

Abstract
Background
South Pacific Gyre (SPG) is the largest and clearest gyre in the world, where the concentration of surface chlorophyll a and primary production are extremely low. Aquimarina longa SW024T was isolated from surface water of the SPG center. To understand how this bacterium could survive in this ultra-oligotrophic oceanic environment and its function in biogeochemical cycle, we sequenced the genome of A. longa SW024T and performed extensive genomic analyses.
Methods
Genomic DNA was extracted and sequenced using Illumina Hiseq 2000 and Miseq platform. Genome annotation, genomic comparison and phylogenetic analyses were performed with the use of multiple bioinformatics tools like: BLAST+ 2.2.24, Glimmer3.0, RAST server, Geneious 4.8.5, ClustalW2 and MEGA5. Physiological and morphological features were tested by bacterial culture, electron microscopy, fluorescence microscopy and exopolysaccharides extraction.
Results
Analysis of seven Aquimarina genomes and 30 other genomes of Flavobacteriaceae isolated from seawater showed that most of the strains had low DNA G + C contents, and Aquimarina had larger genomes than other strains. Genome comparison showed varying genomic properties among seven Aquimarina genomes, including genome sizes and gene contents, which may warrant their specific adaptive strategies. Genome of A. longa SW024T was further compared with the genomes of two other Aquimarina species which were also isolated from the SPG and A. longa SW024T appeared to have much more genes related to replication, recombination and repair. As a copiotroph, A. longa SW024T is long in length, and possesses large genome size and diverse transporters. However, it has also evolved many properties to survive in the oligotrophic marine environment. This bacterium grew better on solid medium than in liquid medium, suggesting it may be liable to attach to particle surfaces in order to survive in the nutrient-limiting environment. Gliding motility and the capacity to degrade various polymers possibly allow the bacterium to grow on detritus particles and use polymeric substances as carbon and energy sources. Moreover, genes related to carbon, nitrogen, and sulfur metabolisms were identified, which showed that A. longa SW024T might be involved in various elemental cycles.
Conclusions
Genomic comparison of Aquimarina genus exhibits comprehensive capabilities of the strains to adapt to diverse marine environments. The genomic characteristics of A. longa SW024T reveal that it evolves various strategies to cope with both copiotrophic and ultra-oligotrophic marine environment, which provides a better understanding of the survival abilities of bacteria in prevalent and even extreme oceanic environments. Furthermore, carbon, nitrogen and sulfur utilization of A. longa SW024T may represent its potential functions in the global biogeochemical cycle.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-2005-3) contains supplementary material, which is available to authorized users.
Keywords: Aquimarina longa, Genome analysis, Genome comparison, Oligotrophic bacterium
Background
Among the major taxa of marine bacterioplankton, members of the Bacteroidetes are frequently found enriched on organic matter particles and are specialists for degrading high molecular weight compounds of both dissolved and particulate marine organic matters, implying a major role they play in the marine carbon cycle [1, 2]. Bacteroidetes have been shown to comprise the largest fraction of bacteria consuming chitin, polysaccharides and proteins, but the smallest fraction consuming amino acids [3]. Luo [4] found that Bacteroidetes clades had a greater fraction of genes encoding periplasmic proteins and a lower fraction of genes encoding inner membrane proteins in their metatranscriptomes than in their genomes and metagenomes, corroborating the macromolecule degradation process requiring cell surface associated or extracellular hydrolases. Some representatives of the Bacteroidetes phylum such as Flavobacteriaceae were frequently found attached to aggregates and appeared during an algae-bloom collapse [5]. They were also known to move over surfaces by gliding motility. The genus Aquimarina is a member of the family Flavobacteriaceae [6] and was first described in 2005 [7]. Up to now, a total of 18 species in the genus Aquimarina have been recognized, and all of them were isolated from marine environments.
South Pacific Gyre (SPG) is the largest gyre in the world, which has the lowest surface chlorophyll a (Chl a) concentration [8] and is believed to be the clearest water in the world [9]. Comparing with the gyre edge, the low concentration of Chl a, ammonium, nitrate and phosphate in the gyre center makes the region an ultra-oligotrophic oceanic environment [8]. Aquimarina longa SW024T was a new species isolated from the surface water of a station (U1367) located in the central gyre of SPG [10, 11]. It is long-rod in shape, 0.3 μm in width and 3.0–66.0 μm in length, non-flagellated and motile by gliding. Colonies on marine agar 2216 (MA; Becton Dickinson) are yellow, producing pigment with maximum absorption at 453 nm and 479 nm. Some substrates can be hydrolyzed by this bacterium, including chitin, gelatin, DNA, and Tweens 20, 40 and 80. It is also resistant to many antibiotics, such as benzylpenicillin, carbenicillin, cefuroxime, cephalosporin V, polymyxin B, gentamicin, kanamycin, neomycin, tetracycline, cefoperazone and streptomycin. The specific cellular morphology and the physiological function of this bacterium may provide some advantages for its survival in the ultra-oligotrophic environment.
Nowadays, more and more bacterial genomes have been sequenced and analyzed, however, the genomes of genus Aquimarina which is closely associated with marine environment have not been analyzed systematically, except for A. agarilytica ZC1T, which was isolated from marine red alga and had been proved to have agarolytic activity [12]. In this context, the present study aims to provide a better understanding of the survival mechanisms and biogeochemical role of A. longa SW024T in the ultra-oligotrophic environment by extensive genomic analyses. In addition, comparison with other publicly available genome sequences from members of Aquimarina reveals that they are diverse in their genome sizes and gene contents, which might warrant their specific adaptive strategies.
Methods
Bacterial growth and DNA extraction
A. longa SW024T was routinely grown aerobically in marine broth 2216 (MB; BD) or on MA at 28°C. A series of dilutions (1:2, 1:5, 1:10, 1:20 and 1:50) of MB and MA media were used to determine its growth in nutrient-limiting conditions. After being inoculated in MB or streaked on MA, the bacteria were cultured at 28°C for two weeks. Genomic DNA was extracted from the cells by using phenol-chloroform-isoamylic alcohol extraction protocol described by Marmur [13], and the 16S rRNA genes were sequenced to validate the obtained strains.
Genome sequencing, analysis and annotation
The genome of A. longa SW024T was sequenced using the Illumina Hiseq 2000 with 2 kb and 3 kb mate-pair libraries and Illumina Miseq with a 400 bp paired-end library, achieving about 241-fold coverage. The reads were assembled using GS de novo assembler software.Putative genes were identified using GLIMMER 3.0 [14]. Annotation was performed with BLAST+ 2.2.24 [15] searching against databases, including the National Center for Biotechnology Information (NCBI) non-redundant proteins (NR) [16], Clusters of Orthologous Groups of proteins (COG) [17], Kyoto encyclopedia of genes and genomes (KEGG) [18] and Gene ontology (GO) [19]. The criteria used to assign function to a protein translated by predicted open reading frames (ORFs) were a minimum cut-off of 30% identity and at least three best hits among the NR, COG, KEGG and GO databases.
Genomic comparison
The genomes of A. pacifica SW150T, A. megaterium XH134T and A. macrocephali JAMB N27T were previously sequenced in the lab, in which the first two strains were also isolated from SPG [20, 21] (Table 1). The genome sequences of A. latercula DSM 2041T, A. muelleri DSM 19832T, A. agarilytica ZC1T and 30 other Flavobacteriaceae strains were obtained from NCBI (Table 1, Additional file 1). The ORFs of all these genomes were predicted conformably using RAST server [22] and translated to amino acid by Geneious 4.8.5 [23]. Orthologous proteins were defined as reciprocal best hit proteins with a minimum 50% identity and 70% of the length of the query protein [24], calculated by the BLAST algorithm. Proteins existed in all genomes subsequently were aligned using ClustalW2 [25], the resulting alignments were concatenated to provide the whole-genome alignment. Phylogenetic analysis was performed by using the MEGA5 software package [26] and the neighbor-joining tree [27] was constructed and validated with 1000 bootstraps. Genomic features and function annotation of predicted proteins from A. longa SW024T were first compared with those of the six genomes from the same genus and then further compared with the two strains isolated from SPG, using BLASP with an E-value cut-off of 1e-5. Pan-genome and orthologous cluster analyses were performed with pan-genome analysis pipeline [28].
Table 1.
Summary of genomic information of seven Aquimarina genomes
| Strains | Size (Mb) | G + C content (%) | Contig No. | ORF No. | Orthologous cluster No. | Specific genes | GenBank accession No. | Isolation environment |
|---|---|---|---|---|---|---|---|---|
| A. longa SW024T | 5.50 | 31.45 | 90 | 4822 | 4521 | 1522 | AVQK00000000 | Surface seawater of SPG |
| A. pacifica SW150T | 5.26 | 33.49 | 145 | 4368 | 4230 | 1280 | JACC00000000 | Surface seawater of SPG |
| A. megaterium XH134T | 6.21 | 32.93 | 170 | 5425 | 5206 | 1039 | JACB00000000 | Surface seawater of SPG |
| A. macrocephali JAMB N27T | 6.06 | 32.93 | 169 | 5414 | 5211 | 1088 | JACA00000000 | Marine sediment off Kagoshima, Japan |
| A. latercula DSM 2041T | 6.24 | 32.21 | 31 | 5522 | 5165 | 1831 | AUMK00000000 | Outflow of a marine aquarium in La Jolla, California, USA |
| A. muelleri DSM 19832T | 4.90 | 31.33 | 106 | 4085 | 3942 | 869 | AUML00000000 | Seawater of Amursky Bay, Sea of Japan |
| A. agarilytica ZC1T | 4.25 | 32.81 | 131 | 3571 | 3451 | 1711 | AHHE00000000 | Surface of marine red alga, collected near Nan Ao Island, Guangdong province, China |
Cellular morphology and chitinase activity
A. longa SW024T was cultured in MB for one day, and then observed with a transmission electron microscope (TEM-1200EX, JEOL, Tokyo, Japan). It was also stained with DAPI and viewed by fluorescence microscopy with a ×100 oil immersion lens (Nikon Eclipse 50i, Japan). The intracellular structure was shown using ultramicrotomy and observed with transmission electron microscope. Chitinase activity was observed using chitin agar following the method described by Hsu and Lockwood [29].
Exopolysaccharides (EPS) analyses
EPS extraction and analyses were followed the method described by Balsanelli et al. [30] with some modification. Briefly, A. longa SW024T was grown in MB medium at 28°C and 170 rpm. After 3 days, 10 ml of the bacterial cultures were centrifuged and the supernatant were precipitated with 3 volumes of cold ethanol for 24 hours at 4°C and centrifuged for 10 minutes at 4°C and 8,000 g. The precipitate was dissolved in deionized water and dialyzed against MilliQ water. The dialyzed sample was lyophilized and resuspended in 1 ml of distilled water. Total sugar concentration of the samples was determined with phenol/sulfuric acid [31], using glucose as standard. Three independent experiments were performed and the mean concentration was calculated.
Nucleotide sequence accession numbers
The genome project has been deposited in the Genome On Line Database (GOLD) under the accession number Gi0050938. This Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession number AVQK00000000. The version described in this paper is version AVQK01000000.
Results and discussion
Genome features of A. longa SW024T and other Aquimarina bacteria
The genome of A. longa SW024T was composed of 5,506,799 bp, and the calculated G + C content was 31.45 %. A total of 90 contigs ranging from 616 bp to 487,322 bp (the N50 and N90 contig sizes were 288,357 bp and 66,130 bp, respectively) were obtained and combined into 67 scaffolds ranging from 1130 bp to 837,171 bp (the N50 and N90 scaffold sizes were 309,215 bp and 66,130 bp, respectively). A total of 4822 ORFs were identified within the A. longa SW024T genome (Table 1). Among the predicted genes, 2507 (51.99 %) were found in COG categories, 1455 (30.17 %), 4168 (86.44 %) and 1939 (40.21 %) genes were applicable within the KEGG, NR and GO databases, respectively.
General information of seven Aquimarina genomes used for comparison and analysis was summarized in Table 1. The contig numbers of the draft genomes ranged from 31 to 170. The genome sizes ranged from 4.25 to 6.24 Mb, with a mean size of 5.49 Mb.
Phylogenetic and functional properties of Aquimarina genomes
Although it is more popular to use 16S rRNA gene to explain phylogenetic relationship of bacteria, phylogenetic tree constructed using orthologous proteins appears to be more accurate to elucidate the genetic relationship among different microbes [32]. In addition to all the seven Aquimarina genomes, genomic sequences of 30 other Flavobacteriaceae strains isolated from seawater were also obtained to construct an orthologous proteins tree (Fig. 1, Additional file 1). As expected, genomes of the same genus gathered together, and the genera Mesonia, Gillisia, Gramella and Salegentibacter formed a clade which was closely related with the genus Aquimarina. Although there is diversity among Flavobacteriaceae, the adaptation to the degradation of polymeric substances seems to be a common theme [1, 2].
Fig. 1.
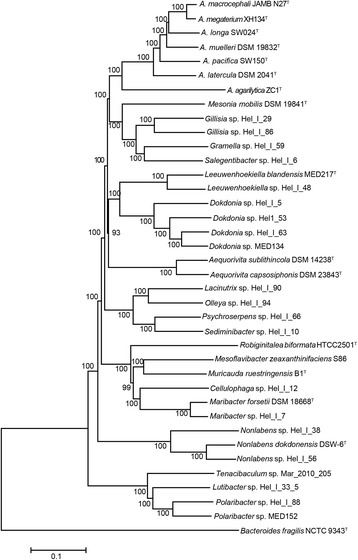
Phylogenetic relationships of the family Flavobacteriaceae. The tree was constructed with concatenated alignment of orthologous proteins using Neighbour-joining method with 1000 bootstrap replications. Type species Bacteroides fragilis NCTC 9343T from Bacteroidaceae served as outgroup
Analysis of the seven Aquimarina genomes and 30 other genomes of Flavobacteriaceae isolated from seawater [Additional file 1] showed that ORF numbers were proportional to their genome sizes, with the larger genomes containing greater number of ORFs. The DNA G + C contents of most of these strains were relatively low, ranging from 30 % to 40 % (Fig. 2). Low G + C content may be an adaptive strategy for bacteria to nitrogen limitation [33], because AT base pairs use less nitrogen than GC pairs. The relative availability and/or energetic expenditure incurred by different nucleotides is another explanation for the low G + C content, by the reason that GTP and CTP are energetically more costly to generate than ATP and UTP. Therefore, low G + C content in this genus may help bacteria save energy and would be favored over their GC-rich counterparts when living in the nutrient limited or energetic constraint marine environments.
Fig. 2.
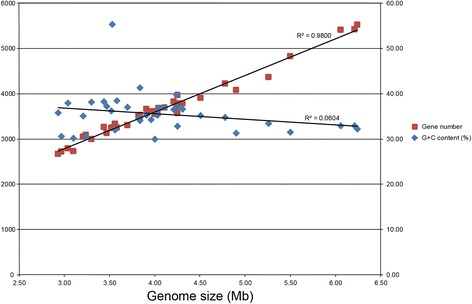
Relationships of genome sizes, ORF numbers and DNA G + C contents. Data of seven Aquimarina strains and 30 other Flavobacteriaceae strains isolated from seawater were chosen
Meanwhile, orthologous clusters among the seven Aquimarina genomes were also analyzed. The number of orthologous clusters contained in each individual genome ranged from 3451 to 5211 (Table 1). Among the strains, A. muelleri DSM 19832T had the smallest number of specific genes (869), while A. latercula DSM 2041T had the largest (1831). Sixty-three percent (8976) of the total clusters (14,249) were specific genes (Fig. 3), and the proportion was higher than that reported in other genera, such as Glaciecola (59 %), Shewanella (48 %) and Streptococcus (18 %) [32, 34, 35]. In addition, the percentage of core genes (1268, approximately 8.9 %) was lower than that of the specific genes (Fig. 3). These results indicated a high degree of gene content variation in Aquimarina genomes, which may be due to the geographic segregation of these strains. Strains of Aquimarina in different geographic locations would be unable to exchange genetic information. Instead, they may exchange DNA among surrounding bacteria, thus leading to the high genetic diversity.
Fig. 3.
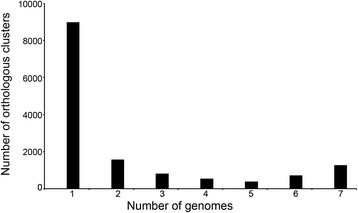
Numbers of orthologous gene clusters that are shared in a given number of Aquimarina genomes. One and seven genomes correspond to the unique and core gene clusters, respectively
The power law and exponential decaying models were used to describe the pan- and core-genome of the genus Aquimarina, respectively. The pan-genome curve can perfectly fit a power law function with an exponent of 0.62, which indicates that the pan-genome of Aquimarina is open and new orthologous clusters will be added when a new genome of Aquimarina is sequenced [36]. The core genome decreased sharply from an average of 4532 to 2300 when the first two genomes were added [Additional file 2].
Functional characterization of COG (Table 2) showed that Aquimarina had higher proportions of genes for translation, ribosomal structure and biogenesis (J), signal transduction mechanisms (T), and secondary metabolites biosynthesis, transport and catabolism (Q) compared to the mean values of 115 genomes calculated by Konstantinidis and Tiedje [37]. The Aquimarina core gene sets were enriched in genes that encode proteins involved in translation, posttranslational modification, as well as metabolism of amino acid, nucleotide, coenzyme and lipid (COG categories J, O, E, F, H and I) when compared with dispensable genes (Fig. 4). These genes are retained in all the genomes since they are related to central metabolisms and are essential to survival. Similar enrichment was also observed in the genera Glaciecola and Shewanella [32, 34]. However, the lower proportions of core gene sets corresponding to transcription (K) and signal transduction mechanisms (T) indicate that different genomes may have evolved different transcriptional and signal transduction systems, or alternatively the transcriptional and signaling genes are not well conserved in this genus.
Table 2.
Percentage of COG categories in each Aquimarina strain
| COG categories | A. longa SW024T | A. pacifica SW150T | A. megaterium XH134T | A. macrocephali JAMB N27T | A. latercula DSM 2041T | A. muelleri DSM 19832T | A. agarilytica ZC1T |
|---|---|---|---|---|---|---|---|
| B | 0.036 | 0.038 | 0.029 | 0.030 | 0.031 | 0.037 | 0.046 |
| C | 4.753 | 4.641 | 4.548 | 4.519 | 4.734 | 4.978 | 5.251 |
| D | 0.756 | 0.761 | 0.583 | 0.667 | 0.611 | 0.780 | 0.883 |
| E | 7.886 | 7.607 | 8.192 | 8.098 | 7.086 | 8.841 | 7.574 |
| F | 2.233 | 2.396 | 2.012 | 2.032 | 2.046 | 2.489 | 2.463 |
| G | 3.277 | 4.108 | 4.052 | 3.518 | 5.559 | 3.306 | 5.112 |
| H | 5.041 | 4.869 | 4.548 | 4.580 | 4.276 | 5.163 | 4.879 |
| I | 3.169 | 3.119 | 3.528 | 3.397 | 3.787 | 3.975 | 3.253 |
| J | 5.906 | 6.162 | 4.956 | 5.126 | 5.162 | 6.241 | 7.342 |
| K | 8.246 | 8.406 | 11.079 | 10.889 | 8.888 | 7.578 | 6.831 |
| L | 6.158 | 4.336 | 3.236 | 4.246 | 3.940 | 4.755 | 5.762 |
| M | 7.742 | 8.939 | 6.239 | 6.127 | 8.125 | 7.355 | 9.201 |
| N | 0.360 | 0.380 | 0.262 | 0.303 | 0.458 | 0.371 | 0.743 |
| O | 3.745 | 3.728 | 3.178 | 3.518 | 3.635 | 4.123 | 4.507 |
| P | 5.149 | 5.401 | 4.519 | 4.610 | 4.795 | 4.866 | 5.390 |
| Q | 2.953 | 3.081 | 3.761 | 3.518 | 3.451 | 4.383 | 2.138 |
| R | 13.936 | 13.998 | 14.519 | 14.680 | 14.508 | 14.042 | 13.151 |
| S | 8.714 | 7.950 | 7.872 | 8.432 | 8.522 | 7.912 | 7.667 |
| T | 6.482 | 6.618 | 8.542 | 7.583 | 6.659 | 5.498 | 4.926 |
| U | 1.296 | 1.445 | 1.137 | 1.092 | 1.100 | 1.226 | 1.115 |
| V | 2.161 | 2.016 | 3.178 | 3.033 | 2.627 | 2.043 | 1.766 |
| W | 0.000 | 0.000 | 0.029 | 0.000 | 0.000 | 0.037 | 0.000 |
Function of COG categories: [B] Chromatin Structure and dynamics; [C] Energy production and conversion; [D] Cell cycle control, cell division, chromosome partitioning; [E] Amino acid transport and metabolism; [F] Nucleotide transport and metabolism; [G] Carbohydrate transport and metabolism; [H] Coenzyme transport and metabolism; [I] Lipid transport and metabolism; [J] Translation, ribosomal structure and biogenesis; [K] Transcription; [L] Replication, recombination and repair; [M] Cell wall/membrane/envelope biogenesis; [N] Cell motility; [O] Posttranslational modification, protein turnover, chaperones; [P] Inorganic ion transport and metabolism; [Q] Secondary metabolites biosynthesis, transport and catabolism; [R] General function prediction only; [S] Function unknown; [T] Signal transduction mechanisms; [U] Intracellular trafficking, secretion, and vesicular transport; [V] Defense mechanisms; [W] Extracellular structures
Fig. 4.
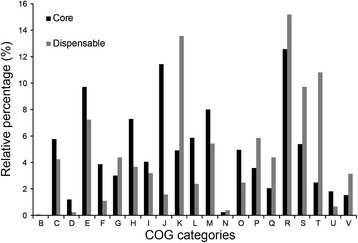
Comparison of the COG categories of the core and dispensable gene sets coding proteins. The function of COG categories is described in Table 2
Comparison of three Aquimarina strains isolated from SPG
A. longa SW024T [10], A. pacifica SW150T [21] and A. megaterium XH134T [20] were isolated from surface seawater of the SPG at stations U1367, U1369 and U1371, respectively, as shown in [11]. The COG-based analysis among the specific and orthologous proteins showed that the largest proportion of orthologous genes of the three bacteria belong to amino acid transport and metabolism (E), while that of specific genes in A. longa SW024T, A. pacifica SW150T and A. megaterium XH134T belong to replication, recombination and repair (L), cell wall/membrane/envelope biogenesis (M) and transcription (K), respectively (Fig. 5). The different genome contents among these bacteria may be likewise correlated with diverse phylogenies, trophic strategies and ocean environments, as described in roseobacters [38]. For instance, A. longa SW024T possesses four photolyases PhrB, while three and only one PhrB coding genes were identified in A. pacifica SW150T and A. megaterium XH134T, respectively. This might be one of the reasons to explain their diverse distributions. With the lowest Chl a in station U1367, bacteria in this place suffer more UV damage than those in other stations and therefore needs more PhrB.
Fig. 5.
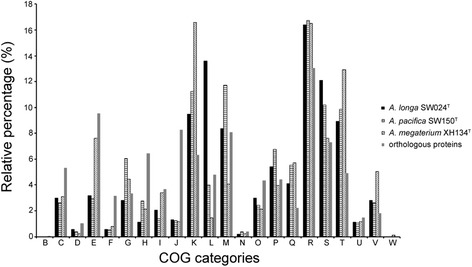
Relative abundance compared to all COG categories of the orthologous and specific proteins. Putative orthologous proteins are defined as reciprocal best hit proteins with a minimum 50 % identity and 70 % of the length of the query protein, calculated by the BLAST algorithm. The function of COG categories is described in Table 2
Bacterial shape and growth of A. longa SW024T
Cells of A. longa SW024T usually appear as filamentous and are relatively long in length (Fig. 6a), the maximum length grown in MB is 66 μm [10], about 100 times longer than the smallest bacteria observed in oligotrophic ocean environment [39]. Moreover, it is thin and the width is only about 0.3 μm (Fig. 6d). A. megaterium XH134T, which was also isolated from SPG, is even longer (up to 77.8 μm) and harbors a larger genome (6.21 Mb, Table 1). Large genome brings large nucleic acids and proteins, therefore the cell must have sufficient room to include all the nucleic acids, proteins, molecular complexes and other gears required for survival and proliferation [40]. The relatively long lengths might confer an advantage for the bacteria attaching to particle surfaces by increasing the contact area. Proteins functioning in cell size control by maintaining cell shape within normal ranges were found in the genome of A. longa SW024T, such as FtsZ, penicillin binding protein 2 (PBP 2) and MreBCD.
Fig. 6.
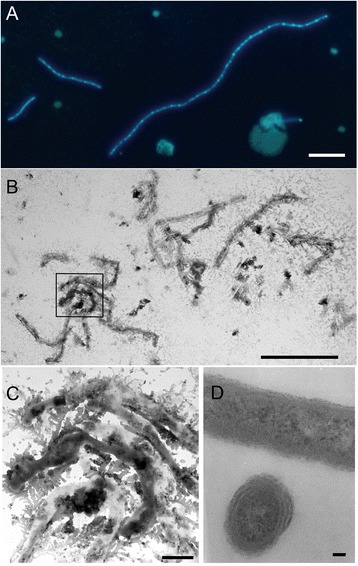
Bacterial shape of A. longa SW024T. Fluorescence microscopy of A. longa SW024T stained with DAPI, bar = 5 μm (a). Transmission electron microscopy of A. longa SW024T culturing in MB medium without staining, bar = 10 μm (b) and magnification of aggregate section in the boxed area, bar = 1 μm (c). Transmission electron microscopy of A. longa SW024T using ultramicrotomy, including transections and longitudinal section, bar = 50 nm (d)
Growth was measured in a series of diluted MB and MA media to examine its survival in nutrient-limiting conditions. The result showed that A. longa SW024T could grow on 1:20 dilution of MA plate, but only in 1:2 dilution of MB liquid medium, after two weeks’ culture. A. longa SW024T formed yellow colonies on agar plates [Additional file 3] and tended to aggregate into flocks in liquid culture (Fig. 6b). Abundant extracellular materials were secreted which may help bacteria gather together (Fig. 6c). The genome owns a large array of genes involved in the synthesis and export of extracellular polysaccharide material (e.g., 33 putative glycosyl transferases). These results suggest that A. longa SW024T may grow in the nutrient-limiting environment by attaching to surfaces of particles.
After being precipitated with ethanol and lyophilized, 0.8 mg mL−1 EPS was obtained from A. longa SW024T cultured in MB medium. This amount is equal to the production of Herbaspirillum seropedicae [30], but less than that of Pseudomonas atlantic (mean production is 2.7 mg mL−1) [41]. In natural aquatic environments, nutrients required to support maximal microbial growth are rarely present in sufficient quantities in the water column. Microbial attachment and aggregates is likely a strategy to increase the rate of substrate uptake [42], and a porous matrix of exopolymer surrounding microbial cells could sequester and concentrate dissolved organic compounds [41]. It thus could be inferred that EPSs synthesized by A. longa SW024T might act as a sponge to trap and concentrate nutrients in flowing liquids and be able to help this bacterium absorb dissolved organic material.
Smaller genomes, fewer gene duplications, and depleted in DNA G + C contents, noncoding nucleotides, and genes encoding transcription, signal transduction and noncytoplasmic proteins have been identified as indicators of genome streamlining and oligotrophy [43, 44], all of which are opponent to A. longa SW024T except the low DNA G + C content. Genome of A. longa SW024T is overrepresented in dehydrogenases (77 predicted dehydrogenases), and is enriched in COGs involved in defense mechanisms (V), transcription (K) and signal transduction (T, Table 2), consisting with the properties of copiotrophs proposed by Lauro et al. [44]. With these genomic and phenotypic features, we assume that A. longa SW024T is a copiotroph but can survive in oligotrophic marine environment probably by attaching to particles.
Gliding motility
A. longa SW024T has a complete set of genes involved in gliding motility (gldA, gldB, gldC, gldD, gldE, gldF, gldG, gldH, gldI, gldJ, gldK, gldL, gldM, gldN, sprA and sprE), which could be beneficial in the exploration of solid surfaces. A. longa SW024T attaches readily to glass slide and displays rapid motility [10]. Previous studies showed that cells with mutations in genes encoding these proteins were completely nonmotile in Flavobacterium johnsoniae ([45] and references therein). Mutants did not exhibit movement on agar or glass surfaces, failed to propel latex spheres, and formed nonspreading colonies. The gliding motility of Aquimarina may be stimulated by the oligotrophic environment, since it is beneficial to search insoluble macromolecular substrates such as starch and chitin, which could be utilized by Aquimarina as carbon resource. Moreover, gliding motility can help bacteria position themselves at optimal conditions of light intensity, oxygen, hydrogen sulfide, temperature and other factors that influence growth [46].
Adaptation strategies to the oligotrophic marine environment
Survival of a bacterium in the ultra-oligotrophic surface seawater depends on effective uptake of the primary elemental ingredients for life, such as nitrogen (N), phosphorus (P), sulfur (S) and iron (Fe). Many proteins produced by A. longa SW024T are involved in these processes (Table 3).
Table 3.
Genes related to oligotrophic marine environment adaption
| Function | Encoded gene product | Gene |
|---|---|---|
| Nitrogen sensing and regulation | nitrogen regulation protein | ntrY |
| nitrogen regulatory protein | glnB | |
| Ammonium assimilation | glutamine synthetase | glnA |
| glutamate synthase, NADH/NADPH small subunit | gltD | |
| glutamate synthase, NADPH/NADH large subnit | gltB | |
| Denitrification | nitrate reductase | napA |
| nitrite reductase | nirK | |
| nitric oxide reductase | norB | |
| nitrous oxide reductase | nosZ | |
| Hydrolysis of dissolved organic phosphorus | alkaline phosphatase | phoD |
| Sensing and responding to changes in external/internal P levels | PhoP family transcriptional regulator | phoP |
| histidine kinase | phoR | |
| Sulfate transport | putative sulfate transporter | cysZ |
| sulfate permease | sulP | |
| Iron uptake | TonB-dependent siderophore receptor | / |
| iron(III) ABC transporter | / | |
| ferrous iron transport protein B | feoB | |
| ferrous iron transport protein A | feoA | |
| ferric enterobactin receptor | / | |
| Primary Na + pump | Na+-translocating NADH/ubiquinone oxidoreductase subunit A | nqrA |
| Na+-translocating NADH/ubiquinone oxidoreductase subunit B | nqrB | |
| Na+-translocating NADH/ubiquinone oxidoreductase subunit C | nqrC | |
| Na+-translocating NADH/ubiquinone oxidoreductase subunit D | nqrD | |
| Na+-translocating NADH/ubiquinone oxidoreductase subunit E | nqrE | |
| Na+-translocating NADH/ubiquinone oxidoreductase subunit F | nqrF | |
| Primary H+ pump | cytochrome d ubiquinol oxidase subunit I | cydA |
| cytochrome d ubiquinol oxidase subunit II | cydB | |
| cytochrome c oxidase subunit I | coxA | |
| cytochrome c oxidase subunit II | coxB | |
| cytochrome c oxidase subunit III | coxC | |
| cytochrome c oxidase subunit IV | coxD | |
| protoheme IX farnesyltransferase | cyoE | |
| Exporting of Na+ ions | Na+/H+ antiporter | nhaA |
| Na+/H+ antiporter | nhaB | |
| Na+/H+ antiporter | nhaC | |
| Na+/H+ antiporter | nhaP | |
| Other sodium dependent | Na+/proline symporter | / |
| Na+/phosphate symporter | / | |
| cation/acetate symporter | actP | |
| Na+/dicarboxylate symporter | / | |
| Na+/nucleoside permease | / | |
| Na+/iodide cotransporter | / | |
| Na+/K+/Ca2+ exchanger | yrbG | |
| Na+/bile acid transporter | / | |
| Na+/multivitamin transporter | / | |
| Na+/glucose cotransporter | / | |
| proton/Na+-glutamate symporter | / | |
| Extracellular osmolarity and salinity stress adaption | MIP family channel protein aquaporin | aqpZ |
A. longa SW024T harbors nitrogen regulation proteins NtrY and PII that are required for sensing and responding to N fluctuation in seawater. The PII signal transduction protein has a central position in the coordination of carbon, nitrogen and energy status of the cells. Most of the target proteins interacting with PII protein perform or regulate crucial reactions in nitrogen assimilatory pathways [47]. In addition, ammonium assimilation in this bacterium is mediated via glutamine synthetase and glutamate synthase. Nitrate is the most abundant N species in ocean environments, a complete pathway of denitrification exists in A. longa SW024T, helping the bacterium acquire energy by this process.
P starvation may limit growth, and potentially constrain nitrogen fixation. Four genes in the genome are involved in P acquisition, and all of them encode putative alkaline phosphatases, which are necessary for hydrolysis of dissolved organic phosphorus [48]. Two-component system PhoR/PhoP in the genome may play a role in sensing and responding to changes in external/internal P levels prior to activating components of the P acquisition tool kit.
Although the classical ABC-type sulfate transport system is missing in A. longa SW024T, it encodes two proteins, i.e., a homolog of CysZ and a putative sulfate permease, both of which could serve as a sulfate transporter. Iron uptake mechanisms include TonB-dependent siderophore receptor, ABC-type iron transporter, ferrous iron transport protein, and ferric enterobactin receptor.
Like other marine bacteria, A. longa SW024T encodes a primary Na+ pump, the Na+-translocating NADH/ubiquinone oxidoreductase, and probably uses a sodium ion gradient as the source of energy for nutrient uptake. In addition, it encodes primary H+ pumps, namely, cytochrome bd complex, and cytochrome c oxidase. Salt acclimation includes several ion transporters which serve as exporters for sodium and chloride, the main toxic ions in seawater, and importers for potassium, which is essential for many cellular processes. Exporting of Na+ ions at the expense of the proton gradient is performed by a variety of Na+/H+ antiporters, including NhaA, NhaB, NhaC and NhaP. In addition, A. longa SW024T harbors Na+/proline symporter, Na+/phosphate symporter, cation/acetate symporter, Na+/dicarboxylate symporter, Na+/nucleoside permease, Na+/iodide cotransporter, Na+/K+/Ca2+ exchanger, Na+/bile acid transporter, Na+/multivitamin transporter, Na+/glucose cotransporter, and proton/Na+-glutamate symporter. Besides, three aquaporins AqpZ could help the bacterium to withstand dramatic changes in extracellular osmolarity and adapt to salinity stress [49].
Resistance to adverse effects
Oxidative DNA damage is a major source of mutation load in living organisms by means of damaging DNA, proteins and membranes of cells [50]. To avoid oxidative damage, A. longa SW024T has set up several antioxidant defense mechanisms comprising antioxidant enzymes as well as antioxidative compounds. Three types of superoxide dismutase (SOD, i.e., Cu-Zn SOD, Mn-SOD, Fe-SOD) which catalyze the dismutation of O2− to O2 and H2O2, have been identified in the A. longa SW024T genome. Genes encoding for two catalase-peroxidases, KatG and KatE, catalyzing the decomposition of hydrogen peroxide to water and oxygen are present in the genome. Moreover, it was reported that translation of the quinone-binding proteins was protected by the katE gene in tobacco leaves during exposure to light stress [51]. These antioxidants might also be crucial for bacteria survival during exposure to other stresses such as UV radiation. The reactive oxygen species-scavenging system in A. longa SW024T also contains three peroxiredoxins (Prx), termed thioredoxin peroxidases, including one PrxQ and two 2-Cys Prx, which catalyze the reduction of various hydroxyperoxides. Prx proteins mainly function when the concentration of H2O2 is low, while catalases mainly detoxify high H2O2 levels [52], although both of them decompose H2O2. Several genes encoding thioredoxins (Trx) and Trx-like proteins are also found in the genome, including TrxA and TrxB. In addition, A. longa SW024T possesses gene encoding for MutY which may prevent mutations arising from oxidatively damaged guanine residues [53].
Inhabiting in such clear surface seawater, A. longa SW024T has evolved several genetic potentials for UV radiation defense. Corresponding genes involved in the biosynthetic pathways of C40 carotenoids (i.e., crtE, crtB, crtI, crtY, crtZ) could be identified in the genome of A. longa SW024T. In addition to protecting cells against damaging radicals resulting from the degradation of heterocycles [54], carotenoids can also function in resisting to photodestruction [55]. Hence, the synthesis of carotenoids may protect A. longa SW024T from UV damage in the clear seawater. Moreover, A. longa SW024T possesses four putative photolyases PhrB, which are DNA repairing flavoproteins that response to blue light and repair cyclobutane pyrimidine (mainly thymine) dimers created by UV light. One BLUF domain protein which has been shown to sense blue light is found in the genome. Similar domain was also found in other marine Flavobacteriaceae bacteira [2]. In addition, genes involved in nucleotide excision repair (NER) are also found in A. longa SW024T, including uvrA, uvrB, uvrC and uvrD [56]. The NER system has an advantage over photolyase in that it can repair UV lesions in the dark. Moreover, the key protein in the transcription coupled repair process is a transcription repair coupling factor (TRCF) encoded by the mfd gene [57]. Mfd recognizes RNA polymerase stalled at a non-coding template site of DNA damage, disrupts the transcription complex to release the transcript and enzyme, and binds UvrA via the UvrA-binding domain 2 that is very similar to domain 2 of UvrB thereby recruiting the NER machinery to the DNA lesion [56]. Further, three 6-O-methylguanine-DNA methyltransferases can also help repair alkylated forms of guanine and thymine that can lead to G:C to A:T transversions in DNA [58].
The in vitro antibiotic sensitivity test demonstrated multidrug resistance pattern of A. longa SW024T, with resistance to 12 antibiotics [10]. A variety of known antibiotic-resistance proteins, such as β-lactamase (AmpC), outer membrane proteins (OmpA, OmpW), and potential drug transporters were found in the genome of A. longa SW024T. The bacterium is resistant to β-lactam antibiotics, i.e., benzylpenicillin, carbenicillin, cefuroxime and cephalosporin. Several β-lactamases encoding genes were identified in the genome. Thus, the inactivation of the antibiotics via degradation by β-lactamases seems to be an intrinsic resistance mechanism. It is also resistant to aminoglycosides, i.e., gentamicin, kanamycin, neomycin and streptomycin. Multidrug efflux pumps also play important roles in A. longa SW024T antimicrobial resistance. A large number of drug transporters and efflux pumps were identified in the genome, including multidrug ABC transporter, SMR family multidrug resistance protein, cation/multidrug efflux pump, ABC efflux pump, Na+ driven efflux pump and MATE efflux pump. These multidrug transporters recognize lipophilic drugs by their physic-chemical properties that allow them to intercalate into the lipid bilayer, and transport these agents from the lipid bilayer to the exterior. Antimicrobial activity is of help for A. longa SW024T to compete with opponents in the same environment for survival, and might also help the bacterium use antibiotics-like substances as energy source and adapt to the ultra-oligotrophic marine environment.
Potential role in biogeochemical cycles
Chitin is the most abundant renewable biopolymers in the marine environment. It has been estimated that 1011 tons of chitin are produced annually in marine systems, primarily in the form of zooplankton exoskeletons, and this polymer must be continually remineralized to support sustained primary production in the oceans [59]. Thus, degradation of chitin may reflect one of the most important extracellular enzymatic processes in the marine environment and create important trophic links within bacterioplankton communities. From the chitinase activity assay, chitin can be hydrolyzed by A. longa SW024T [Additional file 3]. Chitinase is a glycosyl hydrolase which catalyzes the degradation of chitin. Based on amino acid sequence similarity, chitinases are classified into families 18 and 19 of glycosyl hydrolases [60, 61]. A. longa SW024T harbored seven genes encoding chitinase, four of which belong to family 19 (blast using UNIPROTKB database), which is an interesting result because most of the family 19 chitinases were found in higher plants. In recent years, genus Aquimarina was isolated in many oceanic areas, and it may play a role in the cycling of nutrients especially for carbon in the oceans.
Denitrification constitutes one of the main branches of the global nitrogen cycle sustained by bacteria. For nitrogen metabolism, a complete pathway of denitrification was found in the genome of A. longa SW024T, which is the process of converting nitrate (NO3−) to nitrite (NO2−), nitric oxide (NO), nitrous oxide (N2O) and dinitrogen gas (N2), making use of N oxides as terminal electron acceptors for cellular bioenergetics. Moreover, genes involved in the pathway of assimilatory sulfate reduction were also found in the genome, which converts sulfate to sulfide. The ability to consume a wide array of carbon, nitrogen and sulfur substrates indicates that the bacterium might play an important role in biogeochemical cycles.
Conclusions
Genome comparison of Aquimarina strains suggest that the genome contents of these bacteria are in line with their living environments. The general features of A. longa SW024T genome are consistent with its life style in the surface ocean. With large genome size, a large number of ORFs and COG categories comparable to other copiotrophs, A. longa SW024T is assumed to be a copiotroph. Living in the ultra-oligotrophic marine environment, A. longa SW024T is more likely abundant on particles than free-living in the water column, and search for polymers by its gliding ability. A series of adaptation strategies to oligotrophic marine environment including uptake of the primary elemental ingredients such as N, P, S and Fe were identified in the genome. Antioxidative enzymes and compounds as well as other antibiotic activity proteins here might help the bacterium resistant to adverse effects such as DNA damage. Carbon, nitrogen and sulfate metabolism indicate that the bacterium may play a role in biogeochemistry cycle. The analysis of the genome of A. longa SW024T presented here provides a better understanding of its survival mechanisms and ecophysiological functions in the ultra-oligotrophic marine environment.
Availability of supporting data
The data sets supporting the results of this article are included within the article.
Acknowledgements
This work was supported by projects from the National Natural Science Foundation of China (No. 41276141 and No. 41476112) and the National High Technology Research and Development Program of China (863 Program, No. 2012AA091605).
Abbreviations
- BD
Becton dickinson
- BLAST
Basic local alignment search tool
- Chl a
Chlorophyll a
- COG
Clusters of orthologous groups of proteins
- DDBJ
DNA data bank of japan
- EMBL
European molecular biology laboratory
- EPS
Exopolysaccharides
- GO
Gene ontology
- GOLD
Genomes online database
- KEGG
Kyoto encyclopedia of genes and genomes
- MA
Marine Agar 2216
- MB
Marine Broth 2216
- NCBI
National center for biotechnology information
- NER
Nucleotide excision repair
- NR
Non-Redundant
- ORF
Open reading frame
- PBP 2
Penicillin binding protein 2
- Prx
Peroxiredoxin
- SOD
Superoxide dismutase
- SPG
South Pacific Gyre
- TRCF
Transcription repair coupling factor
- Trx
Thioredoxin
Additional files
Summary of genomic information of 30 Flavobacteriaceae genomes. (DOCX 21 kb)
Pan-genome and Core-genome of the genus Aquimarina . Squares are the values obtained for the different strain combinations of Pan-genome. Circles are the values obtained for the different strain combinations of Core-genome. Triangles are the average of such values. The curves are the least squares fit of the power law to the average values. (TIFF 410 kb)
Chitinase activity of A.longa SW024 T . (TIFF 1703 kb)
Footnotes
Tingting Xu and Min Yu contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
TX carried out the laboratory work, participated in the data analyzation and drafted the manuscript. MY participated in the design of the study and helped to draft the manuscript. HL, ZZ and JL participated in the data analyzation. X-HZ conceived the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
Tingting Xu, Email: xutingtingno1@163.com.
Min Yu, Email: yumin@ouc.edu.cn.
Heyu Lin, Email: linheyu1991@qq.com.
Zenghu Zhang, Email: zhangzenghu632@163.com.
Jiwen Liu, Email: liujiwen333@163.com.
Xiao-Hua Zhang, Email: xhzhang@ouc.edu.cn.
References
- 1.Bauer M, Kube M, Teeling H, Richter M, Lombardot T, Allers E, et al. Whole genome analysis of the marine Bacteroidetes ‘Gramella forsetii’ reveals adaptations to degradation of polymeric organic matter. Environ Microbiol. 2006;8:2201–13. doi: 10.1111/j.1462-2920.2006.01152.x. [DOI] [PubMed] [Google Scholar]
- 2.Gonzalez JM, Fernandez-Gomez B, Fernandez-Guerra A, Gomez-Consarnau L, Sanchez O, Coll-Llado M, et al. Genome analysis of the proteorhodopsin-containing marine bacterium Polaribacter sp. MED152 (Flavobacteria) Proc Natl Acad Sci USA. 2008;105:8724–9. doi: 10.1073/pnas.0712027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cottrell M, Kirchman D. Natural assemblages of marine proteobacteria and members of the Cytophaga-Flavobacter cluster consuming low- and high-molecular-weight dissolved organic matter. Appl Environ Microbiol. 2000;66:1692–7. doi: 10.1128/AEM.66.4.1692-1697.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luo H. Predicted protein subcellular localization in dominant surface ocean bacterioplankton. Appl Environ Microbiol. 2012;78:6550–7. doi: 10.1128/AEM.01406-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pinhassi J, Sala MM, Havskum H, Peters F, Guadayol O, Malits A, et al. Changes in bacterioplankton composition under different phytoplankton regimens. Appl Environ Microbiol. 2004;70:6753–66. doi: 10.1128/AEM.70.11.6753-6766.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernardet J-F, Nakagawa Y, Holmes B. Proposed minimal standards for describing new taxa of the family Flavobacteriaceae and emended description of the family. Int J Syst Evol Microbiol. 2002;52:1049–70. doi: 10.1099/00207713-52-3-1049. [DOI] [PubMed] [Google Scholar]
- 7.Nedashkovskaya OI, Kim SB, Lysenko AM, Frolova GM, Mikhailov VV, Lee KH, et al. Description of Aquimarina muelleri gen. nov., sp. nov., and proposal of the reclassification of [Cytophaga] latercula Lewin 1969 as Stanierella latercula gen. nov., comb. nov. Int J Syst Evol Microbiol. 2005;55:225–9. doi: 10.1099/ijs.0.63349-0. [DOI] [PubMed] [Google Scholar]
- 8.Ras J, Claustre H, Uitz J. Spatial variability of phytoplankton pigment distributions in the Subtropical South Pacific Ocean: comparison between in situ and predicted data. Biogeosciences. 2008;5:353–69. doi: 10.5194/bg-5-353-2008. [DOI] [Google Scholar]
- 9.Morel A, Gentili B, Claustre H, Babin M, Bricaud A, Ras J, et al. Optical properties of the “clearest” natural waters. Limnol Oceanogr. 2007;52:217–29. doi: 10.4319/lo.2007.52.1.0217. [DOI] [Google Scholar]
- 10.Yu T, Yin Q, Song X, Zhao R, Shi X, Zhang X-H. Aquimarina longa sp. nov., isolated from seawater, and emended description of Aquimarina muelleri. Int J Syst Evol Microbiol. 2013;63:1235–40. doi: 10.1099/ijs.0.041509-0. [DOI] [PubMed] [Google Scholar]
- 11.Yin Q, Fu B, Li B, Shi X, Inagaki F, Zhang X-H. Spatial variations in microbial community composition in surface seawater from the ultra-oligotrophic center to rim of the South Pacific Gyre. PLoS ONE. 2013;8:e55148. doi: 10.1371/journal.pone.0055148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin B, Lu G, Li S, Hu Z, Chen H. Draft genome sequence of the novel agarolytic bacterium Aquimarina agarilytica ZC1. J Bacteriol. 2012;194:2769. doi: 10.1128/JB.00311-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marmur J. A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J Mol Biol. 1961;3:208–18. doi: 10.1016/S0022-2836(61)80047-8. [DOI] [Google Scholar]
- 14.Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23:673–9. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–5. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–6. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–D80. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu T, Zhang Z, Fan X, Shi X, Zhang X-H. Aquimarina megaterium sp. nov., isolated from seawater. Int J Syst Evol Microbiol. 2014;64:122–7. doi: 10.1099/ijs.0.055517-0. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Z, Yu T, Xu T, Zhang X-H. Aquimarina pacifica sp. nov., isolated from seawater. Int J Syst Evol Microbiol. 2014;64:1991–7. doi: 10.1099/ijs.0.062695-0. [DOI] [PubMed] [Google Scholar]
- 22.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. [DOI] [PMC free article] [PubMed]
- 23.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647-9. [DOI] [PMC free article] [PubMed]
- 24.Qin QL, Li Y, Zhang YJ, Zhou ZM, Zhang WX, Chen XL, et al. Comparative genomics reveals a deep-sea sediment-adapted life style of Pseudoalteromonas sp. SM9913. ISME J. 2011;5:274-84. [DOI] [PMC free article] [PubMed]
- 25.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673-80. [DOI] [PMC free article] [PubMed]
- 26.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731-9. [DOI] [PMC free article] [PubMed]
- 27.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987; 4:406-25. [DOI] [PubMed]
- 28.Zhao Y, Wu J, Yang J, Sun S, Xiao J, Yu J. PGAP: pan-genomes analysis pipeline. Bioinformatics. 2012;28:416-8. [DOI] [PMC free article] [PubMed]
- 29.Hsu S, Lockwood J. Powdered chitin agar as a selective medium for enumeration of actinomycetes in water and soil. Appl Environ Microbiol. 1975;29:422-6. [DOI] [PMC free article] [PubMed]
- 30.Balsanelli E, de Baura VA, de Oliveira Pedrosa F, de Souza EM, Monteiro RA. Exopolysaccharide biosynthesis enables mature biofilm formation on abiotic surfaces by Herbaspirillum seropedicae. PLoS ONE. 2014;9:e110392. [DOI] [PMC free article] [PubMed]
- 31.Dubois M, Gilles KA, Hamilton JK, Rebers PA, Smith F. Colorimetric method for determination of sugars and related substances. Anal Chem (Wash). 1956;28:350-6.
- 32.Qin QL, Xie BB, Yu Y, Shu YL, Rong JC, Zhang YJ, et al. Comparative genomics of the marine bacterial genus Glaciecola reveals the high degree of genomic diversity and genomic characteristic for cold adaptation. Environ Microbiol. 2014;16:1642-53. [DOI] [PubMed]
- 33.Grzymski JJ, Dussaq AM. The significance of nitrogen cost minimization in proteomes of marine microorganisms. ISME J. 2012;6:71-80. [DOI] [PMC free article] [PubMed]
- 34.Konstantinidis KT, Serres MH, Romine MF, Rodrigues JL, Auchtung J, McCue LA, et al. Comparative systems biology across an evolutionary gradient within the Shewanella genus. Proc Natl Acad Sci USA. 2009;106:15909-14. [DOI] [PMC free article] [PubMed]
- 35.Hiller NL, Janto B, Hogg JS, Boissy R, Yu S, Powell E, et al. Comparative genomic analyses of seventeen Streptococcus pneumoniae strains: insights into the pneumococcal supragenome. J Bacteriol. 2007;189:8186-95. [DOI] [PMC free article] [PubMed]
- 36.Tettelin H, Riley D, Cattuto C, Medini D. Comparative genomics: the bacterial pan-genome. Curr Opin Microbiol. 2008;11:472-7. [DOI] [PubMed]
- 37.Konstantinidis KT, Tiedje JM. Trends between gene content and genome size in prokaryotic species with larger genomes. Proc Natl Acad Sci USA. 2004;101:3160-5. [DOI] [PMC free article] [PubMed]
- 38.Newton RJ, Griffin LE, Bowles KM, Meile C, Gifford S, Givens CE, et al. Genome characteristics of a generalist marine bacterial lineage. ISME J. 2010;4:784-98. [DOI] [PubMed]
- 39.Connon SA, Giovannoni SJ. High-throughput methods for culturing microorganisms in very-low-nutrient media yield diverse new marine isolates. Appl Environ Microbiol. 2002;68:3878-85. [DOI] [PMC free article] [PubMed]
- 40.Young KD. The selective value of bacterial shape. Microbiol Mol Biol Rev. 2006;70:660-703. [DOI] [PMC free article] [PubMed]
- 41.Decho AW. Exopolymer microenvironments of microbial flora: multiple and interactive effects on trophic relationships. Limnol Oceanogr. 1993;38:1633-45.
- 42.Logan BE, Hunt JR. Advantages to microbes of growth in permeable aggregates in marine systems. Limnol Oceanogr. 1987;32:1034-48.
- 43.Swan BK, Tupper B, Sczyrba A, Lauro FM, Martinez-Garcia M, Gonzalez JM, et al. Prevalent genome streamlining and latitudinal divergence of planktonic bacteria in the surface ocean. Proc Natl Acad Sci U S A. 2013;110:11463-8. [DOI] [PMC free article] [PubMed]
- 44.Lauro FM, McDougald D, Thomas T, Williams TJ, Egan S, Rice S, et al. The genomic basis of trophic strategy in marine bacteria. Proc Natl Acad Sci U S A. 2009;106:15527-33. [DOI] [PMC free article] [PubMed]
- 45.Braun TF, Khubbar MK, Saffarini DA, McBride MJ. Flavobacterium johnsoniae gliding motility genes identified by mariner mutagenesis. J Bacteriol. 2005;187:6943-52. [DOI] [PMC free article] [PubMed]
- 46.Prescott L, Harley J, Klein D. Microbiology, 5th edition. Ohio:McGraw-Hill Companies, Inc. 2002.
- 47.Forchhammer K. PII signal transducers: novel functional and structural insights. Trends Microbiol. 2008;16:65-72. [DOI] [PubMed]
- 48.Scanlan DJ, Ostrowski M, Mazard S, Dufresne A, Garczarek L, Hess WR, et al. Ecological genomics of marine picocyanobacteria. Microbiol Mol Biol Rev. 2009;73:249-99. [DOI] [PMC free article] [PubMed]
- 49.Delamarche C, Thomas D, Rolland J-P, Froger A, Gouranton J, Svelto M, et al. Visualization of AqpZ-mediated water permeability in Escherichia coli by cryoelectron microscopy. J Bacteriol. 1999;181:4193-7. [DOI] [PMC free article] [PubMed]
- 50.Imlay JA. Pathways of oxidative damage. Annu Rev Microbiol. 2003;57:395-418. [DOI] [PubMed]
- 51.Al-Taweel K, Iwaki T, Yabuta Y, Shigeoka S, Murata N, Wadano A. A bacterial transgene for catalase protects translation of D1 protein during exposure of salt-stressed tobacco leaves to strong light. Plant Physiol. 2007;145:258-65. [DOI] [PMC free article] [PubMed]
- 52.Stork T, Michel KP, Pistorius EK, Dietz KJ. Bioinformatic analysis of the genomes of the cyanobacteria Synechocystis sp. PCC 6803 and Synechococcus elongatus PCC 7942 for the presence of peroxiredoxins and their transcript regulation under stress. J Exp Bot. 2005;56:3193-206. [DOI] [PubMed]
- 53.Lu AL, Li X, Gu Y, Wright PM, Chang DY. Repair of oxidative DNA damage: mechanisms and functions. Cell Biochem Biophys. 2001;35:141-70. [DOI] [PubMed]
- 54.Krinsky NI. Antioxidant functions of carotenoids. Free Radic Biol Med. 1989;7:617-35. [DOI] [PubMed]
- 55.Will III OH, Newland NA, Reppe CR. Photosensitivity of pigmented and nonpigmented strains of Ustilago violacea. Curr Microbiol. 1984;10:295-301.
- 56.Goosen N, Moolenaar GF. Repair of UV damage in bacteria. DNA Repair. 2008;7:353-79. [DOI] [PubMed]
- 57.Selby CP, Sancar A. Molecular mechanism of transcription-repair coupling. Science. 1993;260:53-8. [DOI] [PubMed]
- 58.Mackay WJ, Han S, Samson LD. DNA alkylation repair limits spontaneous base substitution mutations in Escherichia coli. J Bacteriol. 1994;176:3224-30. [DOI] [PMC free article] [PubMed]
- 59.Li X, Roseman S. The chitinolytic cascade in Vibrios is regulated by chitin oligosaccharides and a two-component chitin catabolic sensor/kinase. Proc Natl Acad Sci USA. 2004;101:627–31. doi: 10.1073/pnas.0307645100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Henrissat B. A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J. 1991;280:309–16. doi: 10.1042/bj2800309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Henrissat B, Bairoch A. New families in the classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J. 1993;293:781–8. doi: 10.1042/bj2930781. [DOI] [PMC free article] [PubMed] [Google Scholar]