

Abstract
Background
Lactobacillus fermentum 222 and Lactobacillus plantarum 80, isolates from a spontaneous Ghanaian cocoa bean fermentation process, proved to be interesting functional starter culture strains for cocoa bean fermentations. Lactobacillus fermentum 222 is a thermotolerant strain, able to dominate the fermentation process, thereby converting citrate and producing mannitol. Lactobacillus plantarum 80 is an acid-tolerant and facultative heterofermentative strain that is competitive during cocoa bean fermentation processes. In this study, whole-genome sequencing and comparative genome analysis was used to investigate the mechanisms of these strains to dominate the cocoa bean fermentation process.
Results
Through functional annotation and analysis of the high-coverage contigs obtained through 454 pyrosequencing, plantaricin production was predicted for L. plantarum 80. For L. fermentum 222, genes encoding a complete arginine deiminase pathway were attributed. Further, in-depth functional analysis revealed the capacities of these strains associated with carbohydrate and amino acid metabolism, such as the ability to use alternative external electron acceptors, the presence of an extended pyruvate metabolism, and the occurrence of several amino acid conversion pathways. A comparative genome sequence analysis using publicly available genome sequences of strains of the species L. plantarum and L. fermentum revealed unique features of both strains studied. Indeed, L. fermentum 222 possessed genes encoding additional citrate transporters and enzymes involved in amino acid conversions, whereas L. plantarum 80 is the only member of this species that harboured a gene cluster involved in uptake and consumption of fructose and/or sorbose.
Conclusions
In-depth genome sequence analysis of the candidate functional starter culture strains L. fermentum 222 and L. plantarum 80 revealed their metabolic capacities, niche adaptations and functionalities that enable them to dominate the cocoa bean fermentation process. Further, these results offered insights into the cocoa bean fermentation ecosystem as a whole and will facilitate the selection of appropriate starter culture strains for controlled cocoa bean fermentation processes.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1927-0) contains supplementary material, which is available to authorized users.
Keywords: Lactobacillus fermentum, Lactobacillus plantarum, Cocoa bean fermentation, 454 pyrosequencing, Functional starter cultures
Background
Lactic acid bacteria (LAB) are Gram-positive bacteria that are key players in the majority of food fermentation ecosystems [1, 2]. The genus Lactobacillus represents the largest group among the LAB, encompassing more than 200 species [3, 4]. As this genus possesses a wide metabolic diversity, lactobacilli occur and have been used as functional starter cultures in a wide variety of fermented food products, including cheese, fermented plant-derived foods, fermented meats, wine and beer production, and sourdoughs [5]. Furthermore, lactobacilli play an important role during the cocoa bean fermentation process [6].
At the onset of the cocoa bean fermentation process, a wide LAB species diversity is often present [7–12]. However, only a restricted number of Lactobacillus species predominate this fermentation process, consisting mainly of (i) strictly heterofermentative Lactobacillus fermentum [7–22] and (ii) facultative heterofermentative Lactobacillus plantarum [7–12, 14–21, 23]. It has been proposed that L. plantarum is the dominant member at the onset of the cocoa bean fermentation process, while L. fermentum dominates in later stages [7, 8, 18, 21]. This is in accordance with the cocoa-specific functional roles that have been associated with these two species [24–26].
Homolactic fermentation of cocoa pulp-bean mass carbohydrates such as glucose and fructose, resulting from the hydrolysis of sucrose present in the cocoa pulp-bean mass by pulp or yeast invertase, results in lactic acid, while heterolactic fermentation gives lactic acid, acetic acid and/or ethanol, and carbon dioxide [6]. Fructose and citric acid, present in the cocoa pulp, are also used as alternative external electron acceptors by heterofermentative LAB, enhancing their competitiveness and resulting in the production of mannitol and succinate, or lactate and flavor-active compounds such as 2,3-butanediol or acetoin, respectively [24–26].
To better understand the exact role of cocoa-derived microorganisms, their whole-genome sequence can be investigated, as has been done for the cocoa-specific Acetobacter pasteurianus 386B strain [27]. Whole-genome sequencing of particular strains of the LAB species L. fermentum and L. plantarum originating from other sources (including fermented food ecosystems; Additional files 1 and 2) has been performed previously, thereby elucidating a plethora of specific functionalities and niche adaptations. For instance, genome sequence analysis of particular L. fermentum strains has revealed adaptations to specific host systems, such as the presence of adhesins [28] and probiotic properties [29–31]. Further, comparative genomics has enabled insights into the genomic features that are unique for L. fermentum strains [32]. Genome sequencing of particular L. plantarum strains elucidated niche adaptations [33, 34], probiotic properties [35–38], prophage occurrence [39–41], general metabolic properties [42–46], and bacteriocin production [47]. Therefore, it can be expected that whole-genome sequence analysis of the cocoa-derived L. fermentum 222 and L. plantarum 80 strains, isolates from a spontaneous Ghanaian cocoa bean fermentation process and proposed as interesting functional starter culture strains [7, 25, 26], will provide detailed insight into their genetic potential, metabolic capacities and functionalities as well as cocoa niche adaptations. Next to their central carbohydrate metabolism, their proteolytic system and amino acid conversion pathways have to be analysed, as these are important functionalities of LAB in various food fermentation ecosystems [48–50].
Results and discussion
454 pyrosequencing, genome sequence assembly, and annotation
454 pyrosequencing of the genomic DNA of L. fermentum 222 and L. plantarum 80 yielded 571,369 reads with a median length of 435 base pairs (bp) and 621,544 reads with a median length of 416 bp, respectively (Table 1). The reads obtained from the genomic DNA of L. fermentum 222 were assembled into 73 contigs (>500 bp) with a 104-fold depth of coverage, whereas the reads obtained from the genomic DNA of L. plantarum 80 were assembled into 67 contigs (>500 bp) with a 65-fold depth of coverage. Furthermore, the genome sequences were estimated to be 2.2 million bp (Mb) with a guanine plus cytosine (G+C) content of 52.08 % for L. fermentum 222 and 3.6 Mb with a G+C content of 44.36 % for L. plantarum 80. For L. fermentum 222, these data were in accordance with other members of this species [29–31, 51]. However, the genome sequence size of L. plantarum 80 was larger compared with other members of this species, generally being around 3.2 Mb [39], indicating that the genome sequence of L. plantarum 80 encompassed one or more plasmids, which is common for this species [33, 37, 40, 41, 52]. Indeed, two genes encoding a plasmid replication initiator (repA) were found, located on a 49,183-bp (LP80_2408) and a 8,306-bp (LP80_3155) contig. Also, several contigs showed high sequence identity with the plasmid-specific PATRIC database, providing further evidence for the presence of plasmids. By automated annotation of the contigs obtained, 1,864 and 3,123 protein-encoding sequences (CDS) were found in the genome sequences of L. fermentum 222 and L. plantarum 80, respectively. Other relevant features deduced from the genome sequences are summarised in Table 1.
Table 1.
General features of the reads, contigs, and draft genome sequences of Lactobacillus fermentum 222 and L. plantarum 80
| Statistics | L. fermentum 222 | L. plantarum 80 |
|---|---|---|
| Reads | ||
| Total bases | 225,141,845 | 234,878,528 |
| Number of reads | 571,369 | 621,544 |
| Average read length | 394.65 | 378.34 |
| Median read length | 435 | 416 |
| Assembly | ||
| Number of contigs | 73 | 67 |
| N50 contigs | 55,702 | 92,828 |
| Depth of coverage | 104 | 65 |
| Average length | 26,717 | 48,130 |
| Longest contig | 159,496 | 312,995 |
| Draft genome sequences | ||
| Estimated genome size (Mb)a | 2.2 | 3.6 |
| Total contig length (bp) | 1,950,408 | 3,224,773 |
| G+C content (%) | 52.08 | 44.36 |
| Number of CDS | 1,864 | 3,123 |
| Coding density (%) | 83.00 | 83.00 |
| Average gene length (bp) | 869 | 857 |
| Number of rRNA operonsb | 6 | 5 |
| Number of tRNAs | 54 | 67 |
aEstimation according to Newbler
bEstimation based on contig coverage
Genome architecture and genetic potential
The genome sequences of L. fermentum 222 and L. plantarum 80 did not contain virulence factors, antibiotic resistance genes, or clustered regularly interspaced short palindromic repeat (CRISPR) sequences, confirming their applicability for food fermentation processes. One prophage-related genomic region was found in the genome sequence of L. fermentum 222 (LFER_1134-LFER_1187) and three such regions were found in the genome sequence of L. plantarum 80 (LP80_860-LP80_887, LP80_1614-LP80_1668, LP80_2307-LP80_2359). Although these regions were identified by PHAST analysis as ‘intact prophage’, these phage-associated regions might not be active or inducible, as has been reported before in LAB [51]. Further, genes involved in bacteriocin biosynthesis were not found in the genome sequence of L. fermentum 222. The genome sequence of L. plantarum 80 contained a plantaricin (pln) locus (LP80_422-LP80_443) on a 237,656-bp contig (Fig. 1), comprising all genes needed for the production of this bacteriocin [53]. BLASTn analysis of this contig using the plasmid-specific PATRIC database revealed that this plantaricin is chromosomally encoded in L. plantarum 80. This plantaricin locus was similar to that of L. plantarum WCFS1 [33], including a transport operon (plnGHSTUVW), all genes encoding a two-peptide bacteriocin JK (plnJKLR), a regulatory operon (plnABCD), a putative operon (plnMNOP), and genes encoding a putative toxin-antitoxin system (plnXY). Lactobacillus plantarum 80 did not contain the genes encoding the two-peptide bacteriocin EF (plnEFI), which were present in the pln locus of L. plantarum WCFS1. Instead, genes encoding putative proteins were found in the pln locus of L. plantarum 80 (Fig. 1; LP80_423, LP80_434-LP80_435). Production of plantaricin JK might enable L. plantarum 80 to outcompete other L. plantarum species present in the cocoa bean fermentation process, as this bacteriocin inhibits mainly the species L. plantarum [54, 55]. The genome sequence of L. fermentum 222 contained a 14-kb exopolysaccharide biosynthesis gene cluster (LFER_1022-LFER_1033), including a gene encoding a dextransucrase (LFER_1022), indicating that this strain is able to produce extracellular homopolysaccharides of the dextran type, possibly to protect itself.
Fig. 1.

Genetic map of the pln loci of Lactobacillus plantarum 80 and L. plantarum WCFS1. For L. plantarum 80, a partial sequence of the 237,656-bp contig encoding the pln locus (LP80_422-LP80_443) is shown. For L. plantarum WCFS1, the chromosomal region containing the pln genes lp_0400-lp_0429 is shown
Concerning adaptation to stress conditions, both genome sequences contained genes encoding an ATP synthase (atpABCDEFGH; LFER_671-LFER_678 for L. fermentum 222 and LP80_834-LP80_841 for L. plantarum 80), which enables regulation of the internal pH [56]. Further, the heat-shock operon hrcA-grpE-dnaKJ as well as several other heat-shock protein-encoding genes were found in both genome sequences (Table 2). Also, cold-shock protein-encoding genes as well as genes encoding Clp ATPases and proteases, which are involved in stress responses [57], were found in both genome sequences (Table 2). These cold-shock proteins might be part of the stress adaptation system of L. plantarum strains, as previously suggested for L. plantarum WCFS1 [33]. Finally, the genome sequence of L. plantarum 80 contained a katA gene (LP80_2743) encoding a heme-dependent catalase, which protects the cell from oxidative stress [58].
Table 2.
Mechanisms of Lactobacillus fermentum 222 and Lactobacillus plantarum 80 involved in stress responses. The locus tags are depicted for each mechanism as well as the gene names when appropriate
| Mechanism | L. fermentum 222 | L. plantarum 80 |
|---|---|---|
| Heat-shock operon hrcA-grpE-dnaKJ | LFER_1617-LFER_1620 | LP80_2199-LP80_2202 |
| Heat-shock proteins | LFER_1794 | LP80_658; LP80_2456; LP80_2821 |
| Cold-shock proteins | LFER_598 | LP80_47; LP80_193; LP80_2125 |
| Clp ATPases and proteases | clpL (LFER_111); clpB (LFER_572); clpP (LFER_1165, LFER_1522); clpE (LFER_1495); clpX (LFER_1563) | clpB (LP80_2653); clpC (LP80_170, LP80_2748); clpE (LP80_2599); clpP (LP80_1719, LP80_2338); clpX (LP80_923); clpQY; (LP80_1233-LP80_1234); lon (LP80_936) |
Metabolic pathway reconstruction
Pathway analysis of Lactobacillus fermentum 222
Central carbohydrate metabolism
All genes encoding the enzymes involved in the heterolactic fermentation pathway, responsible for the production of lactic acid, acetic acid and/or ethanol, were retrieved in the genome sequence of L. fermentum 222 (Fig. 2; Additional file 3). Further, different carbohydrate transport mechanisms were found, such as phosphoenolpyruvate (PEP)-dependent sugar phosphotransferase systems (PTS) and permeases. Pyruvate, originating from the heterolactic fermentation or the citrate metabolism (see below), may be used by L. fermentum 222 for NAD+ regeneration by production of diacetyl and/or acetoin, as the genes encoding α-acetolactate synthase (als), α-acetolactate decarboxylase (aldB), 2,3-butanediol dehydrogenase (butC), and diacetyl reductase (butA) were all found in the genome sequence (Fig. 2; Additional file 3). Alternatively, pyruvate may be converted into acetyl-CoA by the pyruvate dehydrogenase complex, with acetate or ethanol as the final products. Finally, a gene encoding pyruvate oxidase (pox) was found, enabling this strain to convert pyruvate into carbon dioxide and acetyl phosphate when oxygen is available. This requires reactive oxygen species tolerance, for which a gene encoding a thiol peroxidase (tpx; LFER_707) was found.
Fig. 2.
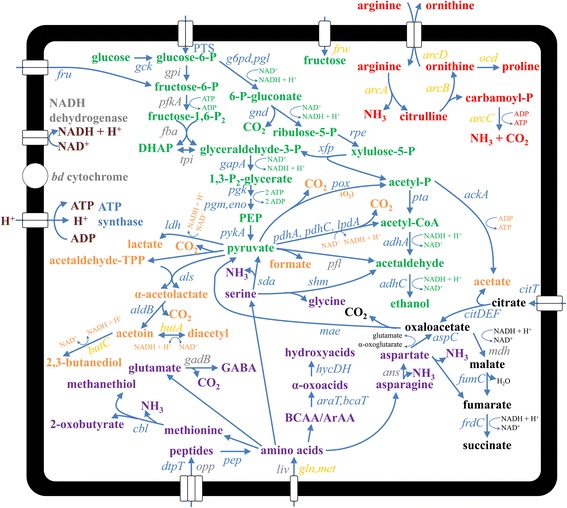
Metabolic pathways of Lactobacillus fermentum 222 and Lactobacillus plantarum 80. The names of the genes that were present in both genomes are depicted in blue, those that were present in L. fermentum 222 only are depicted in yellow, and those that were present in L. plantarum 80 only are depicted in grey. Metabolites involved in the homo- and heterolactic fermentation pathways are depicted in green, those involved in pyruvate metabolism are depicted in purple, those involved in citrate metabolism are depicted in black, those involved in the proteolytic system and amino acid conversion pathways are depicted in orange, those involved in the arginine deiminase pathway are depicted in red, and those involved in the respiratory machinery are depicted in dark red. Enzymes and locus tags that were associated with the gene names are listed in Additional file 3. DHAP Dihydroxyacetone phosphate, PEP Phosphoenolpyruvate, BCAA, Branched-chain amino acid, ArAA Aromatic amino acid, GABA γ-aminobutyric acid, TPP Thiamine pyrophosphate
A citrate operon, involved in the conversion of citric acid [26], was found in L. fermentum 222 (LFER_302-309; Figs. 2 and 3; Additional file 3), consisting of genes coding for a citrate lyase and its regulator (citRDEF), an oxaloacetate decarboxylating malate dehydrogenase (mae), a putative citrate transporter (LFER_304), and an accessory protein (citG*). In comparison with the two citrate operons that are encoded in the genome sequence of L. fermentum F-6, the only L. fermentum strain for which the whole-genome sequence is available harbouring citrate operons, the citrate operon of L. fermentum 222 showed differences with the ones of L. fermentum F-6. Indeed, the first citrate operon of L. fermentum F-6 seemed to be incomplete, as the mae gene was missing and the second operon had an atypical citrate transporter (Fig. 3) that was also found in L. fermentum 3872 [28], which is unlikely to be specific for citrate based on the sequence annotation of this gene. As such, the gene encoding this transporter was neither homologous to the one present in the citrate operon of L. fermentum 222 nor to the one in the other citrate operon of L. fermentum F-6. Besides the citrate transporter present in the citrate operon, two additional genes encoding a putative citrate transporter (LFER_11 and LFER_1857) were found in the genome sequence of L. fermentum 222. Yet, not all genes encoding the enzymes to convert oxaloacetate (produced by citrate lyase) into succinate were found, because only genes encoding fumarase (fumC) and fumarate reductase (frdC) were present, while a gene encoding malate dehydrogenase was missing.
Fig. 3.

Organisation of the citrate operons in Lactobacillus fermentum 222, L. fermentum F-6, and L. plantarum 80. The appropriate genomic regions encoding the proteins involved in citrate uptake and conversion are shown for each genome sequence. For L. fermentum F-6, both citrate operons are depicted. Flanking genes outside the citrate operon are shown in black
Pathways that enable the use of oxygen or glycerol as alternative external electron acceptors were not found in the genome sequence of L. fermentum 222. Although this strain is able to use fructose as an alternative external electron acceptor [26] and the genome sequence contained a gene coding for a fructose permease (frw), a gene encoding a mannitol dehydrogenase could not be found. However, a fructose-specific PEP-dependent PTS (fru) was present, which is associated with mannitol production from fructose in homofermentative LAB [59]. Based on the genome sequence annotation, no further evidence was found that this transporter was involved in mannitol production.
Proteolytic system and amino acid conversion pathways
The genome sequence of L. fermentum 222 harboured genes encoding a peptide transporter (dtpT) and several amino acid ATP-binding cassette (ABC) transporters specific for glutamine (gln), methionine (met), and several other amino acids that could not be further specified based on the genome sequence annotation (Fig. 2; Additional file 3). Further, a plethora of genes encoding peptidases were retrieved, such as pepF and pepO encoding endopeptidases, pepI, pepP, pepQ, and pepX encoding proline-specific peptidases, pepD and pepV encoding dipeptidases, pepT encoding a tripeptidase, and pepC, pepN, and pepM encoding aminopeptidases (Additional file 3). The acquisition of a large variety of peptidases by Lactobacillales is related to their adaptation to nutritionally rich environments [60], which might also be the case for members of the cocoa bean fermentation process, as cocoa pulp consists of 0.5–0.7 % proteins [6].
Aspartate transaminase-encoding genes were found (aspC), enabling L. fermentum 222 to produce oxaloacetate from aspartate. This oxaloacetate might contribute to the pyruvate metabolism through the pathway associated with citrate metabolism (see above). Indeed, it has been shown for other Lactobacillus species that diacetyl and acetoin can be produced through aspartate catabolism [61]. Genes encoding aspartase (aspA), fumarase (fum), and succinate dehydrogenase (sdh) were found as well, offering an alternative pathway for aspartate catabolism (Fig. 2; Additional file 3). This pathway may contribute to NAD+ regeneration, and, therefore, offer a more efficient degradation of carbohydrates [62].
Genes encoding the transaminases araT and bcaT, which catalyse the hydrolysis of branched-chain amino acids as well as aromatic amino acids, were retrieved (Fig. 2; Additional file 3). The concomitant α-oxoacids produced might be further reduced to hydroxyacids by the 2-hydroxyacid dehydrogenases (hycDH) present, thereby contributing to NAD+ regeneration and thus allowing more efficient energy production through heterofermentation, as hypothesised before [48]. Genes associated with the conversion of α-oxoacids by oxidative decarboxylation into carboxylic acids or by decarboxylation into aldehydes were not found. Neither were enzymes involved in decarboxylation of histidine and aromatic amino acids found, the latter indicating that L. fermentum 222 is unable of biogenic amine production. Further, evidence was found for the presence of other amino acid conversion pathways, such as the occurrence of genes encoding cystathionine β-lyase (cbl) that is involved in methionine catabolism, serine deaminase (sda) that enables the production of pyruvate and ammonia from serine, and serine hydroxymethyltransferase (shm) that is involved in the production of the flavour compound acetaldehyde (Fig. 2; Additional file 3).
The genome sequence of L. fermentum 222 contained an operon encoding the arginine deiminase (ADI) pathway, consisting of genes encoding arginine deiminase (arcA), ornithine transcarbamoyltransferase (arcB), carbamate kinase (arcC), an arginine/ornithine antiporter (arcD), and a transaminase (arcT; Fig. 2; Additional file 3). This pathway might provide L. fermentum 222 with additional energy, provided through amino acid conversions, as well as protection against acid stress conditions [63, 64] and has been shown to be of importance for competitiveness in food fermentation processes [64–66].
Further, a gene encoding ornithine cyclodeaminase (ocd) was present, enabling direct biosynthesis of proline from ornithine, thereby producing ammonia (Fig. 2; Additional file 3). The production of additional ammonia may further protect against acid stress, whereas proline can act as an osmotic protectant [67].
Pathway analysis of Lactobacillus plantarum 80
Central carbohydrate metabolism
The genome sequence of L. plantarum 80 contained all genes encoding the enzymes involved in the homo- and heterolactic fermentation pathways (Fig. 2; Additional file 3) as well as various genes encoding carbohydrate uptake systems, enabling this strain to switch between both metabolic pathways, depending on the energy source available. Metabolic pathway analysis revealed that pyruvate can be converted into acetyl-CoA by a pyruvate dehydrogenase complex (pdhAC, lpdA) or by pyruvate-formate lyase (pfl), the latter yielding formate (Fig. 2; Additional file 3). Further, a gene encoding pyruvate oxidase (pox) was present, enabling L. plantarum 80 to convert pyruvate into carbon dioxide and acetyl phosphate under oxic conditions. Genes encoding α-acetolactate synthase (als) and α-acetolactate decarboxylase (aldB) were found, but diacetyl reductase- or 2,3-butanediol dehydrogenase-encoding genes were absent, indicating that acetoin would be the only flavour compound that can be formed out of pyruvate by L. plantarum 80, although 2,3-butanediol nor acetoin were found during monoculture fermentations with L. plantarum 80 in cocoa pulp simulation medium for LAB [26].
Genes associated with uptake and conversion mechanisms of citrate, such as a gene encoding citrate lyase (citDEF), oxaloacetate decarboxylating malate dehydrogenase (mae), a citrate transporter (citT), and accessory proteins (citGX), were all retrieved in the genome sequence of L. plantarum 80 (Fig. 2; Additional file 3). However, these were not clustered together (Fig. 3), as genes encoding transposases (LP80_92, LP80_93), an ABC transporter (mtsABC), a fumarase (fumC) and a fumarate reductase (frdC) were found between them. Further, a gene encoding a malate dehydrogenase (mdh) was found, which might enable L. plantarum 80 to use citrate as an alternative external electron acceptor by converting citrate into succinate by the reductive tricarboxylic acid cycle, next to its ability to convert citrate into pyruvate, which has been shown experimentally [27]. Lactobacillus plantarum 80 might be able to use oxygen as an alternative external electron acceptor, as a NADH-dependent oxidase-encoding gene (nox) was found. Genes associated with the ability to use glycerol or fructose as alternative external electron acceptors were absent.
The genome sequence of L. plantarum 80 harboured genes encoding a respiration machinery, which is an energetically favourable metabolic pathway that leads to less oxidative and acid stress in LAB [68]. Indeed, genes encoding a NADH-dependent dehydrogenase, a bd-type cytochrome (cydABCD), an ATP synthase (see above), and a heme-dependent catalase (katA; see above) were found (Fig. 2; Additional file 3). Although several L. plantarum strains possess a similar electron transport chain [69, 70], there are no experimental data indicating that L. plantarum 80 could perform aerobic respiration when provided with exogenous sources of heme and menaquinones.
Proteolytic system and amino acid conversion pathways
Genes encoding extracellular proteases were not found in the genome sequence of L. plantarum 80, which seems to be a general feature for L. plantarum strains [33]. Genes encoding a DtpT proton motive force-dependent peptide transporter and an Opp transporter (oppABCDF) were found as well as peptidase-encoding genes, including the genes pepD, pepE, and pepV encoding dipeptidases, the genes pepN and pepC encoding aminopeptidases, the genes pepX, pepR, and pepP encoding proline-specific peptidases, and the genes pepF, pepQ, pepO, and pepR encoding endopeptidases (Fig. 2; Additional file 3). This elaborate peptide uptake and degradation machinery might be an important mechanism for L. plantarum 80 to thrive in the protein-containing fermenting cocoa pulp-bean mass. Further, several genes encoding various amino acid transporters were present, such as livABCDE, enabling L. plantarum 80 to take up a variety of amino acids from the environment.
The genome sequence of L. plantarum 80 contained genes encoding an asparaginase (ans), an aspartate transaminase (aspC), and an aspartase (aspA; Fig. 2; Additional file 3). This pathway enables production of oxaloacetate and/or fumarate, which can subsequently be used for energy production and/or NAD+ regeneration by the enzymes encoded in the citrate operon (Fig. 3). Although a gene encoding glutamate decarboxylase was found (gadB), this pathway was unlikely to be operational, as the genes encoding the glutamate-γ-aminobutyric acid antiporter (gadC) and the transcriptional regulator (gadR) were not found, which are required for glutamate decarboxylation activity in other LAB [71].
A gene encoding the α-oxoglutarate-dependent aminotransferase BcaT was found in the genome sequence of L. plantarum 80, enabling the production of α-oxoacids from branched-chain amino acids. Further degradation of the α-oxoacids into hydroxyacids by this strain is feasible, as the genome sequence harboured a gene encoding a 2-hydroxyacid dehydrogenase (Fig. 2; Additional file 3). Enzymes involved in the conversion of α-oxoacids by oxidative decarboxylation into carboxylic acids or by decarboxylation into aldehydes were not found in the genome sequence. Further, the aromatic aminotransferase AraT was present in the genome sequence, which enables L. plantarum 80 to transaminate aromatic amino acids. In addition, aromatic amino acid decarboxylation enzymes were not found, indicating that L. plantarum 80 is not able to produce biogenic amines.
Genes encoding cystathionine β-lyase (metC) and cysteine kinase (cysK) were found in the same locus, which indicated that the methionine and cysteine catabolic pathways of L. plantarum 80 are linked, as has been shown previously for other LAB [72, 73]. Further, genes associated with the catabolism of serine and threonine, i.e., a serine deaminase (sda) and a serine hydroxymethyltransferase (shm), were retrieved in the genome sequence of L. plantarum 80 (Fig. 2; Additional file 3). Genes involved in histidine decarboxylation or the ADI pathway were absent.
Comparative genome sequence analysis
Comparative genomics of Lactobacillus fermentum 222
The genome sequence of L. fermentum 222 harboured 182 strain-specific genes, i.e., genes without a homolog in other known L. fermentum genome sequences (Additional file 1). Most of these genes encoded hypothetical proteins or were disrupted, although some had a predicted function. The strain-specific genes included two putative citrate transporters (LFER_11 and LFER_1857, discussed above) and a gene cluster (LFER_810-LFER_816) containing a diacetyl reductase-encoding gene (LFER_814; Fig. 2; Additional file 3). The complete cit gene cluster that was not interrupted by other genes or transposable elements (discussed above) and the occurrence of two unique additional (putative) citrate transporters might be the result of specific adaptations that enable this strain to consume citrate in an efficient way during the cocoa bean fermentation process [25, 74]. Further, a gene cluster involved in amino acid uptake and catabolism, consisting of genes coding for an amino acid transporter (LFER_135), an aspartase (LFER_136, discussed above), an amino acid permease (LFER_137), and an aminotransferase (LFER_138, discussed above) was unique for L. fermentum 222, providing evidence that amino acid conversion pathways might be an important feature for functional starter culture strains that have to improve the cocoa bean fermentation process.
The core genome of the species L. fermentum, based on eleven complete and draft genome sequences available (Additional file 1), consisted of 400 genes. The pan-genome of the species L. fermentum was found to encompass 4,475 genes and seemed to be open, i.e., the addition of more strains in the comparative analysis would result in a larger pan-genome [75]. The latter result was in agreement with the fact that this species has been found in various environments, including mammalian body sites [28, 31, 76] and food (fermentation) environments [2, 30, 31]. A phylogenetic tree based on the core gene sequences revealed that L. fermentum 222 was not grouped together with any other L. fermentum strain (Fig. 4), which might indicate its specific adaptation to the cocoa bean fermentation ecosystem.
Fig. 4.

Phylogenetic tree based on all core gene sequences of Lactobacillus fermentum available. Multiple sequence alignments of concatenated core gene sequences were calculated using the EDGAR framework, including plasmid sequences. The genome sequence of L. plantarum 80 was used as an outgroup
Comparative genomics of Lactobacillus plantarum
A total of 223 strain-specific genes were found in the genome sequence of L. plantarum 80 compared to the currently available L. plantarum genome sequences (Additional file 2). Among these strain-specific genes was a gene cluster (LP80_2381-LP80_2387) consisting of the PTS-encoding sorABCDE genes, a dehydrogenase-encoding gene (sorF), and a gene encoding the transcriptional regulator (sorR). In Lactobacillus casei, this gene cluster is involved in transport of sorbose and subsequent production of fructose 6-phosphate, which can be channeled into the glycolytic pathway [77]. This mechanism can also be used to transport fructose, as shown for L. casei and Klebsiella pneumoniae [78]. These genes might thus offer L. plantarum 80 unique features to be able to dominate the fructose-containing cocoa pulp-bean mass during cocoa bean fermentation. Furthermore, a unique cluster of four genes was found, consisting of an oxidoreductase (LP80_2995), a ribose 5-phosphate isomerase (LP80_2996), a fructose-specific PTS (LP80_2997), and a transcriptional regulator (LP80_2998). These traits underline the fructophilic character of L. plantarum 80 during cocoa bean fermentation processes [26].
Based on a comparative analysis of 25 L. plantarum genome sequences (Additional file 2), the core- and pan-genome of this species encompassed 877 and 8,657 genes, respectively. Construction of a phylogenetic tree based on core gene sequences only (Fig. 5) revealed that two strains (L. plantarum EGD-AQ4 and L. plantarum AY01) were not part of a larger cluster that contained all other strains. These two strains originated both from fermented food products [38, 45]. Although the phylogenetic distance between the strains was small (Fig. 5), the cocoa bean fermentation isolate L. plantarum 80 was grouped together with L. plantarum Lp90, an Italian wine isolate [79]. The latter strain possesses the ADI pathway [80] and is tolerant to wine fermentation conditions, such as high sulphite concentrations [81].
Fig. 5.

Phylogenetic tree based on all core gene sequences of Lactobacillus plantarum available. Multiple sequence alignments of concatenated core gene sequences were calculated using the EDGAR framework, including plasmid sequences. The genome sequence of L. fermentum 222 was used as an outgroup
Conclusions
Genomics-based pathway reconstruction of the carbohydrate metabolism and amino acid conversion mechanisms of candidate functional LAB starter culture strains for the cocoa bean fermentation process revealed important insights into their lifestyle, niche adaptations, and metabolic capacities. Although some studies report on comparative genomics of LAB [32, 59, 82], this is the first extensive (comparative) genome sequence analysis of these species, revealing their core- and pan-genome. Identification of the genetic potential of cocoa-derived candidate functional starter cultures obtained from this and other studies [27] in metagenomic data sets will allow further insights into the contribution of functional starter culture strains to cocoa bean fermentation processes [83]. This information is crucial for an improved and rationalised selection of functional starter culture strains for the cocoa bean fermentation process.
Methods
Bacterial strains and growth conditions
Lactobacillus fermentum 222 and L. plantarum 80 were originally isolated from a spontaneous cocoa bean heap fermentation process carried out in Ghana [7]. The strains were stored at −80 °C in de Man-Rogosa-Sharpe (MRS) medium (Oxoid, Basingstoke, United Kingdom), supplemented with 25 % (v/v) glycerol as a cryoprotectant. To obtain cell pellets, the pure strains were grown overnight in MRS medium at 37 °C, followed by cell harvesting through centrifugation (21,036 × g, 15 min, 4 °C) of 2-ml cultures.
DNA extraction and 454 pyrosequencing
Total genomic DNA was extracted from four cell pellets of both L. fermentum 222 and L. plantarum 80, using a previously described protocol with modifications [84]. A thawed pellet was washed in 1 ml of TES buffer [50 mM Tris base, 1 mM ethylene diamine tetra-acetic acid (EDTA), 6.7 % (m/v) sucrose, pH 8.0] and centrifuged at 2,795 × g for 10 min at 4 °C. The resulting pellet was collected and 300 μl of STET buffer [50 mM Tris base, 50 mM EDTA, 8 % (m/v) sucrose, 5 % (m/v) Triton X-100, pH 8.0] was added. The suspension was incubated in the presence of lysozyme (VWR International, Darmstadt, Germany), which was dissolved in TES buffer in a final concentration of 5 U/μl, at 37 °C for 1 h. Thereafter, a chemical treatment with a preheated (37 °C) solution of 20 % (m/v) sodium dodecyl sulphate (SDS; VWR International) in TE buffer (50 mM Tris base, 1 mM EDTA, pH 8.0) and a mechanical treatment with a pinch of glass beads (150–210 μm; Sigma-Aldrich, Steinheim, Germany) was applied, by vortexing the lysate for 60 s and subsequently incubating the mixture at 37 °C for 10 min, followed by a 10-min treatment at 65 °C to inactivate the enzymes. This lysate was extracted with one volume of phenol/chloroform/isoamylalcohol (49.5:49.5:1.0; Sigma-Aldrich). Phases were separated by centrifugation (18,900 × g, 5 min) using Phase Lock Gel™ tubes (Eppendorf AG, Hamburg, Germany). DNA was precipitated with 1 ml of isopropanol and 70 μl of NaCl (5 M) on ice and was collected by centrifugation (22,000 × g, 30 min, 4 °C). The resulting DNA pellet was washed with 500 μl of a 70 % (v/v) ethanol solution, dried under vacuum, and resuspended in 50 μl of TE buffer. Two μl RNase (10 mg/ml; Sigma-Aldrich) was added, followed by incubation at 37 °C for 10 min. For each strain, the four solutions were pooled and the DNA precipitation and washing steps were repeated. To confirm the identity of the bacterial strains grown, the near full-length 16S rRNA gene was amplified based on the genomic DNA extracted, purified, and sequenced, as described previously [85]. The quality of the genomic DNA was assessed by gel electrophoresis; its quantity was estimated by a fluorescence-based method using the Quant-iT dsDNA assay kit (Invitrogen, Carlsbad, CA, USA) and the DTX800 multimode detector (Beckman Coulter, Pasadena, CA, USA). Next, the isolated genomic DNA was used as template for shotgun pyrosequencing on a genome sequencer (GS) FLX system (Roche Applied Science, Mannheim, Germany) using Titanium chemistry, which was performed by a commercial facility (VIB Nucleomics Core, Leuven, Belgium). A DNA library was constructed for each strain according to the GS FLX rapid library preparation kit (Roche Applied Science). The optimal DNA copy per bead ratio was determined by an emulsion PCR titration, using a GS FLX Titanium SV emPCR kit (Lib-L; Roche Applied Science). A final emulsion PCR for the sequencing runs was performed using the GS FLX Titanium LV emPCR kit (Lib-L; Roche Applied Science). One pyrosequencing run was carried out with the DNA libraries, with each strain occupying half a PicoTiterPlate.
Genome sequence assembly and annotation
Reads were assembled using Newbler version 2.7 (Roche Applied Science) with default parameters. Automated gene prediction and annotation of the assembled contigs longer than 500 bp was carried out using a local installation of the bacterial genome sequence annotation system GenDB version 2.2, as described previously [27]. Briefly, GLIMMER 2.1 [86] and CRITICA [87] were applied to predict the gene sequences present. Ribosomal binding sites were identified with RBSfinder [88] and tRNA genes were searched for using tRNAscan-SE [89]. The proteins deduced were functionally characterised by REGANOR [90] using automated searches in public databases, including SWISS-PROT [91], Pfam [92], KEGG [93], and TIGRFAM [94]. Additionally, SignalP [95], helix-turn-helix [96], and TMHMM [97] were applied to find signal peptides, helix-turn-helix DNA binding motifs, and transmembrane regions, respectively. Each gene was functionally classified by assigning a cluster of orthologous groups (COG) and a gene ontology (GO) number [98, 99]. To further supplement the annotation, plasmids were identified using the PATRIC database plasmid_seq, virulence factors were searched for by BLASTn (based on nucleotide sequence) analysis using the human pathogenic bacteria virulence factor database (VFDB; [100]), antibiotic resistance genes were searched for by BLASTn analysis using the antibiotic resistance gene annotation database (ARG-ANNOT; [101]), CRISPRs were searched for with CRISPRFinder [102] and CRT [103], prophage sequences were identified by PHAST analysis [104], and BAGEL3 [105] was used to find genes encoding bacteriocins. Based on these annotations, metabolic pathways were reconstructed.
Data availability
The annotated genome sequences were deposited in the DDBJ/EMBL/GenBank database as sequencing project PRJEB5182 with EMBL accession numbers CBZV010000001-CBZV010000073 for L. fermentum 222 and sequencing project PRJEB5195 with EMBL accession numbers CBZW010000001-CBZW010000067 for L. plantarum 80.
Comparative genome sequence analysis
Comparative analysis of the genome sequences of L. fermentum 222 and L. plantarum 80 with available complete and draft genome sequences of strains of the same species (Additional files 1 and 2) was performed by the EDGAR framework [106] using default parameters. This included identification and classification of orthologous genes, identification of strain-specific genes, and calculation of the pan- and core-genomes. Also within this framework, phylogenetic analyses were performed using PHYLIP [107], thereby relying on an alignment of the core genes of a selection of complete and draft genome sequences of members of the species L. fermentum (Additional file 1) and L. plantarum (Additional file 2), that was generated using MUSCLE [108] and GBLOCKS [109]. As the publicly available draft genome sequence of L. fermentum FTDC 8312 was not annotated, its genome sequence was retrieved from the NCBI data repository, followed by automated annotation using the GenDB platform as described above. For the phylogenetic analysis of the species L. fermentum, L. plantarum 80 was used as an outgroup; for the phylogenetic analysis of the species L. plantarum, L. fermentum 222 was used as an outgroup.
Acknowledgements
The authors thank Dr. Jochen Blom and Prof. Dr. Alexander Goesmann (Institute of Bioinformatics and Systems Biology, Justus-Liebig-University, Giessen, Germany) for performing the EDGAR computational analyses and the VIB Nucleomics core for carrying out the 454 pyrosequencing. Part of this research was financed by the Research Council of the Vrije Universiteit Brussel (OZR, GOA, SRP, and IOF projects). The authors further acknowledge their finances from the Research Foundation Flanders (FWO-Vlaanderen) and the Hercules foundation. KI was the receiver of a post-graduate grant of the Agency for Innovation by Science and Technology (IWT). The funders had no role in the design of the study, the collection and analysis of the data, the writing of the manuscript, or the decision to submit the manuscript for publication.
Additional files
Overview of strains of Lactobacillus fermentum that have a complete or draft genome sequence publicly available and that were included in the comparative genome sequence analysis performed using the EDGAR framework. (DOCX 12 kb)
Overview of strains of Lactobacillus plantarum that have a complete or draft genome sequence publicly available and that were included in the comparative genome sequence analysis performed using the EDGAR framework [ 110 - 111 ]. (DOCX 14 kb)
Enzymes involved in key metabolic pathways of Lactobacillus fermentum 222 and L. plantarum 80. (DOCX 20 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KI carried out DNA extraction, conducted genome assembly, performed sequence annotation, bioinformatics analyses, and comparative genome analyses. SW and LDV designed and coordinated the study and participated in the analysis of the results. KI, SW, and LDV wrote the manuscript. All authors read and approved the final manuscript.
Authors’ information
Not applicable.
Availability of data and materials
Not applicable.
Contributor Information
Koen Illeghems, Email: killeghe@vub.ac.be.
Luc De Vuyst, Email: ldvuyst@vub.ac.be.
Stefan Weckx, Phone: +32 2 6293863, Email: stweckx@vub.ac.be, Email: Stefan.Weckx@vub.ac.be.
References
- 1.Bourdichon F, Casaregola S, Farrokh C, Frisvad JC, Gerds ML, Hammes WP, Harnett J, Huys G, Laulund S, Ouwehand A, et al. Food fermentations: microorganisms with technological beneficial use. Int J Food Microbiol. 2012;154:87–97. doi: 10.1016/j.ijfoodmicro.2011.12.030. [DOI] [PubMed] [Google Scholar]
- 2.Hutkins RW. Microbiology and technology of fermented foods. 1. Oxford: Blackwell Publishing; 2006. [Google Scholar]
- 3.Giraffa G, Chanishvili N, Widyastuti Y. Importance of lactobacilli in food and feed biotechnology. Res Microbiol. 2010;161:480–487. doi: 10.1016/j.resmic.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Holzapfel WH, Wood BJB. Lactic acid bacteria: biodiversity and taxonomy. 1. Hoboken: Wiley; 2014. [Google Scholar]
- 5.Leroy F, De Vuyst L. Lactic acid bacteria as functional starter cultures for the food fermentation industry. Trends Food Sci Tech. 2004;15:67–78. doi: 10.1016/j.tifs.2003.09.004. [DOI] [Google Scholar]
- 6.De Vuyst L, Lefeber T, Papalexandratou Z, Camu N. The functional role of lactic acid bacteria in cocoa bean fermentation. In: Mozzi F, Raya RR, Vignolo GM, editors. Biotechnology of lactic acid bacteria: novel applications. Ames: Wiley-Blackwell; 2010. pp. 301–325. [Google Scholar]
- 7.Camu N, De Winter T, Verbrugghe K, Cleenwerck I, Vandamme P, Takrama JS, Vancanneyt M, De Vuyst L. Dynamics and biodiversity of populations of lactic acid bacteria and acetic acid bacteria involved in spontaneous heap fermentation of cocoa beans in Ghana. Appl Environ Microbiol. 2007;73:1809–1824. doi: 10.1128/AEM.02189-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Camu N, González Á, De Winter T, Van Schoor A, De Bruyne K, Vandamme P, Takrama JS, Addo SK, De Vuyst L. Influence of turning and environmental contamination on the dynamics of populations of lactic acid and acetic acid bacteria involved in spontaneous cocoa bean heap fermentation in Ghana. Appl Environ Microbiol. 2008;74:86–98. doi: 10.1128/AEM.01512-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garcia-Armisen T, Papalexandratou Z, Hendryckx H, Camu N, Vrancken G, De Vuyst L, Cornelis P. Diversity of the total bacterial community associated with Ghanaian and Brazilian cocoa bean fermentation samples as revealed by a 16S rRNA gene clone library. Appl Microbiol Biotechnol. 2010;87:2281–2292. doi: 10.1007/s00253-010-2698-9. [DOI] [PubMed] [Google Scholar]
- 10.Papalexandratou Z, Camu N, Falony G, De Vuyst L. Comparison of the bacterial species diversity of spontaneous cocoa bean fermentations carried out at selected farms in Ivory Coast and Brazil. Food Microbiol. 2011;28:964–973. doi: 10.1016/j.fm.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 11.Papalexandratou Z, Falony G, Romanens E, Jimenez JC, Amores F, Daniel H-M, De Vuyst L. Species diversity, community dynamics, and metabolite kinetics of the microbiota associated with traditional Ecuadorian spontaneous cocoa bean fermentations. Appl Environ Microbiol. 2011;77:7698–7714. doi: 10.1128/AEM.05523-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papalexandratou Z, Vrancken G, De Bruyne K, Vandamme P, De Vuyst L. Spontaneous organic cocoa bean box fermentations in Brazil are characterized by a restricted species diversity of lactic acid bacteria and acetic acid bacteria. Food Microbiol. 2011;28:1326–1338. doi: 10.1016/j.fm.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 13.Crafack M, Mikkelsen MB, Saerens S, Knudsen M, Blennow A, Lowor S, Takrama J, Swiegers JH, Petersen GB, Heimdal H, et al. Influencing cocoa flavour using Pichia kluyveri and Kluyveromyces marxianus in a defined mixed starter culture for cocoa fermentation. Int J Food Microbiol. 2013;167:103–116. doi: 10.1016/j.ijfoodmicro.2013.06.024. [DOI] [PubMed] [Google Scholar]
- 14.de Melo Pereira GV, Miguel MG, Ramos CL, Schwan RF. Microbiological and physicochemical characterization of small-scale cocoa fermentations and screening of yeast and bacterial strains to develop a defined starter culture. Appl Environ Microbiol. 2012;78:5395–5405. doi: 10.1128/AEM.01144-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Melo Pereira GV, Magalhães KT, de Almeida EG, da Silva CI, Schwan RF. Spontaneous cocoa bean fermentation carried out in a novel-design stainless steel tank: influence on the dynamics of microbial populations and physical-chemical properties. Int J Food Microbiol. 2013;161:121–133. doi: 10.1016/j.ijfoodmicro.2012.11.018. [DOI] [PubMed] [Google Scholar]
- 16.Hamdouche Y, Guehi T, Durand N, Kedjebo KBD, Montet D, Meile JC. Dynamics of microbial ecology during cocoa fermentation and drying: towards the identification of molecular markers. Food Control. 2015;48:117–122. doi: 10.1016/j.foodcont.2014.05.031. [DOI] [Google Scholar]
- 17.Ho VTT, Zhao J, Fleet G. Yeasts are essential for cocoa bean fermentation. Int J Food Microbiol. 2013;174:72–87. doi: 10.1016/j.ijfoodmicro.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 18.Kostinek M, Ban-Koffi L, Ottah-Atikpo M, Teniola D, Schillinger U, Holzapfel W, Franz C. Diversity of predominant lactic acid bacteria associated with cocoa fermentation in Nigeria. Curr Microbiol. 2008;56:306–314. doi: 10.1007/s00284-008-9097-9. [DOI] [PubMed] [Google Scholar]
- 19.Lefeber T, Gobert W, Vrancken G, Camu N, De Vuyst L. Dynamics and species diversity of communities of lactic acid bacteria and acetic acid bacteria during spontaneous cocoa bean fermentation in vessels. Food Microbiol. 2011;28:457–464. doi: 10.1016/j.fm.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 20.Meersman E, Steensels J, Mathawan M, Wittocx P-J, Saels V, Struyf N, Bernaert H, Vrancken G, Verstrepen KJ. Detailed analysis of the microbial population in Malaysian spontaneous cocoa pulp fermentations reveals a core and variable microbiota. PLoS ONE. 2013;8:e81559. doi: 10.1371/journal.pone.0081559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nielsen DS, Teniola OD, Ban-Koffi L, Owusu M, Andersson TS, Holzapfel WH. The microbiology of Ghanaian cocoa fermentations analysed using culture-dependent and culture-independent methods. Int J Food Microbiol. 2007;114:168–186. doi: 10.1016/j.ijfoodmicro.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 22.Papalexandratou Z, Lefeber T, Bahrim B, Lee OS, Daniel H-M, De Vuyst L. Hanseniaspora opuntiae, Saccharomyces cerevisiae, Lactobacillus fermentum, and Acetobacter pasteurianus predominate during well-performed Malaysian cocoa bean box fermentations, underlining the importance of these microbial species for a successful cocoa bean fermentation process. Food Microbiol. 2013;35:73–85. doi: 10.1016/j.fm.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 23.Ardhana MM, Fleet GH. The microbial ecology of cocoa bean fermentations in Indonesia. Int J Food Microbiol. 2003;86:87–99. doi: 10.1016/S0168-1605(03)00081-3. [DOI] [PubMed] [Google Scholar]
- 24.Adler P, Bolten CJ, Dohnt K, Hansen CE, Wittmann C. Core fluxome and metafluxome of lactic acid bacteria under simulated cocoa pulp fermentation conditions. Appl Environ Microbiol. 2013;79:5670–5681. doi: 10.1128/AEM.01483-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lefeber T, Janssens M, Camu N, De Vuyst L. Kinetic analysis of strains of lactic acid bacteria and acetic acid bacteria in cocoa pulp simulation media toward development of a starter culture for cocoa bean fermentation. Appl Environ Microbiol. 2010;76:7708–7716. doi: 10.1128/AEM.01206-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lefeber T, Janssens M, Moens F, Gobert W, De Vuyst L. Interesting starter culture strains for controlled cocoa bean fermentation revealed by simulated cocoa pulp fermentations of cocoa-specific lactic acid bacteria. Appl Environ Microbiol. 2011;77:6694–6698. doi: 10.1128/AEM.00594-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Illeghems K, De Vuyst L, Weckx S. Complete genome sequence and comparative analysis of Acetobacter pasteurianus 386B, a strain well-adapted to the cocoa bean fermentation ecosystem. BMC Genomics. 2013;14:526. doi: 10.1186/1471-2164-14-526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karlyshev AV, Raju K, Abramov VM. Draft genome sequence of Lactobacillus fermentum strain 3872. Genome Announc. 2013;1:e01006–13. doi: 10.1128/genomeA.01006-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grover S, Sharma VK, Mallapa RH, Batish VK. Draft genome sequence of Lactobacillus fermentum Lf1, an Indian isolate of human gut origin. Genome Announc. 2013;1:e00883–13. doi: 10.1128/genomeA.00883-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jayashree S, Pooja S, Pushpanathan M, Vishnu U, Sankarasubramanian J, Rajendhran J, Gunasekaran P. Genome sequence of Lactobacillus fermentum strain MTCC 8711, a probiotic bacterium isolated from yogurt. Genome Announc. 2013;1:e00770–13. doi: 10.1128/genomeA.00770-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiménez E, Langa S, Martín V, Arroyo R, Martín R, Fernández L, Rodríguez JM. Complete genome sequence of Lactobacillus fermentum CECT 5716, a probiotic strain isolated from human milk. J Bacteriol. 2010;192:4800. doi: 10.1128/JB.00702-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morita H, Toh H, Fukuda S, Horikawa H, Oshima K, Suzuki T, Murakami M, Hisamatsu S, Kato Y, Takizawa T, et al. Comparative genome analysis of Lactobacillus reuteri and Lactobacillus fermentum reveal a genomic island for reuterin and cobalamin production. DNA Res. 2008;15:151–161. doi: 10.1093/dnares/dsn009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kleerebezem M, Boekhorst J, van Kranenburg R, Molenaar D, Kuipers OP, Leer R, Tarchini R, Peters SA, Sandbrink HM, Fiers MWEJ, et al. Complete genome sequence of Lactobacillus plantarum WCFS1. Proc Natl Acad Sci U S A. 2003;100:1990–1995. doi: 10.1073/pnas.0337704100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ladero V, Alvarez-Sieiro P, Redruello B, del Rio B, Linares DM, Martin MC, Fernández M, Alvarez MA. Draft genome sequence of Lactobacillus plantarum strain IPLA 88. Genome Announc. 2013;1:e00524–13. doi: 10.1128/genomeA.00524-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grover S, Sharma VK, Mallapa RH, Batish VK. Draft genome sequence of Lactobacillus plantarum strain Lp91, a promising Indian probiotic isolate of human gut origin. Genome Announc. 2013;1:e00976–13. doi: 10.1128/genomeA.00976-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karlyshev AV, Abramov VM. Draft genome sequence of Lactobacillus plantarum 2165. Genome Announc. 2014;2:e01179–13. doi: 10.1128/genomeA.01179-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li X, Gu Q, Lou X, Zhang X, Song D, Shen L, Zhao Y. Complete genome sequence of the probiotic Lactobacillus plantarum strain ZJ316. Genome Announc. 2011;1:e00094–13. doi: 10.1128/genomeA.00094-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qureshi A, Itankar Y, Ojha R, Mandal M, Khardenavis A, Kapley A, Purohit HJ. Genome sequence of Lactobacillus plantarum EGD-AQ4, isolated from fermented product of northeast India. Genome Announc. 2014;2:e01122–13. doi: 10.1128/genomeA.01122-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Axelsson L, Rud I, Naterstad K, Blom H, Renckens B, Boekhorst J, Kleerebezem M, van Hijum S, Siezen RJ. Genome sequence of the naturally plasmid-free Lactobacillus plantarum strain NC8 (CCUG 61730) J Bacteriol. 2012;194:2391–2392. doi: 10.1128/JB.00141-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crowley S, Bottacini F, Mahony J, van Sinderen D. Complete genome sequence of Lactobacillus plantarum strain 16, a broad-spectrum antifungal-producing lactic acid bacterium. Genome Announc. 2013;1:e00533–13. doi: 10.1128/genomeA.00533-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Z-Y, Liu C, Zhu Y-Z, Zhong Y, Zhu Y-Q, Zheng H-J, Zhao G-P, Wang S-Y, Guo X-K. Complete genome sequence of Lactobacillus plantarum JDM1. J Bacteriol. 2009;191:5020–5021. doi: 10.1128/JB.00587-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.D’Auria G, Džunková M, Moya A, Tomáška M, Kološta M, Kmet V. Genome sequence of Lactobacillus plantarum 19L3, a strain proposed as a starter culture for Slovenská bryndza ovine cheese. Genome Announc. 2014;2:e00292–14. doi: 10.1128/genomeA.00292-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jang JY, Lim HI, Park HW, Choi H-J, Kim T-W, Kang M, Lee J-H. Draft genome sequence of Lactobacillus plantarum wikim18, isolated from Korean kimchi. Genome Announc. 2014;2:e00467–14. doi: 10.1128/genomeA.00467-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim E-K, Park YM, Lee OY, Lee W-J. Draft genome sequence of Lactobacillus plantarum strain WJL, a Drosophila gut symbiont. Genome Announc. 2013;1:e00937–13. doi: 10.1128/genomeA.00937-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li X-R, Gong F-M, Zheng H-J, Zhang Z-H, Luo Y-Y, Liu C-J. Draft genome sequence of Lactobacillus plantarum strain AY01, isolated from the raw material of fermented goat milk cheese. Genome Announc. 2013;1:e00737–13. doi: 10.1128/genomeA.00737-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Naz S, Tareb R, Bernardeau M, Vaisse M, Lucchetti-Miganeh C, Rechenmann M, Vernoux J-P. Genome sequence of Lactobacillus plantarum strain UCMA 3037. Genome Announc. 2013;1:e00251–13. doi: 10.1128/genomeA.00251-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laursen MF, Bahl MI, Licht TR, Gram L, Knudsen GM. A single exposure to a sublethal pediocin concentration initiates a resistance-associated temporal cell envelope and general stress response in Listeria monocytogenes. Environ Microbiol. 2014 doi: 10.1111/1462-2920.12534. [DOI] [PubMed] [Google Scholar]
- 48.Christensen JE, Dudley EG, Pederson JA, Steele JL. Peptidases and amino acid catabolism in lactic acid bacteria. Antonie Van Leeuwenhoek. 1999;76:217–246. doi: 10.1023/A:1002001919720. [DOI] [PubMed] [Google Scholar]
- 49.Fernández M, Zúñiga M. Amino acid catabolic pathways of lactic acid bacteria. Crit Rev Microbiol. 2006;32:155–183. doi: 10.1080/10408410600880643. [DOI] [PubMed] [Google Scholar]
- 50.Liu M, Nauta A, Francke C, Siezen RJ. Comparative genomics of enzymes in flavor-forming pathways from amino acids in lactic acid bacteria. Appl Environ Microbiol. 2008;74:4590–4600. doi: 10.1128/AEM.00150-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pfeiler EA, Klaenhammer TR. The genomics of lactic acid bacteria. Trends Microbiol. 2007;15:546–553. doi: 10.1016/j.tim.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 52.Wang Y, Chen C, Ai L, Zhou F, Zhou Z, Wang L, Zhang H, Chen W, Guo B. Complete genome sequence of the probiotic Lactobacillus plantarum ST-III. J Bacteriol. 2011;193:313–314. doi: 10.1128/JB.01159-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Diep DB, Straume D, Kjos M, Torres C, Nes IF. An overview of the mosaic bacteriocin pln loci from Lactobacillus plantarum. Peptides. 2009;30:1562–1574. doi: 10.1016/j.peptides.2009.05.014. [DOI] [PubMed] [Google Scholar]
- 54.Navarro L, Rojo-Bezares B, Sáenz Y, Díez L, Zarazaga M, Ruiz-Larrea F, Torres C. Comparative study of the pln locus of the quorum-sensing regulated bacteriocin-producing L. plantarum J51 strain. Int J Food Microbiol. 2008;128:390–394. doi: 10.1016/j.ijfoodmicro.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 55.Anderssen EL, Diep DB, Nes IF, Eijsink VGH, Nissen-Meyer J. Antagonistic activity of Lactobacillus plantarum C11: two new two-peptide bacteriocins, plantaricins EF and JK, and the induction factor plantaricin A. Appl Environ Microbiol. 1998;64:2269–2272. doi: 10.1128/aem.64.6.2269-2272.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cotter PD, Hill C. Surviving the acid test: responses of Gram-positive bacteria to low pH. Microbiol Mol Biol Rev. 2003;67:429–453. doi: 10.1128/MMBR.67.3.429-453.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frees D, Savijoki K, Varmanen P, Ingmer H. Clp ATPases and ClpP proteolytic complexes regulate vital biological processes in low GC, Gram-positive bacteria. Mol Microbiol. 2007;63:1285–1295. doi: 10.1111/j.1365-2958.2007.05598.x. [DOI] [PubMed] [Google Scholar]
- 58.De Angelis M, Gobbetti M. Stress responses of lactobacilli. In: Tsakalidou E, Papadimitriou K, editors. Stress responses of lactic acid bacteria. Heidelberg: Springer; 2011. pp. 219–249. [Google Scholar]
- 59.Wisselink HW, Weusthuis RA, Eggink G, Hugenholtz J, Grobben GJ. Mannitol production by lactic acid bacteria: a review. Int Dairy J. 2002;12:151–161. doi: 10.1016/S0958-6946(01)00153-4. [DOI] [Google Scholar]
- 60.Makarova K, Slesarev A, Wolf Y, Sorokin A, Mirkin B, Koonin E, Pavlov A, Pavlova N, Karamychev V, Polouchine N, et al. Comparative genomics of the lactic acid bacteria. Proc Natl Acad Sci U S A. 2006;103:15611–15616. doi: 10.1073/pnas.0607117103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kieronczyk A, Skeie S, Langsrud T, Le Bars D, Yvon M. The nature of aroma compounds produced in a cheese model by glutamate dehydrogenase positive Lactobacillus INF15D depends on its relative aminotransferase activities towards the different amino acids. Int Dairy J. 2004;14:227–235. doi: 10.1016/j.idairyj.2003.07.001. [DOI] [Google Scholar]
- 62.Ryu HW, Kang KH, Pan JG, Chang HN. Characteristics and glycerol metabolism of fumarate-reducing Enterococcus faecalis RKY1. Biotechnol Bioeng. 2001;72:119–124. doi: 10.1002/1097-0290(20010105)72:1<119::AID-BIT15>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 63.Cunin R, Glansdorff N, Pierard A, Stalon V. Biosynthesis and metabolism of arginine in bacteria. Microbiol Rev. 1986;50:314–352. doi: 10.1128/mr.50.3.314-352.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vrancken G, Rimaux T, Weckx S, De Vuyst L, Leroy F. Environmental pH determines citrulline and ornithine release through the arginine deiminase pathway in Lactobacillus fermentum IMDO 130101. Int J Food Microbiol. 2009;135:216–222. doi: 10.1016/j.ijfoodmicro.2009.07.035. [DOI] [PubMed] [Google Scholar]
- 65.Rimaux T, Rivière A, Illeghems K, Weckx S, De Vuyst L, Leroy F. Expression of the arginine deiminase pathway genes in Lactobacillus sakei is strain dependent and is affected by the environmental pH. Appl Environ Microbiol. 2012;78:4874–4883. doi: 10.1128/AEM.07724-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Champomier Vergès M-C, Zuñiga M, Morel-Deville F, Pérez-Martínez G, Zagorec M, Ehrlich SD. Relationships between arginine degradation, pH and survival in Lactobacillus sakei. FEMS Microbiol Lett. 1999;180:297–304. doi: 10.1016/S0378-1097(99)00492-9. [DOI] [PubMed] [Google Scholar]
- 67.Poolman B, Glaasker E. Regulation of compatible solute accumulation in bacteria. Mol Microbiol. 1998;29:397–407. doi: 10.1046/j.1365-2958.1998.00875.x. [DOI] [PubMed] [Google Scholar]
- 68.Lechardeur D, Cesselin B, Fernandez A, Lamberet G, Garrigues C, Pedersen M, Gaudu P, Gruss A. Using heme as an energy boost for lactic acid bacteria. Curr Opin Biotechnol. 2011;22:143–149. doi: 10.1016/j.copbio.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 69.Brooijmans RJW, de Vos WM, Hugenholtz J. Lactobacillus plantarum WCFS1 electron transport chains. Appl Environ Microbiol. 2009;75:3580–3585. doi: 10.1128/AEM.00147-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pedersen MB, Gaudu P, Lechardeur D, Petit M-A, Gruss A. Aerobic respiration metabolism in lactic acid bacteria and uses in biotechnology. Ann Rev Food Sci Tech. 2012;3:37–58. doi: 10.1146/annurev-food-022811-101255. [DOI] [PubMed] [Google Scholar]
- 71.Nomura M, Kobayashi M, Ohmomo S, Okamoto T. Inactivation of the glutamate decarboxylase gene in Lactococcus lactis subsp. cremoris. Appl Environ Microbiol. 2000;66:2235–2237. doi: 10.1128/AEM.66.5.2235-2237.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Altermann E, Russell WM, Azcarate-Peril MA, Barrangou R, Buck BL, McAuliffe O, Souther N, Dobson A, Duong T, Callanan M. Complete genome sequence of the probiotic lactic acid bacterium Lactobacillus acidophilus NCFM. Proc Natl Acad Sci U S A. 2005;102:3906–3912. doi: 10.1073/pnas.0409188102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fernández M, van Doesburg W, Rutten GAM, Marugg JD, Alting AC, van Kranenburg R, Kuipers OP. Molecular and functional analyses of the metC gene of Lactococcus lactis, encoding cystathionine β-lyase. Appl Environ Microbiol. 2000;66:42–48. doi: 10.1128/AEM.66.1.42-48.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moens F, Lefeber T, De Vuyst L. Oxidation of metabolites highlights the microbial interactions and role of Acetobacter pasteurianus during cocoa bean fermentation. Appl Environ Microbiol. 2014;80:1848–1857. doi: 10.1128/AEM.03344-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mira A, Martín-Cuadrado AB, D’Auria G, Rodríguez-Valera F. The bacterial pan-genome: a new paradigm in microbiology. Int Microbiol. 2010;13:45–57. doi: 10.2436/20.1501.01.110. [DOI] [PubMed] [Google Scholar]
- 76.Lin W-H, Yu B, Jang S-H, Tsen H-Y. Different probiotic properties for Lactobacillus fermentum strains isolated from swine and poultry. Anaerobe. 2007;13:107–113. doi: 10.1016/j.anaerobe.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 77.Yebra MJ, Pérez-Martínez G. Cross-talk between the L-sorbose and D-sorbitol (D-glucitol) metabolic pathways in Lactobacillus casei. Microbiology. 2002;148:2351–2359. doi: 10.1099/00221287-148-8-2351. [DOI] [PubMed] [Google Scholar]
- 78.Yebra MJ, Veyrat A, Santos MA, Pérez-Martínez G. Genetics of L-sorbose transport and metabolism in Lactobacillus casei. J Bacteriol. 2000;182:155–163. doi: 10.1128/JB.182.1.155-163.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Spano G, Chieppa G, Beneduce L, Massa S. Expression analysis of putative arcA, arcB and arcC genes partially cloned from Lactobacillus plantarum isolated from wine. J Appl Microbiol. 2004;96:185–193. doi: 10.1046/j.1365-2672.2003.02132.x. [DOI] [PubMed] [Google Scholar]
- 80.Spano G, Massa S, Arena ME, de Nadra MCM. Arginine metabolism in wine Lactobacillus plantarum: in vitro activities of the enzymes arginine deiminase (ADI) and ornithine transcarbamylase (OTCase) Ann Microbiol. 2007;57:67–70. doi: 10.1007/BF03175052. [DOI] [Google Scholar]
- 81.Fiocco D, Capozzi V, Pepe D, Crisetti E, Spano G. Sulphite stress induce small heat shock genes in wine Lactobacillus plantarum. Res J Microbiol. 2007;2:838–844. doi: 10.3923/jm.2007.838.844. [DOI] [Google Scholar]
- 82.Altermann E, Klaenhammer TR. Group-specific comparison of four lactobacilli isolated from human sources using differential BLAST analysis. Genes Nutr. 2011;6:319–340. doi: 10.1007/s12263-010-0191-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Illeghems K, Weckx S, De Vuyst L. Applying meta-pathway analyses through metagenomics to identify the functional properties of the major bacterial communities of a single spontaneous cocoa bean fermentation process sample. Food Microbiol. 2015;50:54–63. doi: 10.1016/j.fm.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 84.Gevers D, Huys G, Swings J. Applicability of rep-PCR fingerprinting for identification of Lactobacillus species. FEMS Microbiol Lett. 2001;205:31–36. doi: 10.1111/j.1574-6968.2001.tb10921.x. [DOI] [PubMed] [Google Scholar]
- 85.Ravyts F, Barbuti S, Frustoli MA, Parolari G, Saccani G, de Vuyst L, Leroy F. Competitiveness and antibacterial potential of bacteriocin-producing starter cultures in different types of fermented sausages. J Food Protect. 2008;71:1817–1827. doi: 10.4315/0362-028x-71.9.1817. [DOI] [PubMed] [Google Scholar]
- 86.Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27:4636–4641. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Badger JH, Olsen GJ. CRITICA: coding region identification tool invoking comparative analysis. Mol Biol Evol. 1999;16:512–524. doi: 10.1093/oxfordjournals.molbev.a026133. [DOI] [PubMed] [Google Scholar]
- 88.Suzek BE, Ermolaeva MD, Schreiber M, Salzberg SL. A probabilistic method for identifying start codons in bacterial genomes. Bioinformatics. 2001;17:1123–1130. doi: 10.1093/bioinformatics/17.12.1123. [DOI] [PubMed] [Google Scholar]
- 89.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Linke B, McHardy AC, Neuweger H, Krause L, Meyer F. REGANOR: a gene prediction server for prokaryotic genomes and a database of high quality gene predictions for prokaryotes. Appl Bioinformatics. 2006;5:193–198. doi: 10.2165/00822942-200605030-00008. [DOI] [PubMed] [Google Scholar]
- 91.Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O’Donovan C, Phan I, et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer ELL, et al. The Pfam protein families database. Nucleic Acids Res. 2004;32:D138–D141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Haft DH, Selengut JD, White O. The TIGRFAMs database of protein families. Nucleic Acids Res. 2003;31:371–373. doi: 10.1093/nar/gkg128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 96.Dodd IB, Egan JB. Improved detection of helix-turn-helix DNA-binding motif in protein sequences. Nucleic Acids Res. 1990;18:5019–5026. doi: 10.1093/nar/18.17.5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 98.Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, et al. The gene ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32:D258–D261. doi: 10.1093/nar/gkh066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen L, Xiong Z, Sun L, Yang J, Jin Q. VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 2012;40:D641–D645. doi: 10.1093/nar/gkr989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gupta SK, Padmanabhan BR, Diene SM, Lopez-Rojas R, Kempf M, Landraud L, Rolain J-M. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob Agents Chemother. 2013;58:212–220. doi: 10.1128/AAC.01310-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–W57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bland C, Ramsey T, Sabree F, Lowe M, Brown K, Kyrpides N, Hugenholtz P. CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics. 2007;8:209. doi: 10.1186/1471-2105-8-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39:W347–W352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.van Heel AJ, de Jong A, Montalbán-López M, Kok J, Kuipers OP. BAGEL3: automated identification of genes encoding bacteriocins and (non-)bactericidal posttranslationally modified peptides. Nucleic Acids Res. 2013;41:W448–W453. doi: 10.1093/nar/gkt391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Blom J, Albaum S, Doppmeier D, Puhler A, Vorholter F-J, Zakrzewski M, Goesmann A. EDGAR: a software framework for the comparative analysis of prokaryotic genomes. BMC Bioinformatics. 2009;10:154. doi: 10.1186/1471-2105-10-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Felsenstein J. PHYLIP (phylogeny inference package) version 3.695. Seattle: Department of Genome Sciences, University of Washington; 2013. [Google Scholar]
- 108.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 110.Newell PD, Chaston JM, Wang Y, Winans NJ, Sannino D, Wong AC, Dobson AJ, Kagle J, Douglas AE. In vivo function and comparative genomic analyses of the Drosophila gut microbiota identify candidate symbiosis factors. Front Microbiol. 2014;5:576. doi: 10.3389/fmicb.2014.00576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Siezen RJ, Francke C, Renckens B, Boekhorst J, Wels M, Kleerebezem M, van Hijum SAFT. Complete resequencing and reannotation of the Lactobacillus plantarum WCFS1 genome. J Bacteriol. 2012;194:195–196. doi: 10.1128/JB.06275-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The annotated genome sequences were deposited in the DDBJ/EMBL/GenBank database as sequencing project PRJEB5182 with EMBL accession numbers CBZV010000001-CBZV010000073 for L. fermentum 222 and sequencing project PRJEB5195 with EMBL accession numbers CBZW010000001-CBZW010000067 for L. plantarum 80.