

Abstract
Well-defined molecular catalysts for the reduction of N2 to NH3 with protons and electrons remain very rare despite decades of interest, and are currently limited to systems featuring Mo or Fe. This report details the synthesis of a molecular Co complex that generates superstoichiometric yields of NH3 (>200% NH3 per Co-N2 precursor) via the direct reduction of N2 with protons and electrons. While the NH3 yields reported herein are modest by comparison to previously described Fe and Mo systems, they intimate that other metals are likely to be viable as molecular N2 reduction catalysts. Additionally, comparison of the featured tris(phosphine)borane Co-N2 complex with structurally related Co-N2 and Fe-N2 species shows how remarkably sensitive the N2 reduction performance of potential pre-catalysts are. These studies enable consideration of structural and electronic effects that are likely relevant to N2 conversion activity, including π-basicity, charge state, and geometric flexibility.
Introduction
The conversion of dinitrogen (N2) to ammonia (NH3) is integral for life.1 Despite extensive study, there are many unanswered questions regarding the rational design of molecular N2-to-NH3 conversion catalysts. It may be that the ability of a complex to activate terminally bound N2 (as reported by the N—N stretching frequency) relates to the propensity of that complex to functionalize the N2 moiety. For example, HCo(N2)(PPh3)3 (ν(N-N) = 2088 cm−1) quantitatively releases N2 upon treatment with acid, with no evidence of N2 functionalization;2,3 however, if this cobalt complex is deprotonated to generate the more activated complex [(PPh3)3Co(N2)][Li(Et2O)3] (ν(N-N) = 1900 cm−1), treatment with acid does produce some NH3 and N2H4 (0.21 and 0.22 equivalents respectively).3 Extensive efforts have been made to study the activation and functionalization of N2 bound to metal centers of varying electronic properties.4 In some cases, systems have been shown to activate bound N2 to the extent that the N—N bond is fully cleaved.5 In other cases, it has been shown that treatment of strongly activated N2 complexes with acid or H2 leads to reduced nitrogenous products.2,3,4 However, this guiding principle alone has been insufficient to design many synthetic species capable of the catalytic conversion of N2 to NH3.6–8 In this regard it is prudent to study the few systems known to catalyze this reaction with an emphasis on identifying those properties critical to the observed N2 reduction activity.
We have recently reported that a tris(phosphino)borane-ligated Fe complex is capable of catalyzing the conversion of N2 to NH3 at −78 °C.7 We have postulated that the success of this system in activating N2 stoichiometrically and mediating its catalytic conversion to NH3 may arise from a highly flexible Fe-B interaction.9,10 Such flexibility, trans to the N2 binding site, may allow a single Fe center to access both trigonal bipyramidal and pseudo-tetrahedral coordination geometries, alternately stabilizing π-acidic or π-basic nitrogenous moieties sampled along an N2 fixation pathway.4d,11 Consistent with this hypothesis, we have studied isostructural (P3E)-ligated Fe systems and found a measurable dependence of activity on the identity of the E atom, with the least flexible E = Si system furnishing divergently low NH3 yields and the more flexible E = C or B systems affording moderate yields of NH3.7,8 However, the lower NH3 production by the E = Si precursor may alternatively be attributed to other factors. Potential factors include (i) a lesser degree of N2 activation than that observed in the E = C or B species (vide infra); (ii) faster poisoning of the E = Si system, for example by more rapid formation of an inactive terminal hydride;7,8 (iii) faster degradation of the E = Si system, for example by dechelation of the ligand.
To complement our previous ligand modification studies, we chose to alter the identity of the transition metal. Moving from Fe to Co predictably modulates the π-basicity and electronic configuration of the metal center while maintaining the ligand environment. In principle, this allows the extrication of electronic effects, such as π-backbonding, from structural features, such as geometric flexibility, via comparison of the Fe and Co systems. We therefore sought to explore the N2 reduction activity of Co complexes of TPB (TPB = [o-(iPr2P)C6H4]3B), SiP3 (SiP3 = [o-(iPr2P)C6H4]3Si), and CP3 (CP3 = [o-(iPr2P)C6H4]3C). While correlating NH3 yields with molecular structure is no doubt informative in terms of understanding the behavior of nitrogen fixing systems, correlation does not imply causation and the results described herein should be read with that in mind.
Results and Discussion
The previously reported12 (TPB)Co(N2) complex (Scheme 1, 1) provided a logical entry point to study the N2 chemistry of (TPB)Co complexes. The cyclic voltammogram of 1 in THF displays a quasi-reversible reduction wave at −2.0 V vs. Fc/Fc+ and a feature corresponding to an oxidation process at −0.2 V vs. Fc/Fc+ (Figure 1). These features are reminiscent of the cyclic voltammogram of (TPB)Fe(N2), which shows a reduction event at −2.2 V vs. Fc/Fc+ and an oxidation event at −1.5 V vs. Fc/Fc+.10
Scheme 1.

Chemical oxidation and reduction of (TPB)Co(N2).
Figure 1.
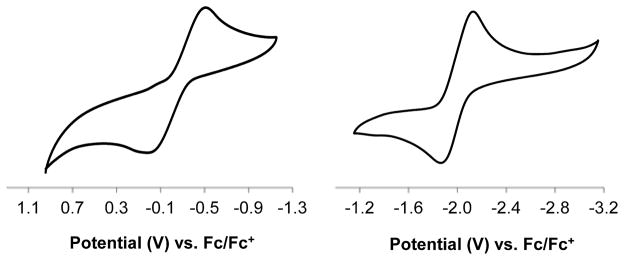
Cyclic voltammagram of (TPB)Co(N2) (1) scanning oxidatively (left) and reductively (right) at 100 mV/sec in THF with 0.1 M TBAPF6 electrolyte.
Treatment of 1 with 1 equivalent of NaC10H8 followed by 2 equivalents of 12-crown-4 (12-c-4) generates diamagnetic [Na(12-c-4)2)][(TPB)Co(N2)] as red crystals (Scheme 1, 2). The ν(N-N) stretch of 2 is lower in energy than that of 1 (Table 1) and the solid-state structure of 2 (Figure 2, left) displays contracted Co-N, Co-B, and Co-P distances compared to 1, consistent with increased backbonding to each of these atoms. The one-electron oxidation of 1 can be achieved by addition of 1 equivalent of [H•(OEt2)2][BArF4] at low temperature followed by warming, which generates red-purple [(TPB)Co][BArF4] (Scheme 1, 3, BArF4 = tetrakis(3,5-bistrifluoromethylphenyl)borate). The structure of 3 (Figure 2, right) confirms that [(TPB)Co][BArF4] does not bind N2 in the solid state. The lack of dinitrogen binding at room temperature for 3 is consistent with the behavior of the isostructural Fe complex, [(TPB)Fe][BArF4].13 SQUID magnetometry measurements indicate that 3 is high spin (S = 1) in the solid state with no evidence for spin crossover (Figure 3).
Table 1.
Select Characterization Data for (P3E)M Complexes (M = Co, Fe; E = B, C, Si)
| Entry | Complex | M—E (Å) | ν(N-N) (cm−1)f |
|---|---|---|---|
| Aa | (TPB)Co(N2) (1) | 2.319(1) | 2089 |
| B | [(TPB)Co(N2)]− (2) | 2.300(3) | 1978 |
| C | [(TPB)Co]+ (3) | 2.256(2) | — |
| Db | (SiP3)Co(N2) (4) | 2.2327(7) | 2063 |
| E | (CP3)Co(N2) (5) | 2.135(4) | 2057 |
| F | [(CP3)Co(N2)]+ (6) | 2.054(2) | 2182 |
| Ga | (TPB)CoBr | 2.4629(8) | — |
| Hc | (TPB)FeBr | 2.458(5) | — |
| Ic | (TPB)Fe(N2) | — | 2011 |
| Jc | [(TPB)Fe(N2)]- | 2.293(3) | 1905 |
| Kd | [(CP3)Fe(N2)]+ | 2.081(3) | 2128 |
| Ld | (CP3)Fe(N2) | 2.152(3) | 1992 |
| Md | [(CP3)Fe(N2)]- | 2.165(2) | 1905 |
| Ne | [(SiP3)Fe(N2)]- | 2.236(1) | 1920 |
Figure 2.
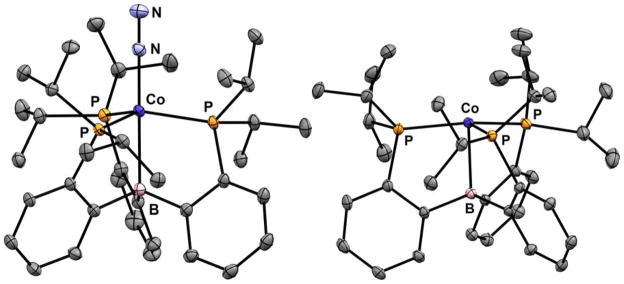
Solid-state crystal structures of 2 (left) and 3 (right; also see SI). Thermal ellipsoids shown at 50% probability. Countertions, solvent molecules, and H atoms omitted for clarity.
Figure 3.
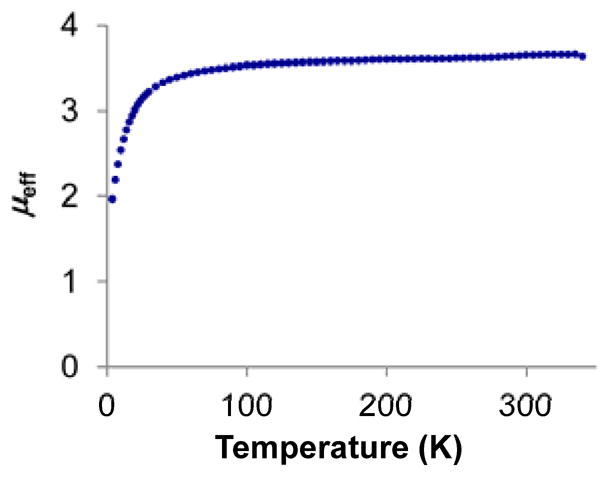
Temperature dependence of the magnetic susceptibility of [(TPB)Co][BArF4] (3) as measured by SQUID magnetometry.
The synthesis of (SiP3)Co(N2) (4) has been reported previously.14 The isoelectronic alkyl species, (CP3)Co(N2) (5), was obtained in 83% yield as a deep red solid from the reaction of CP3H, CoCl2•(THF)1.5, and MeMgCl under an N2 atmosphere (Scheme 2). Complex 5 (ν(N-N) = 2057 cm−1) is diamagnetic, possesses C3 symmetry in solution, and binds N2 as confirmed by a solid-state structure. The cyclic voltammogram of 5 in THF displays a quasi-reversible oxidation wave at −1.1 V vs. Fc/Fc+ (Figure 4, left). Treatment of 5 with 1 equivalent of [Fc][BArF4] at low temperature allowed for the isolation of the one electron oxidation product, [(CP3)Co(N2)][BArF4] (6, ν(N-N) = 2182 cm−1), in 86% yield after recrystallization (Scheme 2). The coordinated N2 ligand of 6 is labile and can be displaced under vacuum (Figure 4, middle) to generate a vacant or possibly solvent-coordinated [(CP3)Co(L)]+ species. The EPR spectrum of 6 at 80 K under N2 is consistent with an S = ½ species (Figure 4, right).
Scheme 2.

Synthesis and oxidation of (CP3)Co(N2)
Figure 4.
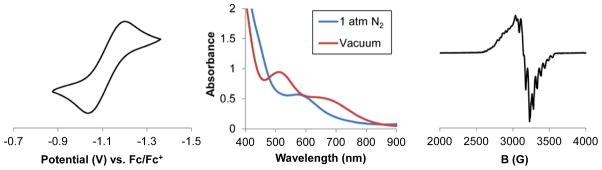
(left) Cyclic voltammagram of (CP3)Co(N2) (5) scanning oxidatively at 100 mV/sec in THF with 0.1 M TBAPF6 electrolyte. (middle) UV-vis spectra of 6 under 1 atm N2 (solid line) and under static vacuum (dotted line: after three freeze-pump-thaw cycles). Spectra were collected on a 1 mM solution of 6 in THF at 298 K. (right) X-band EPR spectrum of 6 collected under 1 atm N2 in 2-Me-THF at 80 K. No low-field features were detected.
Consideration of the M-E interatomic distances presented in Table 1 reveals that the (TPB)Co platform exhibits a significant degree of flexibility of the M-B interaction, similar to that observed for the (TPB)Fe platform. Within each platform the M-B distance varies by > 0.16 Å between the neutral halide (Table 1, G and H) and anionic N2 complexes (B and J). Likewise, the M-C interaction among (CP3)Co complexes exhibits flexibility comparable to the analogous Fe series. For both platforms the M-C distance increases by ~0.07 Å upon one electron reduction of the cationic N2 complexes (Table 1, F to E and K to L).
A comparison of the trends in interatomic metrics between the isoelectronic {(TPB)Co(N2)}n and {(CP3)Co(N2)}n redox series reveals divergent geometric behavior. Upon reduction from 1 to 2, the Co-B distance decreases by 0.02 Å, resulting in a significant decrease in the pyramidalization about Co (Δτ= 0.13).16 The opposite is true for the reduction of 6 to 5, which results in an increase in the Co-C distance and an increase in the pyramidalization (Δτ = −0.13). A plausible rationale is that the Z-type borane ligand in (TPB)Co complexes enforces a trigonal bipyramidal geometry upon reduction, by drawing the Co atom into the P3 plane with an attractive Co-B interaction. The X-type alkyl ligand in (CP3)Co complexes instead causes a distortion away from a trigonal pyramidal geometry upon reduction, with a comparatively repulsive Co-C interaction forcing the Co above the P3 plane.
The reactivity of these (P3E)Co complexes with sources of protons and electrons in the presence of N2 was investigated. In analogy to the [Na(12-c-4)2][(TPB)Fe(N2)] complex, treatment of a suspension of 2 in Et2O at −78 °C with excess [H•(OEt2)2][BArF4] followed by excess KC8 under an atmosphere of N2 leads to the formation of 2.4 ± 0.3 equivalents of NH3 (240% per Co, average of six iterations with independent NH3 yields of 2.3, 2.1, 2.2, 2.5, 2.8, 2.2 equiv, Table 2, A). Yields of NH3 were determined by the indophenol method;17 no hydrazine was detected by a standard UV-vis quantification method.18 We acknowledge that these results are close to a stoichiometric yield of NH3, and include the results of individual experiments here to demonstrate that the yields are reproducibly above 2.1 equivalents. While modest with respect to establishing bona fide catalysis, these yields are consistently greater than 200% NH3 normalized to Co in 2 and represent an order of magnitude improvement over the only previous report of N2 to NH3 conversion mediated by well-defined Co complexes (NH3 yield 0.21 equiv per Co-N2 vide supra).3,19
Table 2.
Ammonia Generation from N2 Mediated by Co Precursorsa
|
| ||
|---|---|---|
| Entry | Co complex | NH3 equiv/Co |
| A | [(TPB)Co(N2)][Na(12-c-4)2] (2) | 2.4 ± 0.3b |
| B | (TPB)Co(N2) (1) | 0.8 ± 0.3 |
| C | [(TPB)Co][BArF4] (3) | 1.6 ± 0.2 |
| D | (TPB)CoBr | 0.7 ± 0.4 |
| E | (SiP3)Co(N2) (4) | <0.1 |
| F | (CP3)Co(N2) (5) | 0.1 ± 0.1 |
| G | [(NArP3)CoCl][BPh4] | <0.1 |
| H | (PBP)Co(N2) | 0.4 ± 0.2 |
| I | Co(PPh3)2I2 | 0.4 ± 0.1 |
| J | CoCp2 | 0.1 ± 0.1 |
| K | Co2(CO)8 | <0.1 |
Co precursors at −78 °C under an N2 atmosphere treated with an Et2O solution containing 47 equiv [H•(OEt2)2][BArF4] followed by an Et2O suspension containing 60 equiv KC8. Yields are reported as an average of 3 iterations; data for individual experimental iterations presented in Supporting Information.
Average of 6 iterations.
Notably, no NH3 is formed when either 2, [H•(OEt2)2][BArF4], or KC8 is omitted from the standard conditions, indicating that all three components are necessary for NH3 production. In an effort to study the fate of 2 under the reaction conditions, we treated 2 with 10 equiv [H•(OEt2)2][BArF4] and 12 equiv KC8, and observed signs of ligand decomposition by 31P NMR (see SI). If the observed reactivity indeed represents modest catalysis, ligand decomposition under the reaction conditions provides a plausible rationale for the limited turnover number. As a control, free TPB ligand was subjected to the standard conditions as a precatalyst, leading to no detectable NH3 production.
Interestingly, though anionic 2 and cationic 3 both generated substantial NH3 under the standard conditions, submitting neutral 1 to these conditions provided attenuated yields of NH3, comparable to the yields obtained with (TPB)CoBr (Table 2, B–D). Furthermore, complexes 4 and 5, which are isoelectronic to 2, are not competent for the reduction of N2 with protons and electrons, producing ≤ 0.1 equivalents of NH3 and no detectable hydrazine under identical conditions (Table 2, E and F). This result appears to underscore the importance of the nature of the M-E interaction in facilitating N2 fixation by (P3E)M complexes.
To further explore the generality of N2 conversion activity for Co complexes under these conditions, we screened a number of additional Co species. We targeted, for instance, a Co complex of the tris(phosphino)amine ligand, NArP3 (NArP3 = [2-(iPr2P)-4-(CH3)-C6H3]3N).20 Synthesis of a (NArP3)Co complex completes a family of tris(phosphino) Co complexes featuring L, X and Z type axial donors. [(NArP3)CoCl][BPh4] (Table 2, G) was isolated as purple crystals in 90% yield from the reaction of the NArP3 ligand with CoCl2 and NaBPh4. An X-ray diffraction study revealed a pseudo-tetrahedral geometry at the Co center, with minimum interaction with the apical N of the ligand (d = 2.64 Å). As expected for tetrahedral Co(II), [(NArP3)CoCl][BPh4] is high-spin S = 3/2, with a solution magnetic moment of 3.97 μB in CD2Cl2 at 23 °C. We also tested the known bis(phosphino)boryl Co-N2 complex21 (Table 2, H), as well as various other common Co complexes (Table 2, I–K). Interestingly, of all the Co precursors subjected to the standard conditions, only (TPB)-ligated Co complexes generated >0.5 equivalents of NH3 per metal center. At this point, we can begin to delineate the structural/electronic factors correlated to NH3 production by (P3E)M complexes.
Among (P3E)Fe complexes, NH3 production appears to be correlated both with flexibility of the M-E interaction and with degree of N2 activation; more flexible and more activating platforms providing greater yields of NH3. Moving from Fe to Co, the degrees of N2 activation are systematically lower, which is expected due to the decreased spatial extent of the Co 3d orbitals (due to increased Zeff).22 Nevertheless, NH3 production is still correlated among these (P3E)Co complexes with N2 activation. However, comparing the Fe to the Co complexes demonstrates that, in an absolute sense, the degree of N2 activation is not predictive of the yield of NH3 (Figure 5). For example, [Na(12-c-4)2][(SiP3)Fe(N2)] shows a higher degree of N2 activation than 2, yet 2 demonstrates higher N2-to-NH3 conversion activity. The relative activity of these two complexes is predicted, on the other hand, by the flexibility of the M-E interactions trans to bound N2. Indeed, among the factors considered here, only M-E interaction flexibility appears to predict the comparatively high N2 conversion activity of 2.
Figure 5.
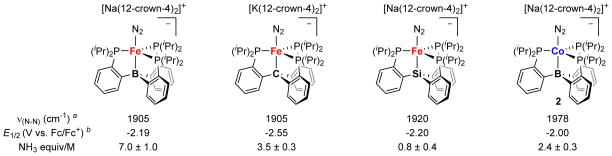
Vibrational spectroscopy, electrochemistry, and catalytic competence data for select [(P3E)M(N2)]− complexes. Data for M = Fe, E = B is from refs. 7 and 10; data for M = Fe, E = C is from ref. 8; data for M = Fe, E = Si is taken refs. 8 and 15; and data for M = Co, E = B is from this work. aIR from solid state samples bOxidation potentials determined by cyclic voltammetry in THF. Note: NH3 yields based on the addition of ~ 50 equiv [H•(OEt2)2][BArF4] and ~ 60 equiv KC8 in Et2O (see refs. provided for specific details).
The potential at which the anionic states of the complexes depicted in Figure 5 are achieved do not follow a clear trend regarding their relative N2 conversion activity. However, a comparison of the Fe and Co systems does demonstrate that the accessibility of highly reduced, anionic [(P3E)M(N2)]− complexes is favorably correlated to NH3 production. It may be the case that the relative basicity of the β-N atom (Nβ) plays an important role in N2 conversion activity, with anionic species being appreciably more basic. Considering complexes 2 and 5, neutral 5 affords <5% NH3 per Co-N2 subunit under the standard reaction conditions whereas the isoelectronic and isostructural, yet anionic, 2 produces >200% NH3 (Table 2). The enhanced basicity of Nβ in the anion would in turn favor protonation to produce a “Co(N2H)” intermediate relative to other reaction pathways. We have performed calculations (DFT; see SI for details) to compare the theoretically predicted electrostatic potential maps of 2 and 5. As shown in Figure 6, Nβ in anionic 2 shows a far greater degree of negative charge relative to the same atom in neutral 5.
Figure 6.

Electrostatic potential maps of anionic 2 and neutral 5 (isovalue = 0.015, color map in Hartrees), and atomic charges for Nβ.
Conclusion
We have demonstrated the ability of a molecular Co-dinitrogen complex to facilitate the conversion of N2 to NH3 at −78 °C in the presence of proton and electron sources (2.4 equivalents of NH3 generated per Co center on average). Prior to this report, the only well-defined molecular systems (including nitrogenase enzymes) capable of directly mediating the catalytic conversion of N2 to NH3 contained either Mo or Fe. While the measured NH3 production by the featured cobalt complex is very modest with respect to catalysis, the yields measured do consistently indicate that some degree of catalysis is viable. The propensity of the (P3E)M complexes we have studied to perform productive nitrogen fixation does not appear to depend solely on the ability of the precursor complex to activate N2. The observations collected herein indicate that anionic charge, and hence basicity of the bound N2 ligand, in addition to flexibility of the M-E interaction trans to the bound N2 ligand, correlate with more favorable NH3 production. Of course, correlation does not presume causation and the factors that lead to different NH3 yields may be numerous. While some of the design features important to consider in catalysts of the (P3E)M(N2) type have been highlighted here, other factors, including comparative rates of H2 evolution and catalyst degradation/poisoning rates, warrant further studies.
Experimental
General considerations
All manipulations were carried out using standard Schlenk or glovebox techniques under an N2 atmosphere. Solvents were deoxygenated and dried by thoroughly sparging with N2 followed by passage through an activated alumina column in a solvent purification system by SG Water, USA LLC. Nonhalogenated solvents were tested with sodium benzophenone ketyl in tetrahydrofuran in order to confirm the absence of oxygen and water. Deuterated solvents were purchased from Cambridge Isotope Laboratories, Inc., degassed, and dried over activated 3-Å molecular sieves prior to use.
[H(OEt2)2][BArF4],23 KC8,24 (TPB)Co(N2),12 (TPB)CoBr,12 (SiP3)Co(N2),14 NArP3,20 (PBP)Co(N2),21 CP3H,8 and Co(PPh3)2I225 were prepared according to literature procedures. All other reagents were purchased from commercial vendors and used without further purification unless otherwise stated. Et2O for NH3 generation reactions was stirred over Na/K (≥ 2 hours) and filtered before use.
Physical Methods
Elemental analyses were performed by Midwest Microlab, LLC (Indianapolis, IN). 1H and 13C chemical shifts are reported in ppm relative to tetramethylsilane, using 1H and 13C resonances from residual solvent as internal standards. 31P chemical shifts are reported in ppm relative to 85% aqueous H3PO4. Solution phase magnetic measurements were performed by the method of Evans.26 IR measurements were obtained as solutions or thin films formed by evaporation of solutions using a Bruker Alpha Platinum ATR spectrometer with OPUS software. Optical spectroscopy measurements were collected with a Cary 50 UV-vis spectrophotometer using a 1-cm two-window quartz cell. Electrochemical measurements were carried out in a glovebox under an N2 atmosphere in a one compartment cell using a CH Instruments 600B electrochemical analyzer. A glassy carbon electrode was used as the working electrode and platinum wire was used as the auxiliary electrode. The reference electrode was Ag/AgNO3 in THF. The ferrocene couple (Fc/Fc+) was used as an internal reference. THF solutions of electrolyte (0.1 M tetra-n-butylammonium hexafluorophosphate, TBAPF6) and analyte were also prepared under an inert atmosphere. X-band EPR spectra were obtained on a Bruker EMX spectrometer.
X-ray diffraction studies were carried out at the Caltech Division of Chemistry and Chemical Engineering X-ray Crystallography Facility on a Bruker three-circle SMART diffractometer with a SMART 1K CCD detector. Data was collected at 100K using Mo Kα radiation (λ= 0.71073 Å). Structures were solved by direct or Patterson methods using SHELXS and refined against F2 on all data by full-matrix least squares with SHELXL-97. All non-hydrogen atoms were refined anisotropically. All hydrogen atoms were placed at geometrically calculated positions and refined using a riding model. The isotropic displacement parameters of all hydrogen atoms were fixed at 1.2 (1.5 for methyl groups) times the Ueq of the atoms to which they are bonded.
[Na(12-crown-4)2][(TPB)Co(N2)] (2)
To a −78 °C solution of (TPB)CoBr (70.5 mg, 0.0967 mmol) in THF (2 mL) was added a freshly prepared solution of NaC10H8 (23.5 mg C10H8, 0.222 mmol) in THF (3 mL). The solution was brought to room temperature and allowed to stir for six hours. Addition of 12-crown-4 (51.1 mg, 0.290 mmol) and removal of solvent in vacuo provided a dark red solid. Et2O was added and subsequently removed in vacuo. The residue was suspended in C6H6 and filtered and the solids were washed with C6H6 (2 × 2 mL) and pentane (2 × 2 mL) to furnish a red solid (68.8 mg, 0.0660 mmol, 68%). Single crystals were grown by vapor diffusion of pentane onto a THF solution of the title compound that had been layered with Et2O. 1H NMR (400 MHz, THF-d8) δ 7.41 (3H), 6.94 (3H), 6.66 (3H), 6.44 (3H), 3.64 (32H), 2.29 (br), 1.37 (6H), 1.20 (6H), 0.93 (6H), −0.26 (6H). 11B NMR (128 MHz, THF-d8) δ 9.32. 31P NMR (162 MHz, THF-d8) δ 62.03. IR (thin film, cm-1): 1978 (N2). Anal. Calcd. for C52H86BCoN2NaO8P3 : C, 59.32; H, 8.23; N, 2.66. Found: C, 59.05; H, 7.99; N, 2.47.
[(TPB)Co][BArF4] (3)
To a −78 °C solution of (TPB)Co(N2) (1) (91.5 mg, 0.135 mmol) in Et2O (2 mL) was added solid [H(OEt2)2][BArF4] (134.0 mg, 0.132 mmol). The reaction was brought to room temperature and vented to allow for the escape of H2. The purple-brown solution was stirred for 1 hr. The solution was layered with pentane (5 mL) and stored at −35 °C to furnish red-purple single crystals of the title compound (162.9 mg, 0.0952 mmol, 82%) which were washed with pentane (3 × 2 mL). 1H NMR (400 MHz, C6D6) δ 26.25, 23.80, 8.64, 8.44 ([BArF4]), 7.88 ([BArF4]), 6.33, −2.16, −3.68. UV-Vis (Et2O, nm {L cm−1 mol−1}): 585 {1500}, 760 {532}. Anal. Calcd. for C68H66B2CoF24P3 : C, 53.99; H, 4.40. Found: C, 53.94; H, 4.51.
(CP3)Co(N2) (5)
(CP3)H (100 mg, 0.169 mmol) and CoCl2 • 1.5 THF (40 mg, 0.169 mmol) were mixed at room temperature in THF (10 mL). This mixture was allowed to stir for one hour, yielding a homogeneous cyan solution. This solution was chilled to −78 °C, and a solution of MeMgCl in tetrahydrofuran (0.5 M, 0.560 mmol) was added in three 370 μL portions over three hours. The mixture was allowed to warm slowly to room temperature, and then was concentrated to ca. 1 mL. 1,4-dioxane (2 mL) was added, and the resultant suspension was stirred vigorously for at least 2 hours before filtration. The filtrate was concentrated to a tacky red-brown solid, which was extracted with 1:1 C6H6 : pentane (10 mL), filtered over celite and lyophilized to yield the product as a red powder (96 mg, 0.141 mmol, 83%). Crystals suitable for X-ray diffraction were grown via slow evaporation of a pentane solution. 1H NMR (300 MHz, C6D6) δ 7.28 (br, 3H), 6.82 (m, 9H), 2.82 (oct., -CH, 3H), 2.09 (sept., -CH, 3H), 1.49 (m, 18H), 1.06 (dd, -CHCH3, 9H), 0.30 (dd, -CHCH3, 9H). 31P{1H} (121 MHz, C6D6): δ 47.39. IR (thin film, cm−1): 2057 (N2). Anal. Calcd. for C37H54CoN2P3 : C, 65.48; H, 8.02; N, 4.13. Found: C, 64.14; H, 8.36; N, 4.03.
[(CP3)Co(N2)][BArF4] (6)
5 (75 mg, 0.11 mmol) and [Cp2Fe][BArF4] (122 mg, 0.12 mmol) were dissolved separately in diethyl ether (ca. 3 mL each) and the ethereal solutions were cooled to −78°C. The chilled solution of [Cp2Fe][BArF4] was added dropwise to the solution of 5, and the resultant mixture was allowed to stir at low temperature for one hour. At this point, the mixture was allowed to warm to room temperature before filtration over celite and concentration to ca. 2 mL. The concentrated filtrate was layered with pentane, and placed in a freezer at −35°C to induce crystallization. Decanting the mother liquor off crystalline solids and washing thoroughly with pentane yields [(CP3)Co(N2)][BArF4] (6) as dark green-brown crystals (147 mg, 0.095 mmol, 86%). Crystals suitable for X-ray diffraction were grown by slow diffusion of pentane vapors into an ethereal solution of 6 at −35°C. μeff (5:1 d8-toluene:d8-THF, Evans’ method, 23 °C): 3.49 μB. 1H NMR (300 MHz, C6D6) δ17.22, 9.94, 8.24 ([BArF4]), 7.72([BArF4]), 3.13, 2.57, 1.5 – −2 (br, m), −3.68.IR (cm−1): 2182 (N2, thin film), 2180 (N2, solution). Elemental analysis shows low values for N consistent with a labile N2 ligand, Anal. Calcd. for C69H66BCoF24N2P3 : C, 53.75; H, 4.31; N, 1.82. Found: C, 53.86; H, 4.31; N, 0.27. Note: The magnetic moment for 6 in solution may be complicated by some degree of solvent exchange for N2 at the cobalt center as described in the text.
[(NArP3)CoCl][BPh4]
THF (5 mL) was added to a solid mixture of NArP3 (58 mg, 91.2 mmol), CoCl2 (12 mg, 92.4 mmol) and NaBPh4 (31 mg, 90.6 mmol). The reaction was stirred for 4 hours at room temperature during which the color evolved from yellow to green to purple. The solvent was removed in vacuo and the residue was taken up in dichloromethane. The suspension was filtered over a plug of Celite and the filtrate was dried yielding a purple powder (86 mg, 82.1 mmol, 90%). Single crystals were grown by slow evaporation of a saturated solution of [(NArP3)CoCl][BPh4] in diethyl ether/dichloromethane (1:2 v:v). 1H NMR (CD2Cl2, 300 MHz) δ177.77, 37.50, 23.78, 13.48, 12.96, 7.37, 7.08, 6.92, 4.41, 1.50, −3.60, −9.81; UV-Vis (THF, nm {L cm−1 mol−1}): 564 {452}, 760 {532}; μeff (CD2Cl2, Evans’ method, 23 °C): 3.97 μB. Anal. Calcd. for C63H80BClCoNP3 : C, 72.10; H, 7.68; N, 1.33. Found: C, 71.97; H, 7.76; N, 1.30.
Ammonia Quantification
A Schlenk tube was charged with HCl (3 mL of a 2.0 M solution in Et2O, 6 mmol). Reaction mixtures were vacuum transferred into this collection flask. Residual solid in the reaction vessel was treated with a solution of [Na][O-t-Bu] (40 mg, 0.4 mmol) in 1,2-dimethoxyethane (1 mL) and sealed. The resulting suspension was allowed to stir for 10 min before all volatiles were again vacuum transferred into the collection flask. After completion of the vacuum transfer, the flask was sealed and warmed to room temperature. Solvent was removed in vacuo, and the remaining residue was dissolved in H2O (1 mL). An aliquot of this solution (20 _L) was then analyzed for the presence of NH3 (present as [NH4][Cl]) by the indophenol method.17 Quantification was performed with UV–vis spectroscopy by analyzing absorbance at 635 nm.
Standard NH3 Generation Reaction Procedure with [(TPB)Co(N2)][Na(12-crown-4)2] (2)
[(TPB)Co(N2)][Na(12-crown-4)2] (2.2 mg, 0.002 mmol) was suspended in Et2O (0.5 mL) in a 20 mL scintillation vial equipped with a stir bar. This suspension was cooled to −78 °C in a cold well inside of a N2 glovebox. A solution of [H·(OEt2)2][BArF4] (95 mg, 0.094 mmol) in Et2O (1.5 mL) similarly cooled to −78 °C was added to this suspension in one portion with stirring. Residual acid was dissolved in cold Et2O (0.25 mL) and added subsequently. This mixture was allowed to stir 5 minutes at −78 °C, before being transferred to a precooled Schlenk tube equipped with a stir bar. The original reaction vial was washed with cold Et2O (0.25 mL) which was added subsequently to the Schlenk tube. KC8 (16 mg, 0.119 mmol) was suspended in cold Et2O (0.75 mL) and added to the reaction mixture over the course of 1 minute. The Schlenk tube was then sealed, and the reaction was allowed to stir for 40 min at −78 °C before being warmed to room temperature and stirred for 15 min. ASSOCIATED CONTENT
Supplementary Material
Acknowledgments
This work was supported by the NIH (GM 070757) and the Gordon and Betty Moore Foundation, and through the NSF via a GRFP award to TJDC.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. Spectroscopic data, experimental details, additional data for NH3 production experiments, and XRD tables. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Smil V. Enriching the Earth. MIT Press; Cambridge: 2001. [Google Scholar]
- 2.Hidai M, Takahashi T, Yokotake I, Uchida Y. Chem Lett. 1980:645. [Google Scholar]
- 3.Yamamoto A, Miura Y, Ito T, Chen H, Iri K, Ozawa F, Miki K, Sei T, Tanaka N, Kasai N. Organometallics. 1983;2:1429. [Google Scholar]
- 4.(a) Fryzuk MD. Acc Chem Res. 2009;42:127. doi: 10.1021/ar800061g. [DOI] [PubMed] [Google Scholar]; (b) Schrock RR. Angew Chem Int Ed. 2008;47:5512. doi: 10.1002/anie.200705246. [DOI] [PubMed] [Google Scholar]; (c) Chirik PJ. Dalton Trans. 2007:16. doi: 10.1039/b613514e. [DOI] [PubMed] [Google Scholar]; (d) Peters JC, Mehn MP. In: Activation of Small Molecules: Organometallic and Bioinorganic Perspectives. Tolman WB, editor. Wiley-VCH; New York: 2006. pp. 81–119. [Google Scholar]; (e) Crossland JL, Tyler DR. Coord Chem Rev. 2010;254:1883. [Google Scholar]; (f) Chatt J, Dilworth JR, Richards RL. Chem Rev. 1978;78:589. [Google Scholar]; (g) Siedschlag RB, Bernales V, Vogiatzis KD, Planas N, Clouston LJ, Bill E, Gagliardi L, Lu CC. J Am Chem Soc. 2015;137:4638. doi: 10.1021/jacs.5b01445. [DOI] [PubMed] [Google Scholar]
- 5.(a) Laplaza CE, Cummins CC. Science. 1995;268:861. doi: 10.1126/science.268.5212.861. [DOI] [PubMed] [Google Scholar]; (b) Zanotti-Gerosa A, Solari E, Giannini L, Floriani C, Chiesi-Villa A, Rizzoli C. J Am Chem Soc. 1998;120:437. [Google Scholar]; (c) Nikiforov GB, Vidyaratne I, Gambarotta S, Korobkov I. Angew Chem Int Ed. 2009;48:7415. doi: 10.1002/anie.200903648. [DOI] [PubMed] [Google Scholar]; (d) Vidyaratne I, Crewdson P, Lefebvre E, Gambarotta S. Inorg Chem. 2007;46:8836. doi: 10.1021/ic701219h. [DOI] [PubMed] [Google Scholar]; (e) Clentsmith GKB, Bates VME, Hitchcock PB, Cloke FGN. J Am Chem Soc. 1999;121:10444. [Google Scholar]; (f) Hebden TJ, Schrock RR, Takase MK, Müller P. Chem Commun. 2012;48:1851. doi: 10.1039/c2cc17634c. [DOI] [PubMed] [Google Scholar]; (g) Rodriguez MM, Bill E, Brennessel WW, Holland PL. Science. 2011;334:780. doi: 10.1126/science.1211906. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Curley JJ, Cook TR, Reece SY, Müller P, Cummins CC. J Am Chem Soc. 2008;130:9394. doi: 10.1021/ja8002638. [DOI] [PubMed] [Google Scholar]; (i) Fryzuk MD, Kozak CM, Bowdridge MR, Patrick BO, Rettig SJ. J Am Chem Soc. 2002;124:8389. doi: 10.1021/ja025997f. [DOI] [PubMed] [Google Scholar]
- 6.(a) Yandulov DV, Schrock RR. Science. 2003;301:76. doi: 10.1126/science.1085326. [DOI] [PubMed] [Google Scholar]; (b) Schrock RR. Angew Chem Int Ed. 2008;47:5512. doi: 10.1002/anie.200705246. [DOI] [PubMed] [Google Scholar]; (c) Arashiba K, Miyake Y, Nishibayashi Y. Nat Chem. 2010;3:120. doi: 10.1038/nchem.906. [DOI] [PubMed] [Google Scholar]; (d) Kuriyama S, Arashiba K, Nakajima K, Tanaka H, Kamaru N, Yoshizawa K, Nishibayashi Y. J Am Chem Soc. 2014;136:9719. doi: 10.1021/ja5044243. [DOI] [PubMed] [Google Scholar]; (e) Ung G, Peters JC. Angew Chem Int Ed. 2015;54:532. doi: 10.1002/anie.201409454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson JS, Rittle J, Peters JC. Nature. 2013;501:84. doi: 10.1038/nature12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Creutz SE, Peters JC. J Am Chem Soc. 2014;136:1105. doi: 10.1021/ja4114962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moret ME, Peters JC. J Am Chem Soc. 2011;133:18118. doi: 10.1021/ja208675p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moret ME, Peters JC. Angew Chem Int Ed. 2011;50:2063. doi: 10.1002/anie.201006918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For a recent review see: Hoffman BM, Lukoyanov D, Yang ZY, Dean DR, Seefeldt LC. Chem Rev. 2014;114:4041. doi: 10.1021/cr400641x.
- 12.Suess DLM, Tsay C, Peters JC. J Am Chem Soc. 2012;134:14158. doi: 10.1021/ja305248f. [DOI] [PubMed] [Google Scholar]
- 13.Anderson JS, Moret ME, Peters JC. J Am Chem Soc. 2013;135:534. doi: 10.1021/ja307714m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whited MT, Mankad NP, Lee Y, Oblad PF, Peters JC. Inorg Chem. 2009;48:2507. doi: 10.1021/ic801855y. [DOI] [PubMed] [Google Scholar]
- 15.Lee Y, Mankad NP, Peters JC. Nat Chem. 2010;2:558. doi: 10.1038/nchem.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vela J, Cirera J, Smith JM, Lachicotte RJ, Flaschenriem CJ, Alvarez S, Holland PL. Inorg Chem. 2007;46:60. doi: 10.1021/ic0609148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weatherburn MW. Anal Chem. 1967;39:971. [Google Scholar]
- 18.Watt GW, Chrisp JD. Anal Chem. 1952;24:2006. [Google Scholar]
- 19.There is an additional early report of the generation of one equivalent of NH3 by a Co-porphyrin complex under an N2 containing atmosphere upon treatment with sodium borohydride (see Fleischer EB, Krishnamurthy M. J Am Chem Soc. 1972;94:1382. doi: 10.1021/ja00759a072.However a later study firmly concluded that the NH3 detected under these conditions was not derived from N2 (see Chatt J, Elson CM, Leigh GJ. J Am Chem Soc. 1973;95:2408.
- 20.MacBeth CE, Harkins SB, Peters JC. Can J Chem. 2005;83:332. [Google Scholar]
- 21.Lin TP, Peters JC. J Am Chem Soc. 2013;135:15310. doi: 10.1021/ja408397v. [DOI] [PubMed] [Google Scholar]
- 22.Miessler GL, Tarr DA. Inorganic Chemistry. 4. Prentice Hall; New York: 2011. pp. 37–43. [Google Scholar]
- 23.Brookhart M, Grant B, Volpe AF., Jr Organometallics. 1992;11:3920–3922. [Google Scholar]
- 24.Wietz IS, Rabinovitz MJ. J Chem Soc, Perkin Trans. 1993;1:117. [Google Scholar]
- 25.Cotton FA, Faut OD, Goodgame DML, Holm RH. J Am Chem Soc. 1961;83:1780. [Google Scholar]
- 26.Evans DF. J Chem Soc. 1959:2003. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.