT-PCR and RACE experiments and wrote the manuscript. HvA extracted all total RNA for sequencing and submitted gRNA-Seq data to ArrayExpress. FvN and DD performed global RNA sequencing and JV performed differential RNA sequencing. KF provided initial dRNA-Seq data analysis and scripts for transformation of dRNA-Seq data. TC conceived the study, acquired funding and provided research facilities. All authors read and approved the final version of the manuscript.
Authors’ information
AS, HvA, TC: Laboratory of Pharmaceutical Microbiology, Ghent University, Ghent, Belgium.
KF: Core Unit Systems Medicine, University of Würzburg, Würzburg, Germany
FvN, DD: Laboratory of Pharmaceutical Biotechnology, Ghent University, Ghent, Belgium.
JV: Institute for Molecular Infection Biology, University of Würzburg, Würzburg, Germany.
Contributor Information
Andrea M. Sass, Phone: +32 (0) 9 264 80 93, Email: Andrea.Sass@UGent.be
Heleen Van Acker, Email: Heleen.VanAcker@UGent.be.
Konrad U. Förstner, Email: konrad.foerstner@uni-wuerzburg.de
Filip Van Nieuwerburgh, Email: Filip.VanNieuwerburgh@UGent.be.
Dieter Deforce, Email: Dieter.Deforce@UGent.be.
Jörg Vogel, Email: joerg.vogel@uni-wuerzburg.de.
Tom Coenye, Email: Tom.Coenye@UGent.be.
References
- 1.Peeters C, Zlosnik JEA, Spilker T, Hird TJ, LiPuma JJ, Vandamme P. Burkholderia pseudomultivorans sp. nov., a novel Burkholderia cepacia complex species from human respiratory samples and the rhizosphere. Syst Appl Microbiol. 2013;36(7):483–489. doi: 10.1016/j.syapm.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 2.Mahenthiralingam E, Urban TA, Goldberg JB. The multifarious, multireplicon Burkholderia cepacia complex. Nat Rev Microbiol. 2005;3(2):144–156. doi: 10.1038/nrmicro1085. [DOI] [PubMed] [Google Scholar]
- 3.LiPuma JJ. Update on the Burkholderia cepacia complex. Curr Opin Pulm Med. 2005;11(6):528–533. doi: 10.1097/01.mcp.0000181475.85187.ed. [DOI] [PubMed] [Google Scholar]
- 4.Coenye T. Social interactions in the Burkholderia cepacia complex: biofilms and quorum sensing. Future Microbiol. 2010;5(7):1087–1099. doi: 10.2217/fmb.10.68. [DOI] [PubMed] [Google Scholar]
- 5.Holden MTG, Seth-Smith HMB, Crossman LC, Sebaihia M, Bentley SD, Cerdeño-Tárraga AM, Thomson NR, Bason N, Quail MA, Sharp S, et al. The genome of Burkholderia cenocepacia J2315, an epidemic pathogen of cystic fibrosis patients. J Bacteriol. 2009;91(1):261–277. doi: 10.1128/JB.01230-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharma CM, Vogel J. Differential RNA-seq: the approach behind and the biological insight gained. Curr Opin Microbiol. 2014;19:97–105. doi: 10.1016/j.mib.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 7.Lin YF, A D, Guan S, Mamanova L, McDowall KJ. A combination of improved differential and global RNA-seq reveals pervasive transcription initiation and events in all stages of the life-cycle of functional RNAs in Propionibacterium acnes, a major contributor to wide-spread human disease. BMC Genomics. 2013;14(1):620. doi: 10.1186/1471-2164-14-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldman SR, Sharp JS, Vvedenskaya IO, Livny J, Dove SL, Nickels BE. NanoRNAs prime transcription initiation in vivo. Mol Cell. 2011;42(6):817–825. doi: 10.1016/j.molcel.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kröger C, Dillon SC, Cameron AD, Papenfort K, Sivasankaran SK, Hokamp K, Chao Y, Sittka A, Hebrard M, Handler K, et al. The transcriptional landscape and small RNAs of Salmonella enterica serovar Typhimurium. Proc Natl Acad Sci U S A. 2012;109(20):E1277–1286. doi: 10.1073/pnas.1201061109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiß S, Sittka A, Chabas S, Reiche K, Hackermüller J, Reinhardt R, et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010;464(7286):250–255. doi: 10.1038/nature08756. [DOI] [PubMed] [Google Scholar]
- 11.Juhas M, Stark M, von Mering C, Lumjiaktase P, Crook DW, Valvano MA, Eberl L. High confidence prediction of essential genes in Burkholderia cenocepacia. PLOS ONE. 2012;7(6):e40064. doi: 10.1371/journal.pone.0040064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Güell M, Yus E, Lluch-Senar M, Serrano L. Bacterial transcriptomics: what is beyond the RNA horiz-ome? Nat Rev Microbiol. 2011;9(9):658–669. doi: 10.1038/nrmicro2620. [DOI] [PubMed] [Google Scholar]
- 13.Ao W, Gaudet J, Kent WJ, Muttumu S, Mango SE. Environmentally induced foregut remodeling by PHA-4/FoxA and DAF-12/NHR. Science. 2004;305(5691):1743–1746. doi: 10.1126/science.1102216. [DOI] [PubMed] [Google Scholar]
- 14.Hawley DK, McClure WR. Compilation and analysis of Escherichia coli promoter DNA sequences. Nucleic Acids Res. 1983;11(8):2237–2255. doi: 10.1093/nar/11.8.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bailey TL, Johnson J, Grant CE, Noble WS. The MEME suite. Nucleic Acids Res. 2015;43:W39–W49. doi: 10.1093/nar/gkv416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma Q, Zhang H, Mao X, Zhou C, Liu B, Chen X, Xu Y. DMINDA: an integrated web server for DNA motif identification and analyses. Nucleic Acids Res. 2014;42:W12–W19. doi: 10.1093/nar/gku315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Romero DA, Hasan AH, Lin Y-f, Kime L, Ruiz-Larrabeiti O, Urem M, Bucca G, Mamanova L, Laing EE, van Wezel GP, et al. A comparison of key aspects of gene regulation in Streptomyces coelicolor and Escherichia coli using nucleotide-resolution transcription maps produced in parallel by global and differential RNA sequencing. Mol Microbiol. 2014;94(5):963–987. doi: 10.1111/mmi.12810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vockenhuber MP, Sharma CM, Statt MG, Schmidt D, Xu Z, Dietrich S, Liesegang H, Mathews DH, Suess B. Deep sequencing-based identification of small non-coding RNAs in Streptomyces coelicolor. RNA Biol. 2011;8(3):468–477. doi: 10.4161/rna.8.3.14421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sass AM, Schmerk C, Agnoli K, Norville PJ, Eberl L, Valvano MA, Mahenthiralingam E. The unexpected discovery of a novel low-oxygen-activated locus for the anoxic persistence of Burkholderia cenocepacia. ISME J. 2013;7(8):1568–1581. doi: 10.1038/ismej.2013.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Breaker RR. Riboswitches and the RNA world. Cold Spring Harb Perspect Biol. 2012;4(2):a003566. doi: 10.1101/cshperspect.a003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Storz G, Vogel J, Wassarman KM. Regulation by small RNAs in bacteria: Expanding frontiers. Mol Cell. 2011;43(6):880–891. doi: 10.1016/j.molcel.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Milon P, Rodnina MV. Kinetic control of translation initiation in bacteria. Crit Rev Biochem Mol Biol. 2012;47(4):334–348. doi: 10.3109/10409238.2012.678284. [DOI] [PubMed] [Google Scholar]
- 23.Zheng X, Hu G-Q, She Z-S, Zhu H. Leaderless genes in bacteria: clue to the evolution of translation initiation mechanisms in prokaryotes. BMC Genomics. 2011;12(1):361. doi: 10.1186/1471-2164-12-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Q, Cheng X, Cheung MK, Kiselev SS, Ozoline ON, Kwan HS. High-density transcriptional initiation signals underline genomic islands in bacteria. PLOS ONE. 2012;7(3):e33759. doi: 10.1371/journal.pone.0033759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wade JT, Grainger DC. Pervasive transcription: illuminating the dark matter of bacterial transcriptomes. Nat Rev Microbiol. 2014;12(9):647–653. doi: 10.1038/nrmicro3316. [DOI] [PubMed] [Google Scholar]
- 26.Parsons YN, Banasko R, Detsika MG, Duangsonk K, Rainbow L, Hart CA, Winstanley C. Suppression-subtractive hybridisation reveals variations in gene distribution amongst the Burkholderia cepacia complex, including the presence in some strains of a genomic island containing putative polysaccharide production genes. Arch Microbiol. 2003;179(3):214–223. doi: 10.1007/s00203-003-0518-7. [DOI] [PubMed] [Google Scholar]
- 27.Dunbar J, Cohn J, Wall M. Consistency of gene starts among Burkholderia genomes. BMC Genomics. 2011;12(1):125. doi: 10.1186/1471-2164-12-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lybecker M, Bilusic I, Raghavan R. Pervasive transcription: detecting functional RNAs in bacteria. Transcription. 2014;5(4):e944039. doi: 10.4161/21541272.2014.944039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomason MK, Storz G. Bacterial antisense RNAs: How many are there, and what are they doing? Annu Rev Genet. 2010;44(1):167–188. doi: 10.1146/annurev-genet-102209-163523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peeters E, Sass A, Mahenthiralingam E, Nelis H, Coenye T. Transcriptional response of Burkholderia cenocepacia J2315 sessile cells to treatments with high doses of hydrogen peroxide and sodium hypochlorite. BMC Genomics. 2010;11(1):90. doi: 10.1186/1471-2164-11-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freeman ZN, Dorus S, Waterfield NR. The KdpD/KdpE two-component system: Integrating K+ homeostasis and virulence. PLoS Pathog. 2013;9(3):e1003201. doi: 10.1371/journal.ppat.1003201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salvetti S, Faegri K, Ghelardi E, Kolsto AB, Senesi S. Global gene expression profile for swarming Bacillus cereus bacteria. Appl Environ Microbiol. 2011;77(15):5149–5156. doi: 10.1128/AEM.00245-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kinsinger RF, Kearns DB, Hale M, Fall R. Genetic requirements for potassium ion-dependent colony spreading in Bacillus subtilis. J Bacteriol. 2005;187(24):8462–8469. doi: 10.1128/JB.187.24.8462-8469.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beenken KE, Dunman PM, McAleese F, Macapagal D, Murphy E, Projan SJ, Blevins JS, Smeltzer MS. Global gene expression in Staphylococcus aureus biofilms. J Bacteriol. 2004;186(14):4665–4684. doi: 10.1128/JB.186.14.4665-4684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gottesman S, Storz G. Bacterial small RNA regulators: Versatile roles and rapidly evolving variations. Cold Spring Harb Perspect Biol. 2010;3(12):a003798. doi: 10.1101/cshperspect.a003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoder-Himes DR, Chain PSG, Zhu Y, Wurtzel O, Rubin EM, Tiedje JM, Sorek R. Mapping the Burkholderia cenocepacia niche response via high-throughput sequencing. P Natl Acad Sci USA. 2009;106(10):3976–3981. doi: 10.1073/pnas.0813403106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weinberg Z, Wang J, Bogue J, Yang J, Corbino K, Moy R, Breaker R. Comparative genomics reveals 104 candidate structured RNAs from bacteria, archaea, and their metagenomes. Genome Biol. 2010;11(3):R31. doi: 10.1186/gb-2010-11-3-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kimelman A, Levy A, Sberro H, Kidron S, Leavitt A, Amitai G, Yoder-Himes DR, Wurtzel O, Zhu Y, Rubin EM, et al. A vast collection of microbial genes that are toxic to bacteria. Genome Res. 2012;22(4):802–809. doi: 10.1101/gr.133850.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, Eddy SR, Gardner PP, Bateman A. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2013;41:D226–D232. doi: 10.1093/nar/gks1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramos CG, Grilo AM, da Costa PJP, Leitão JH. Experimental identification of small non-coding regulatory RNAs in the opportunistic human pathogen Burkholderia cenocepacia J2315. Genomics. 2013;101(2):139–148. doi: 10.1016/j.ygeno.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 41.Coenye T, Van Acker H, Peeters E, Sass A, Buroni S, Riccardi G, Mahenthiralingam E. Molecular mechanisms of chlorhexidine tolerance in Burkholderia cenocepacia biofilms. Antimicrob Agents Chemoth. 2011;55(5):1912–1919. doi: 10.1128/AAC.01571-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Drevinek P, Holden MTG, Ge Z, Jones AM, Ketchell I, Gill RT, Mahenthiralingam E. Gene expression changes linked to antimicrobial resistance, oxidative stress, iron depletion and retained motility are observed when Burkholderia cenocepacia grows in cystic fibrosis sputum. BMC Infect Dis. 2008;8:121. doi: 10.1186/1471-2334-8-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coenye T, Drevinek P, Mahenthiralingam E, Shah SA, Gill RT, Vandamme P, Ussery DW. Identification of putative noncoding RNA genes in the Burkholderia cenocepacia J2315 genome. FEMS Microbiol Lett. 2007;276(1):83–92. doi: 10.1111/j.1574-6968.2007.00916.x. [DOI] [PubMed] [Google Scholar]
- 44.Vogel J, Luisi BF. Hfq and its constellation of RNA. Nat Rev Microbiol. 2011;9(8):578–589. doi: 10.1038/nrmicro2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Acker H, De Canck E, Van Nieuwerburgh F, Sass A, Deforce D, Nelis HJ, Coenye T. The BCESM genomic region contains a regulator involved in quorum sensing and persistence in Burkholderia cenocepacia J2315. Future Microbiol. 2014;9(7):845–860. doi: 10.2217/fmb.14.54. [DOI] [PubMed] [Google Scholar]
- 46.Sass A, Marchbank A, Tullis E, LiPuma JJ, Mahenthiralingam E. Spontaneous and evolutionary changes in the antibiotic resistance of Burkholderia cenocepacia observed by global gene expression analysis. BMC Genomics. 2011;12:373. doi: 10.1186/1471-2164-12-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Förstner KU, Vogel J, Sharma CM. READemption—a tool for the computational analysis of deep-sequencing–based transcriptome data. Bioinformatics. 2014;30(23):3421–3423. doi: 10.1093/bioinformatics/btu533. [DOI] [PubMed] [Google Scholar]
- 48.Hoffmann S, Otto C, Kurtz S, Sharma CM, Khaitovich P, Vogel J, Stadler PF, Hackermüller J. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput Biol. 2009;5(9):e1000502. doi: 10.1371/journal.pcbi.1000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Amman F, Wolfinger M, Lorenz R, Hofacker I, Stadler P, Findeiß S. TSSAR: TSS annotation regime for dRNA-seq data. BMC Bioinformatics. 2014;15(1):89. doi: 10.1186/1471-2105-15-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Van Acker H, Sass A, Bazzini S, De Roy K, Udine C, Messiaen T, Riccardi G, Boon N, Nelis HJ, Mahenthiralingam E, et al. Biofilm-grown Burkholderia cepacia complex cells survive antibiotic treatment by avoiding production of reactive oxygen species. PLoS ONE. 2013;8(3):e58943. doi: 10.1371/journal.pone.0058943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nicol JW, Helt GA, Blanchard SG, Raja A, Loraine AE. The Integrated Genome Browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics. 2009;25(20):2730–2731. doi: 10.1093/bioinformatics/btp472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ermolaeva MD, Khalak HG, White O, Smith HO, Salzberg SL. Prediction of transcription terminators in bacterial genomes. J Mol Biol. 2000;301(1):27–33. doi: 10.1006/jmbi.2000.3836. [DOI] [PubMed] [Google Scholar]
- 54.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 55.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11(3):119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Winsor GL, Khaira B, Van Rossum T, Lo R, Whiteside MD, Brinkman FS. The Burkholderia Genome Database: facilitating flexible queries and comparative analyses. Bioinformatics. 2008;24(23):2803–2804. doi: 10.1093/bioinformatics/btn524. [DOI] [PMC free article] [PubMed] [Google Scholar]