

Cavin1 degradation is primarily mediated by the ubiquitin proteasome system. The phosphoinositide-binding region in cavin1 acts as a molecular switch for cavin1 degradation upon release of cavins in cytosol. This mechanism may help to maintain low levels of free cytosolic cavins at steady state.
Abstract
Caveolae are abundant surface organelles implicated in a range of cellular processes. Two classes of proteins work together to generate caveolae: integral membrane proteins termed caveolins and cytoplasmic coat proteins called cavins. Caveolae respond to membrane stress by releasing cavins into the cytosol. A crucial aspect of this model is tight regulation of cytosolic pools of cavin under resting conditions. We now show that a recently identified region of cavin1 that can bind phosphoinositide (PI) lipids is also a major site of ubiquitylation. Ubiquitylation of lysines within this site leads to rapid proteasomal degradation. In cells that lack caveolins and caveolae, cavin1 is cytosolic and rapidly degraded as compared with cells in which cavin1 is associated with caveolae. Membrane stretching causes caveolar disassembly, release of cavin complexes into the cytosol, and increased proteasomal degradation of wild-type cavin1 but not mutant cavin1 lacking the major ubiquitylation site. Release of cavin1 from caveolae thus leads to exposure of key lysine residues in the PI-binding region, acting as a trigger for cavin1 ubiquitylation and down-regulation. This mutually exclusive PI-binding/ubiquitylation mechanism may help maintain low levels of cytosolic cavin1 in resting cells, a prerequisite for cavins acting as signaling modules following release from caveolae.
INTRODUCTION
Caveolae are specialized bulb-shaped domains at the plasma membrane of many cell types with distinct functions in cell signaling, mechanosensation, extracellular matrix remodeling, and lipid regulation (Parton and del Pozo, 2013). Caveolin1 (CAV1) was the first core protein component of caveolae discovered and is essential for caveolar formation (Rothberg et al., 1992). Subsequently caveolin2 (CAV2) (Scherer et al., 1996) and muscle-specific caveolin3 (CAV3) (Way and Parton, 1995), two homologues of CAV1, were discovered that hetero-oligomerize with CAV1. Caveolin proteins are essential for caveolar formation but are not sufficient to induce caveolar biogenesis in mammalian cells. Research from a number of groups over the past decade identified a new family of peripheral membrane proteins (cavins) that work together with caveolins to generate caveolae (Hill et al., 2008; Bastiani et al., 2009; Hansen et al., 2009; McMahon et al., 2009). Cavin1/PTRF, the first member of this family, is ubiquitously expressed in all tissues correlating with CAV1 expression and is essential for caveolar formation (Hill et al., 2008). Cavin2/SDPR has been shown to play a role in generating membrane curvature (Hansen et al., 2009). Also, cavin2 influences caveolar formation in lung endothelia and affects the morphology of caveolae in cells lacking cavin2 (Hansen et al., 2013). Cavin3/PRKCDBP affects caveolar endocytosis (McMahon et al., 2009) and is proposed to be involved in signaling through caveolae with recent studies indicating functional links between cavin3 and circadian rhythm and ERK/AKT signaling (Schneider et al., 2012; Hernandez et al., 2013). Cavin4 and CAV3 are muscle-specific isoforms, and cavin4 has been shown to play a role in cardiac hypertrophy (Way and Parton, 1995; Tagawa et al., 2008; Rodriguez et al., 2011).
While caveolins are the major integral membrane proteins of caveolae, cavins have been proposed to form the cytoplasmic coat on the surface of caveolae (Ludwig et al., 2013; Kovtun et al., 2014). Caveolae respond to mechanical stress such as membrane stretch by flattening of caveolae and release of cavin coat proteins (Sinha et al., 2011; Gambin et al., 2014). The precise mechanism of caveolar disassembly is not yet clear, but a recent study (Gambin et al., 2014) suggests that caveolar disassembly causes release of distinct cavin1–cavin2 and cavin1–cavin3 subcomplexes. The fate of the released cavin subcomplexes in the cytoplasm and their possible role in signal transduction has yet to be elucidated, although a number of studies have suggested nuclear and cytosolic roles for the cavin proteins (Jansa et al., 1998; Kovtun et al., 2015; Wei et al., 2015).
Recent molecular studies of the cavin proteins have yielded additional information on the membrane interactions involved in caveolar formation. The crystal structures of N-terminal domains of cavin1 and cavin4 revealed a trimeric helical coiled-coil domain. This region, termed helical region 1 (HR1), is highly conserved and required for cavin oligomerization (Kovtun et al., 2014). HR1 also contains a region of surface-exposed basic residues that are important for binding phosphoinositide (PI) lipids, including phosphatidylinositol(4,5)bisphosphate (PI(4,5)P2). This region is crucial for PI(4,5)P2 binding in vitro, but mutation has only a relatively small effect on caveolar formation in cells. The role of this region in vivo is therefore unclear.
In this study, we show that the putative PI-binding site in cavin1 is a major ubiquitylation site. Ubiquitylation on this site triggers proteasomal degradation of cavin1. We demonstrate that the turnover rate of cavin1 is higher in cells that lack CAV1 compared with cells that express CAV1 and that release of cavin1 from caveolae by mechanical stretch stimulates ubiquitylation and cavin1 turnover through proteasomal degradation. This suggests that the PI-binding region acts as a molecular switch to maintain low levels of cytosolic cavin1, wherein cavin1 release from surface caveolae promotes accessibility of exposed lysines to the ubiquitylation machinery.
RESULTS AND DISCUSSION
Cavin1 turnover is mediated by the proteasome
Previous studies have shown that caveolar disassembly and cavin1 loss upon lipid mobilization in adipocytes is linked to cavin1 degradation, possibly through the proteasome (Briand et al., 2014). We used the PC3 cell line, which expresses high levels of CAV1 but lacks morphological caveolae and does not express any cavin family proteins (Hill et al., 2008), to analyze cavin1 turnover. We analyzed the effect of various proteasomal and lysosomal inhibitors on the turnover of exogenously expressed cavin1–green fluorescent protein (GFP) following the inhibition of protein synthesis by cycloheximide (CHX) treatment (Figure 1). After 6 h of CHX treatment, cavin1-GFP levels are reduced to < 50%. Inhibition of proteasomal degradation by MG132 or clasto-lactacystin β-lactone (Lact) significantly reduced the observed cavin1-GFP turnover. Inhibition of lysosomal degradation by ammonium chloride (NH4Cl) and chloroquine (CQ) had little effect on the decrease in cavin1-GFP protein levels observed after CHX treatment. Similar effects on endogenous cavin1 protein levels were also observed upon inhibitor treatment in the A431 cell line (Supplemental Figure S1A). This suggests that turnover of cavin1 is primarily mediated by the proteasome.
FIGURE 1:
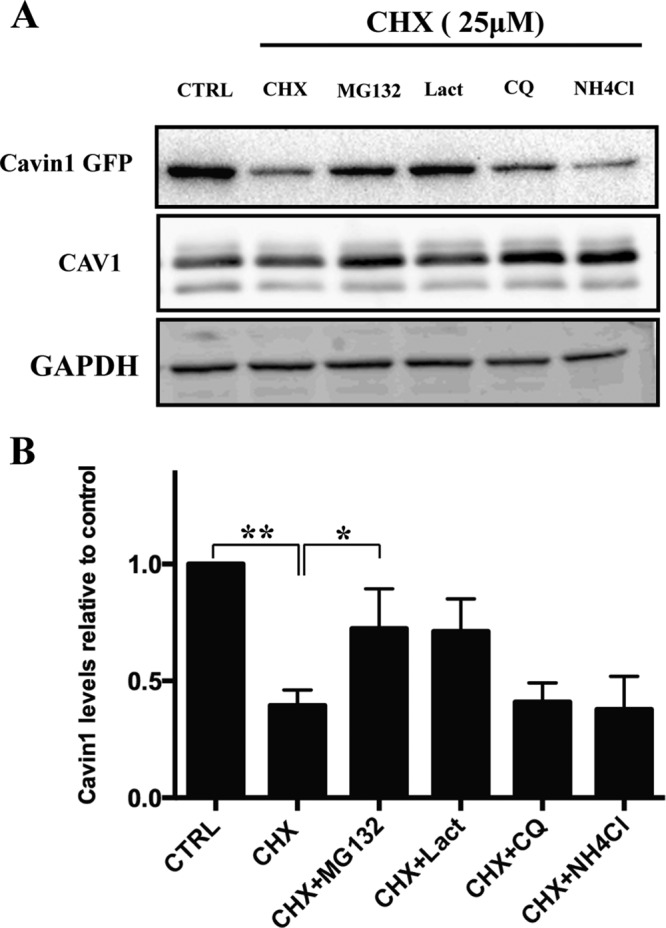
Cavin1 turnover is mediated by the proteasome. (A) PC3 cells overexpressing WT cavin1-GFP were treated with 25 μM CHX in the presence or absence of MG132 (10 μM), Lact (10 μM), CQ (10 μM), or NH4Cl (10 mM) for 6 h and were subsequently immunoblotted for GFP, CAV1, and GAPDH as loading control. (B) Quantification of WT cavin1-GFP levels after inhibitor treatment normalized to untreated samples from three to four independent experiments. Error bars represent SD. *, p < 0.05; **, p < 0.01. Representative uncropped immunoblots are shown in Supplemental Figure S2.
The PI-binding site of cavin1 acts as the major ubiquitylation site responsible for cavin1 turnover
To map the major ubiquitylation sites in cavin1 responsible for its proteasomal degradation, we mutated candidate ubiquitylated lysine residues identified in large-scale mass spectroscopy data sets (Wagner et al., 2011; Figure 2). Six lysine to glutamine point mutants (K47Q, K115Q, K124Q, K163Q, K172Q, K301Q) spanning the full-length cavin1 were generated and analyzed by confocal microscopy. All mutants colocalized with CAV1 in PC3 cells and induced CAV1-positive puncta formation similar to wild-type (WT) cavin1 (Supplemental Figure S1B; Hill et al., 2008; Bastiani et al., 2009; Kovtun et al., 2014). We then analyzed the turnover of these point mutants using the CHX pulse–chase protocol in PC3 cells. No significant difference in protein turnover was observed for any single point mutants as compared with WT cavin1 (Figure 2, B, C, and E). We next mutated the five basic residues of the HR1 domain identified as forming the PI-binding region of cavin1 (K115Q, R117Q, K118Q, K124Q, R127Q; here termed 5Q cavin1) (Kovtun et al., 2014) or two lysine residues in the HR2 domain (K163Q, K172Q; 2Q cavin1) to analyze protein turnover (Figure 2, D and E). Recent studies from our lab showed that although cavin1 5Q mutation partially reduces CAV1 puncta formation it does not abolish colocalization with CAV1 and caveolar formation when expressed in PC3 cells (Kovtun et al., 2014). The 5Q cavin1 mutant showed a dramatically reduced turnover (WT cavin1 t1/2 ∼ 5 h; 5Q cavin1 t1/2 ∼ 14 h) in the presence of CHX, whereas the 2Q cavin1 mutant showed turnover rates similar to WT cavin1. Consistent with this region being the major site involved in triggering proteasomal degradation, no significant difference in 5Q cavin1 levels was observed upon CHX and/or MG132/Lact treatment (Figure 3A). This suggests that lysine residues in the PI-binding region are the major site for cavin1 ubiquitylation and subsequent proteosomal degradation.
FIGURE 2:

Mapping the major ubiquitylation site in cavin1. (A) Schematic representation of cavin1 structural domains (Kovtun et al., 2014), PI(4,5)P2-binding site shown as 5Q, and the position of putative lysine residues chosen for protein turnover study encompassing the 5Q site. (B–D) CHX chase assay for cavin1 point mutants shown in A and respective immunoblot analysis for GFP, CAV1, and GAPDH. (E) Quantification of Western blots for cavin1-GFP levels from two (point mutants) or three to four (WT and 5Q cavin1-GFP) independent experiments normalized to control (untreated samples) for each mutant. Data are presented as mean ± SD. **, p < 0.01. Representative uncropped immunoblots are shown in Supplemental Figure S2. Western blot images shown in Figures 2B and 4C (WT cavin1-GFP turnover upon overexpression in PC3 line) originate from different replicates, but quantifications shown in Figures 2E and 4D are the same.
FIGURE 3:

5Q mutation significantly reduces cavin1 ubiquitylation and is less sensitive to proteasomal inhibitors. (A) PC3 cells were transiently transfected with 5Q cavin1 mutant and were treated with 25 μM CHX in the presence or absence of MG132 (10 μM), Lact (10 μM), CQ (10 μM), or NH4Cl (10 mM) for 6 h and were subsequently immunoblotted for GFP, CAV1, and GAPDH. (B) Quantification of cavin1-GFP levels from immunoblot analysis of three independent experiments. Each bar represents mean and error bars represent SD. ns, no significant difference. (C) A431 cells were transiently transfected with WT cavin1-GFP or 5Q cavin1-GFP and treated with either MG132 or dimethyl sulfoxide. Cell lysates were immunoprecipitated for GFP with GFP nanobeads. Then lysates and immunoprecipitated samples were immunoblotted for GFP, GAPDH, and antiubiquitin antibody to detect total ubiquitylated proteins and Lys-48 linkage–specific antiubiquitin antibody to detect ubiquitylated species of cavin1 specifically attached by the Lys-48 residue of ubiquitin that marks target protein for proteasome degradation. (D) A431 cells were transiently transfected with WT cavin1-GFP or 5Q cavin1-GFP and treated with MG132 for 2 h. Subsequently cells were lysed in buffer C and subjected to ultracentrifugation to separate the membrane fraction as a pellet (P100) and supernatant (S100). Further, GFP immunoprecipitation was performed by dissolving the membrane fraction in buffer B and immunoblotting for GFP, GAPDH, CAV1, and with antiubiquitin to detect ubiquitylated species of cavin1. Representative uncropped immunoblots are shown in Supplemental Figure S2.
Next we directly tested whether cavin1 was ubiquitylated and the major site of ubiquitylation within the HR1 PI-binding region. For this, A431 cells were transfected with either WT cavin1-GFP or 5Q cavin1-GFP, and immunoprecipitation was carried out from cell lysates using GFP-nanotrap beads. Immunoblotting with the antiubiquitin antibody and the Lys-48 linkage–specific polyubiquitin antibody shows that the attachment of ubiquitin to 5Q cavin1 was significantly decreased as compared with WT cavin1 (Figure 3C). Ubiquitylated WT cavin1-GFP shows a slow migrating band (∼130 kDa) also observed when immunoblotted for Lys-48 linkage–specific polyubiquitin antibody that is not present in 5Q cavin1, whereas a lower (∼100 kDa) ubiquitylated band is observed for both WT and 5Q cavin1 (Figure 3C). This suggests that either each lysine residue (K115, K118, K124) in the PI(4,5)P2-binding region is monoubiquitylated or a single lysine residue is polyubiquitylated with three to four ubiquitin linkages. These results show that lysine residues within the PI(4,5)P2-binding region are the major sites of ubiquitylation and that this targets cavin1 for proteasomal degradation.
The PI-binding region of cavin1 is not absolutely essential for cavin1 recruitment to caveolae or caveolar formation (Kovtun et al., 2014), but these results suggest that this region contains the major ubiquitylation site in cavin1. We hypothesize that this region interacts with negatively charged PI(4,5)P2 when cavin1 associates with caveolae, rendering the lysine residues (K115, K118, K124) inaccessible to the proteasome machinery. Caveolar disassembly and release of the cavin complexes into the cytosol may then expose the two major ubiquitylation sites (K115, K124), allowing rapid ubiquitylation. This would have the dual effect of preventing further membrane interaction as well as triggering the proteasomal degradation of cavin1. Note that, although other lysine residues may also be ubiquitylated (the 5Q substitution in cavin1 does not completely abolish ubiquitylation; Figure 3C), our results would suggest that the 5Q site acts as the major trigger for cavin1 degradation.
To further test this hypothesis, we used biochemical membrane fractionation and subsequent immunoblotting for ubiquitin upon overexpression of WT and 5Q cavin1-GFP in the A431 cell line with MG132 treatment for 2 h before cell lysis (Figure 3D). Immunoprecipitation of cavin1-GFP from the cytosolic fraction of WT cavin1 shows slow migrating bands upon immunoblotting with the antiubiquitin antibody that are not present in the membrane fraction (Figure 3D). Immunoprecipitation of 5Q cavin1-GFP shows greatly reduced ubiquitylation in this fraction. These results suggest that ubiquitylation on key lysine residues in the PI(4,5)P2-binding region of cavin1 is mutually exclusive of cavin1 membrane association, and marks cytosolic cavin1 for rapid degradation through the proteasome.
Cavin1 turnover is accelerated in cells lacking CAV1 and upon stretch-induced caveolar disassembly
Previous studies of protein turnover related to caveolae have been mainly focused on the stability and kinetics of CAV1. Endogenous CAV1 has been shown to be a long-lived protein with a half-life of around 36 h, whereas heterologous overexpression of the same protein shows a much shorter half-life (t1/2 ∼ 14 h; Hayer et al., 2010), possibly due to an imbalance of CAV1 and cavins (Parton and del Pozo, 2013). Also, it has been shown that cavin1 is down-regulated in cells lacking CAV1, suggesting that CAV1 plays major role in stable association of cavin1 at the plasma membrane and prevent its degradation (Hill et al., 2008). To study the turnover of cavin1 we used a pulse–chase protocol using the protein synthesis inhibitor CHX. We analyzed cavin1 turnover in the A431 epidermoid carcinoma cell line WTiMEF (WT-immortalized mouse embryonic fibroblasts), MDCK (Madin-Darby canine kidney), the PC3 prostate cancer cell line, and the MCF7 breast cancer cell line. These cell lines differ in their complements of caveolar components; A431, MDCK, and WTiMEF cells express CAV1, cavin1, cavin2, and cavin3, and they form morphological caveolae (Hill et al., 2008; Bastiani et al., 2009). PC3 cells express high levels of CAV1, but they lack morphological caveolae and do not express any cavin family proteins (Hill et al., 2008); the MCF7 cell line expresses neither CAV1 nor any cavin family proteins and lacks morphological caveolae (Lavie et al., 1998). Heterologous expression of cavin1-GFP in the MCF7 cell line shows predominantly cytosolic distribution, whereas coexpression of cavin1-GFP with CAV1-mcherry shows partially punctate localization of cavin1 (Figure 4, E and F).
FIGURE 4:

Analysis of cavin1 turnover in model cell lines. (A) A431 cells were treated with 25 μM CHX for the indicated time period following lysis and immunoblotting for cavin1, CAV1, and GAPDH. WT cavin1-GFP was exogenously expressed in PC3 (C), a CHX chase assay was performed as above, and immunoblotted for GFP, CAV1, and GAPDH. Quantification of protein levels by Western blots at various time points is indicated in B and D. Each point represents the mean of three to four independent experiments, and error bars indicate SD. **, p < 0.01. Subcellular distribution of cavin1-GFP (E) upon overexpression in MCF 7 cells and when coexpressed with CAV1-cherry (F). CHX chase assay upon overexpression of WT cavin1-GFP (G) and 5Q cavin1-GFP (J) in MCF7 cells and with coexpression of WT cavin1-GFP (H)/5Q cavin1-GFP (K) and CAV1-cherry. Quantification of total WT cavin1-GFP (I) and 5Q cavin1-GFP (L) levels at each time point normalized to untreated samples from three independent experiments. Data are presented as mean ± SD. *, p < 0.05; ns, no significant difference. Representative uncropped immunoblots are shown in Supplemental Figure S3. Western blot images shown in Figures 2B and 4C (WT cavin1-GFP turnover upon overexpression in PC3 line) originate from different replicates, but quantifications shown in Figures 2E and 4D are the same.
We analyzed the turnover of endogenous cavin1 in A431, WTiMEF, and MDCK cell lines and the turnover of exogenously expressed cavin1-GFP in PC3 and MCF7 cells. On CHX treatment in the A431 cell line, endogenous cavin1 levels reduce at a faster rate (t1/2 ∼ 8 h) compared with those of CAV1 (t1/2 ∼ 33 h; Figure 4A). Similar turnover rates of cavin1 were observed in WTiMEF and MDCK cell lines (Supplemental Figure S1, C–E). We next analyzed the turnover of cavin1-GFP upon overexpression in PC3 cells as compared with endogenous CAV1. The turnover of cavin1-GFP (t1/2 ∼ 5 h) upon expression in PC3 cells was again faster than that of endogenous CAV1 (t1/2 ∼ 21 h; Figure 1B). In contrast to the above cell lines that express CAV1, cavin1-GFP turnover was significantly faster in MCF7 cells that lack CAV1 (t1/2 ∼ 3.5 h; Figure 4G), but cavin1-GFP levels were stabilized by overexpression of CAV1-mcherry consistent with the hypothesis that cytosolic cavin1 is preferentially degraded compared with caveolar-associated protein (t1/2 cavin1-GFP ∼ 6 h, after CAV1-cherry expression; Figure 4, H and I). No significant difference in 5Q cavin1 GFP turnover (Figure 4L) was observed when expressed in MCF7 cells in the absence (Figure 4J) or presence of CAV1 (Figure 4K).
Previously it has been shown that mechanical stretch causes caveolar flattening and release of cavin proteins into the cytosol. We subjected cells to cyclical stretch on a flexible support (Flexcell; cyclic stretch [90 s] and relax [20 s] cycles for 6 h). As reported previously (Joshi et al., 2012), constitutive cyclical stretch induces transcriptional activation of CAV1/cavin1 expression in cells. Therefore we inhibited new protein synthesis using CHX during the cyclical stretch. On cavin1-GFP overexpression in A431 cells, cyclical stretch accelerates the turnover of both endogenous cavin1 and overexpressed cavin1-GFP, and this is prevented by addition of the proteasomal inhibitor MG132 (Figure 5, A–D). However, turnover of 5Q cavin1-GFP is insensitive to cyclical stretch–induced degradation (Figure 5, E and F). This suggests that cyclical membrane stretch, which transiently releases cavin1 from caveolae (Sinha et al., 2011), accelerates cavin1 turnover, presumably by increasing the opportunity for the (currently unidentified) ubiquitin ligases to target the cytosolic cavin1.
FIGURE 5:

Mechanical stretch stimulates cavin1 turnover. (A) A431 cells expressing WT cavin1-GFP or 5Q cavin1-GFP were subjected to cyclical stretch or rest (control) in the presence or absence of specific inhibitors for 6 h (described in Materials and Methods). Subsequently cells were lysed in lysis buffer A and immunoblotted for endogenous cavin1, cavin1-GFP CAV1, and GAPDH. Quantification of total endogenous cavin1 (B), WT cavin1-GFP (D), and 5Q cavin1-GFP (F) levels was done by normalizing inhibitor-treated samples to untreated samples in respective resting or cyclical stretch conditions. Data are presented as mean ± SD from three independent experiments. **, p < 0.01; ns, no significant difference. Immunoblots for WT cavin1-GFP (C) and 5Q cavin1-GFP (E) levels upon cyclical stretch. Representative immunoblots for endogenous cavin1 and quantification of endogenous cavin1 protein levels from three independent experiments (see panel B) are shown from A431 cells transfected with WT cavin1-GFP. Representative uncropped immunoblots are shown in Supplemental Figure S3. (G) Model of caveolae disassembly and fate of cavins after release into the cytosol.
We recently proposed a model for caveolar assembly in which multiple low-affinity interactions involving homo/hetero-oligomers of cavin proteins, caveolins, and membrane lipids, including PI(4,5)P2, PS and cholesterol, induce caveolar formation at the plasma membrane (Kovtun et al., 2015). Membrane stretch (Sinha et al., 2011) or other stimuli such as lipid flux (Briand et al., 2014) causes caveolar disassembly and release of cavins into the cytosol (Figure 5G). Our data indicate that, in the absence of membrane association, the excess of cytosolic cavin1 undergoes rapid ubiquitylation within the PI(4,5)P2-binding site of the HR1 domain. This modification would reciprocally prevent its membrane rebinding and marks cavin1 for degradation via the proteasome. This will have two related effects: first, it will maintain the cellular homeostasis of cavin1 with its levels dependent on the amount of CAV1 and the ability to form caveolae; and second, it will lead to a low level of free cytosolic cavins at steady state. A potential result of this is that, upon release from caveolae in response to membrane stretch or other potential signals leading to caveolar disassembly such as phosphorylation, cavin1 will be able to mediate downstream signal propagation (Aboulaich et al., 2011). In conclusion, we show that mutually exclusive binding of cavins to membrane phospholipids and the ubiquitylation of the HR1 domain control cavin1 cellular levels and potentially could regulate its involvement in noncaveolar processes.
MATERIALS AND METHODS
Chemicals
Inhibitors used in study were as follows: CHX (C7698; Sigma-Aldrich, St Louis, MO), MG132 (C2211; Sigma-Aldrich), Lact (L7035; Sigma-Aldrich), CQ diphosphate salt (C6628; Sigma-Aldrich), and NH4Cl (ThermoFisher Scientific, Waltham, MA).
Cell lines and transient transfection
WTiMEF, A431, MDCK, and MFC7 cell lines were maintained in DMEM (Life Technologies) supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin. PC3 cells were maintained in RPMI medium (Life Technologies) supplemented with 10% FBS and penicillin/streptomycin. Transient transfections were performed using Lipofectamine 2000 as per the manufacturer’s protocol.
Antibodies
Primary antibodies for immunoblotting were as follows: cavin1 (Sigma Aldrich, cat. no. AV36965, dilution 1:1000), CAV1 (BD Transduction Laboratories, cat. no. 610060, dilution 1:2000), GFP (Roche Diagnostics, cat. no. 11814460001, dilution 1:1000), GAPDH (Life Technologies, Ambion, cat. no. AM4300, dilution 1:5000), mono- and polyubiquitinylated conjugates monoclonal antibody (FK2) (Enzo Life Sciences, cat. no. BML-PW8810), and Lys-48 linkage–specific polyubiquitin antibody (Cell Signaling, cat. no. 4289S). Secondary antibodies for immunoblotting were horseradish peroxidase–conjugated goat anti-rabbit immunoglobulin G (IgG; Sigma Aldrich, cat. no. A0545, dilution 1:15,000) and goat anti-mouse IgG (Sigma Aldrich, cat. no. A4416, dilution 1:15,000), all purchased from Sigma-Aldrich. Fluorescent secondary antibodies were IRDye 800CW goat anti-rabbit IgG (H + L) (Li-Cor, cat. no. 926-32211, dilution 1:10,000) and IRDye 680LT goat anti-mouse IgG (H + L) (Li-Cor, cat. no. 926-68020, dilution 1:10,000). The primary CAV1 antibody for immunofluorescence was from BD Transduction Laboratories (cat. no. 610060, dilution 1:1000). The secondary antibodies for immunofluorescence were: donkey anti-rabbit IgG (H+L), and Alexa Fluor 555 conjugate (2 μg/ml).
Plasmids
Mouse cavin1 cDNA cloned in pEGFP-N1 for the expression of C-terminal EGFP-tagged cavin1 was described in Hill et al. (2008). Cloning of 5Q cavin1, 2Q cavin1, and other point mutants for cavin1 was done using a site-directed mutagenesis kit as per the manufacturer’s protocol. Specific primers for point mutants of mouse cavin1 are as follows (site of base pair mismatch is marked by underline in forward primer: 1) K47Q—FW: TCCGACGAGCTGATCCAGTCGGACCAGGTGAAC, RV: GTTCACCTGGTCCGACTGGATCAGCTCGTCGGA, 2) K115Q—FW: AGCAAGTTGCTGGAGCAGGTGCGCAAGGTCAGC, RV: GCTGACCTTGCGCACCTGCTCCAGCAACTTGCT 3) K124Q—FW: GTCAGCGTCAACGTGCAGACCGTGCGCGGCAGC, RV: GCTGCCGCGCACGGTCTGCACGTTGACGCTGAC, 4) K163Q—FW: TACCAGGATGAAGTCCAGCTGCCGGCCAAACTGA, RV: TCAGTTTGGCCGGCAGCTGGACTTCATCCTGGTA, 5) K172Q—FW: AAACTGAGCGTCAGCCAGTCGCTGAAAGAGTCG, RV: CGACTCTTTCAGCGACTGGCTGACGCTCAGTTT, 6) K301Q—FW: CGGGACAAGCTGCGCCAGTCCTTCACGCCCGAC, RV: GTCGGGCGTGAAGGACTGGCGCAGCTTGTCCCG, 7) KK163,172QQ (2Q) was generated by sequential rounds of mutagenesis of K163Q and then K172Q.
CHX chase and inhibitor assay
A stock solution of CHX (50 mM) in phosphate-buffered saline (PBS) was diluted to the mentioned concentration (indicated in the figure legends) in 10% FBS containing media, and added to cells and incubated at 37°C for the respective time points. For inhibitor study experiments, CHX was initially diluted in media, respective inhibitors were added at desired concentrations, and cells were incubated at 37°C. Cell lysis and immunoblotting was done as described below in the Western blot analysis section.
Immunoprecipitation and crude membrane fractionation
For immunoprecipitation of GFP-tagged cavin proteins, GFP nanotrap beads were used as described earlier (Gambin et al., 2014). Briefly, cells were lysed in 25 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100, and 0.1% SDS (buffer A) and were incubated for 2 h at 4°C with GFP-nanobody beads prewashed with lysis buffer. For immunoprecipitation from the membrane fraction, membrane pellets were dissolved in 25 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100, and 1% SDS (buffer B) before being incubated with GFP-nanobody beads. Beads were then washed with lysis buffer three times, mixed with 4× LDS buffer, and immunoblotted for specific proteins as indicated. For crude membrane fractionation, cells were lysed in 25 mM Tris (pH 7.4) and 150 mM NaCl (buffer C), passed through a 27-G needle six times, and then centrifuged at 80,000 × g for 20 min.
Western blot analysis
Cells were lysed in buffer A with protease and phosphatase inhibitors at 4°C to produce whole-cell lysate. For all experiments except immunoprecipitations, total protein estimation in each condition was performed by the bicinchoninic acid assay (BCA) method and an equal amount of protein was loaded in each well of a 12% SDS PAGE gel. Quantification of Western blots was done using ImageJ/Image lab (Bio-Rad).
Immunofluorescence
Cells were plated on sterilized coverslips at 40% confluency and allowed to grow until they reached 60–70% confluency. Cells were transfected with Lipofectamine 2000 as per the manufacturer’s protocols. Media was changed at 4-h posttransfection, and cells were allowed to grow for 18 h posttransfection. The next day, cells were fixed in 4% PFA in PBS at 4°C for 30 min. Cells were then washed with PBS three times and treated with 25 mM NH4Cl for 5 min. Cells were blocked with 2% BSA in PBS with Tween-20 (0.1%) for 60 min and were then incubated with primary antibody for 60 min. Cells were then washed again for three times with PBS with Tween-20 (0.1%) and incubated with secondary antibody for 45 min. Finally, coverslips were mounted in Mowiol and allowed to dry overnight at room temperature before being imaged on a confocal microscope.
Application of cyclical stretch
A431 cells were plated on collagen-1 coated Bioflex plates (cat. no. BF-3001C, Flexcell International) at 20–30% confluence and were allowed to reach 70–80% confluence before the application of cyclical stretch. Cells were then subjected to cyclical stretch with 15% elongation at a frequency of 60 cycles per min (SINE waveform) using a Flexcell Fx-5000 tension unit (Flexcell International) for 6 h after their respective plasmid transfections. Periodic cyclical stretch (90 s) and relaxation (20 s) was applied for the duration of the experiment (6 h). For the control experiment, cells were plated collagen-1 coated Bioflex plates without application of cyclical stretch and were treated with inhibitors. For the inhibitor experiments, respective wells were treated with CHX/CHX+MG132 for 30 min before cyclical stretch, and cells were maintained in the same medium throughout the cyclical stretch procedure.
Protein half-life calculations
Protein half-life estimation was performed using the exponential decay equation: Nt = N0e−λt. Nt = amount of protein left at time t, N0 = initial amount of protein at start of assay, λ = decay constant. Half-life: t1/2 = ln(2)/λ.
Statistical analysis
Mean, SD, t test, and p value calculations were performed in Prism.
Supplementary Material
Acknowledgments
Confocal microscopy was performed at the Australian Cancer Research Foundation (ACRF)/Institute for Molecular Bioscience Dynamic Imaging Facility for Cancer Biology, established with funding from the ACRF. This work was supported by grants from the National Health and Medical Research Council of Australia (NHMRC) to R.G.P. (grant numbers APP569542 and 1045092) and the Australian Research Council (ARC) to B.M.C. (grant number DP120101298). R.G.P. is supported by an NHMRC Senior Principal Research Fellowship from the NHMRC (APP1058565), and B.M.C. is supported by an NHMRC Career Development Fellowship (APP1061574) and previously held an ARC Future Fellowship Grant (FT100100027).
Glossary
Abbreviations used:
- CAV1/2/3
caveolin1/2/3
- CHX
cycloheximide
- CQ
chloroquine
- FBS
fetal bovine serum
- GFP
green fluorescent protein
- HR1
helical region1
- IgG
immunoglobulin G
- Lact
clasto-lactacystin β-lactone
- MDCK
Madin-Darby canine kidney
- PBS
phosphate-buffered saline
- PI
phosphoinositide
- PI(4,5)P2
phosphatidylinositol(4,5)bisphosphate
- WT
wild type
- WTiMEF
WT-immortalized mouse embryonic fibroblasts.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-06-0359) on August 12, 2015.
REFERENCES
- Aboulaich N, Chui PC, Asara JM, Flier JS, Maratos-Flier E. Polymerase I and transcript release factor regulates lipolysis via a phosphorylation-dependent mechanism. Diabetes. 2011;60:757–765. doi: 10.2337/db10-0744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastiani M, Liu L, Hill MM, Jedrychowski MP, Nixon SJ, Lo HP, Abankwa D, Luetterforst R, Fernandez-Rojo M, Breen MR, et al. MURC/cavin-4 and cavin family members form tissue-specific caveolar complexes. J Cell Biol. 2009;185:1259–1273. doi: 10.1083/jcb.200903053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briand N, Prado C, Mabilleau G, Lasnier F, Le Liepvre X, Covington JD, Ravussin E, Le Lay S, Dugail I. Caveolin-1 expression and cavin stability regulate caveolae dynamics in adipocyte lipid store fluctuation. Diabetes. 2014;63:4032–4044. doi: 10.2337/db13-1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambin Y, Ariotti N, McMahon KA, Bastiani M, Sierecki E, Kovtun O, Polinkovsky ME, Magenau A, Jung W, Okano S, et al. Single-molecule analysis reveals self assembly and nanoscale segregation of two distinct cavin subcomplexes on caveolae. eLife. 2014;3:e01434. doi: 10.7554/eLife.01434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen CG, Bright NA, Howard G, Nichols BJ. SDPR induces membrane curvature and functions in the formation of caveolae. Nat Cell Biol. 2009;11:807–814. doi: 10.1038/ncb1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen CG, Shvets E, Howard G, Riento K, Nichols BJ. Deletion of cavin genes reveals tissue-specific mechanisms for morphogenesis of endothelial caveolae. Nat Communications. 2013;4:1831. doi: 10.1038/ncomms2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayer A, Stoeber M, Ritz D, Engel S, Meyer HH, Helenius A. Caveolin-1 is ubiquitinated and targeted to intralumenal vesicles in endolysosomes for degradation. J Cell Biol. 2010;191:615–629. doi: 10.1083/jcb.201003086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez VJ, Weng J, Ly P, Pompey S, Dong H, Mishra L, Schwarz M, Anderson RG, Michaely P. Cavin-3 dictates the balance between ERK and Akt signaling. eLife. 2013;2:e00905. doi: 10.7554/eLife.00905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MM, Bastiani M, Luetterforst R, Kirkham M, Kirkham A, Nixon SJ, Walser P, Abankwa D, Oorschot VM, Martin S, et al. PTRF-cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell. 2008;132:113–124. doi: 10.1016/j.cell.2007.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansa P, Mason SW, Hoffmann-Rohrer U, Grummt I. Cloning and functional characterization of PTRF, a novel protein which induces dissociation of paused ternary transcription complexes. EMBO J. 1998;17:2855–2864. doi: 10.1093/emboj/17.10.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi B, Bastiani M, Strugnell SS, Boscher C, Parton RG, Nabi IR. Phosphocaveolin-1 is a mechanotransducer that induces caveola biogenesis via Egr1 transcriptional regulation. J Cell Biol. 2012;199:425–435. doi: 10.1083/jcb.201207089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtun O, Tillu VA, Ariotti N, Parton RG, Collins BM. Cavin family proteins and the assembly of caveolae. J Cell Sci. 2015;128:1269–1278. doi: 10.1242/jcs.167866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtun O, Tillu VA, Jung W, Leneva N, Ariotti N, Chaudhary N, Mandyam RA, Ferguson C, Morgan GP, Johnston WA, et al. Structural insights into the organization of the cavin membrane coat complex. Dev Cell. 2014;31:405–419. doi: 10.1016/j.devcel.2014.10.002. [DOI] [PubMed] [Google Scholar]
- Lavie Y, Fiucci G, Liscovitch M. Up-regulation of caveolae and caveolar constituents in multidrug-resistant cancer cells. J Biol Chem. 1998;273:32380–32383. doi: 10.1074/jbc.273.49.32380. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Howard G, Mendoza-Topaz C, Deerinck T, Mackey M, Sandin S, Ellisman MH, Nichols BJ. Molecular composition and ultrastructure of the caveolar coat complex. PLoS Biol. 2013;11:e1001640. doi: 10.1371/journal.pbio.1001640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon KA, Zajicek H, Li WP, Peyton MJ, Minna JD, Hernandez VJ, Luby-Phelps K, Anderson RG. SRBC/cavin-3 is a caveolin adapter protein that regulates caveolae function. EMBO J. 2009;28:1001–1015. doi: 10.1038/emboj.2009.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton RG, del Pozo MA. Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol. 2013;14:98–112. doi: 10.1038/nrm3512. [DOI] [PubMed] [Google Scholar]
- Rodriguez G, Ueyama T, Ogata T, Czernuszewicz G, Tan Y, Dorn GW, Bogaev R, Amano K, Oh H, Matsubara H. Molecular genetic and functional characterization implicate muscle-restricted coiled-coil gene (MURC) as a causal gene for familial dilated cardiomyopathy. Circ Cardiovasc Genet. 2011;4:349–358. doi: 10.1161/CIRCGENETICS.111.959866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothberg KG, Heuser JE, Donzell WC, Ying YS, Glenney JR, Anderson RG. Caveolin, a protein component of caveolae membrane coats. Cell. 1992;68:673–682. doi: 10.1016/0092-8674(92)90143-z. [DOI] [PubMed] [Google Scholar]
- Scherer PE, Okamoto T, Chun M, Nishimoto I, Lodish HF, Lisanti MP. Identification, sequence, and expression of caveolin-2 defines a caveolin gene family. Proc Natl Acad Sci USA. 1996;93:131–135. doi: 10.1073/pnas.93.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider K, Köcher T, Andersin T, Kurzchalia T, Schibler U, Gatfield D. CAVIN-3 regulates circadian period length and PER:CRY protein abundance and interactions. EMBO Rep. 2012;13:1138–1144. doi: 10.1038/embor.2012.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha B, Koster D, Ruez R, Gonnord P, Bastiani M, Abankwa D, Stan RV, Butler-Browne G, Vedie B, Johannes L, et al. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell. 2011;144:402–413. doi: 10.1016/j.cell.2010.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagawa M, Ueyama T, Ogata T, Takehara N, Nakajima N, Isodono K, Asada S, Takahashi T, Matsubara H, Oh H. MURC, a muscle-restricted coiled-coil protein, is involved in the regulation of skeletal myogenesis. Am J Physiol Cell Physiol. 2008;295:C490–C498. doi: 10.1152/ajpcell.00188.2008. [DOI] [PubMed] [Google Scholar]
- Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, Choudhary C. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics. 2011;10:M111.013284. doi: 10.1074/mcp.M111.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way M, Parton RG. M-caveolin, a muscle-specific caveolin-related protein. FEBS Lett. 1995;376:108–112. doi: 10.1016/0014-5793(95)01256-7. [DOI] [PubMed] [Google Scholar]
- Wei Z, Zou X, Wang H, Lei J, Wu Y, Liao K. The N-terminal leucine-zipper motif in PTRF/cavin-1 is essential and sufficient for its caveolae-association. Biochem Biophys Res Commun. 2015;456:750–756. doi: 10.1016/j.bbrc.2014.12.035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.