

Abstract
Endometrial adenocarcinoma in the uterine corpus is a malignant cancer that occurs in menopausal women and aged rodents. Because of the similarities in pathogenesis and morphology of endometrial adenocarcinoma in rodents and humans, prediction of the modes of action (MOA) in uterine carcinogenesis is important for extrapolation of rodent data to humans. Three MOAs have been accepted as major pathways for uterine carcinogenesis in rodents: 1) estrogenic activity, 2) increased serum 17beta-estradiiol (E2) to progesterone (P4) ratio and 3) modulation of estrogen metabolism to produce 4-hydroxyestradiol via P450 induction. Inhibition of estrogen excretion and increased aromatase in situ in the tumor are also a potential pathway. Here, chemicals showing uterine carcinogenicity were chosen from approximately 300 pesticides evaluated in Japan within the past decade, and their mechanisms were predicted using parameters from mechanistic and toxicity studies. Seven pesticides increased uterine tumor formation in rats, and the pathways of 4 pesticides could be predicted based on various mechanistic studies. The MOAs of cyenopyrafen and benthiavalicarb-isopropyl were predicted to be modulation of estrogen metabolism, while those of pyriminobac-methyl and spirodiclofen were predicted to be increased E2 to P4 ratio. The driven pathways of metazosulfuron and isopyrazam could not be predicted using several mechanistic studies. No mechanistic studies have been reported for sedaxane, which has a chemical structure and toxicological profile similar to isopyrazam. Our results indicated that appropriate mechanistic studies are useful for mechanism prediction in risk assessment. From this analysis, a flowchart showing a decision tree for predictive MOAs in uterine carcinogenesis was proposed.
Keywords: mechanism, uterine carcinogenesis, prediction, pesticide
Introduction
Endometrial adenocarcinoma in the uterine corpus is a malignant cancer that occurs in menopausal women. Estrogen plays an essential role in the development of endometrial adenocarcinoma, particularly the endometrioid type, which is the most common tumor in the female genital system1. Endometrial adenocarcinoma is rare in rodents, and similar to be case in humans, estrogen has been shown to be crucial for uterine carcinogenesis in rodents2. Moreover, the developmental process, morphology, and molecular events in endometrial adenocarcinomas are highly similar between women and rodents1, 3. Endometrial adenocarcinomas develop morphologically through multiple stages in rats and women. In middle-aged female rats, atypical hyperplasia of endometrial epithelial cells occurs in the endometrium, with the number and size of such foci increasing over time4. In the next step, a well-differentiated endometrial adenocarcinoma with morphological similarities to severe atypical hyperplasia but featuring invasion into the muscular layer or spread to the serosa in the uteri can be found. With increasing morphological malignancy, such as cellular atypia or increased mitosis, the tumor spreads to the abdominal cavity or metastasizes to the lungs. Early molecular events have not been well defined in rodents; however, p53 mutations are not common and independence from estrogen receptor signaling is considered to be later events in rodents4 and endometrioid-type cancer in humans1.
In the pathogenesis or modes of action (MOAs) of endometrial adenocarcinoma, 3 major pathways have been considered in both women and rodents. First and second pathways are estrogens- and an increased ratio of 17beta-estradiol (E2) to progesterone (P4) ratio mediated pathways, respectively. In women, long-term treatment with estrogens and continuous increases in the ratio of estrogen to progesterone due to ovarian hormone imbalances are risk factors for endometrial cancer in the corpus1. Polycystic ovary syndrome (POS), a disorder occurring in the ovaries before middle age, results in an increased E2 to P4 ratio5. Recently, a cohort study in Denmark reported that women with POS had a 4-fold increase in the risk of endometrial cancer compared with women without POS6. In rodents, similar pathways to those in humans, including continuous treatment with estrogenic chemical and prolonged increases in the E2 to P4 ratio, resulting in persistent estrus by vaginal cytology, have been reported4. Modulation of estrogen metabolism through cytochrome P450 induction is a third common MOA occurring in both humans and rodents7, 8. In the liver and other tissues, estrogens are primarily metabolized to 2- or 4-hydroxylestradiol (2-HE, 4HE), which are defined as catechol estrogens, by estradiol 2- or 4-hydroxylase, respectively9. These hydroxylases are known to be altered by their corresponding drug metabolism enzymes; for example, CYP1B1 is known to convert E2 to 4HE in rats10 and humans11. Previous studies have demonstrated that catechol estrogens and/or changes in CYP1B1 are risk factors for endometrial cancer in humans12, 13. In rats, high-dose treatment with indole-3-carbinol, a cruciferous vegetable, has been reported to promote the development of endometrial adenocarcinomas through CYP1A1, CYP1A2, and CYP1B1 in the liver8. Consequently, increases in estrogen 4-hydroxylase expression in the liver indicate that estrogen metabolism is due to increased activity of the 4HE pathway. In rodents, 4HE is known to induce uterine cancer, although its estrogenic activity in vivo is weaker than the parent molecule8.
Furthermore, two other pathways may also lead to development of endometrial carcinoma. The first is increased blood E2 levels due to inhibitory excretion from the body by chemicals. Tetrabromobisphenol A (TBBPA), a brominated flame retardant, increases the incidence of uterine adenocarcinomas in rats in NTP 2-year bioassays14, and related mechanistic studies have indicated that saturation of elimination pathways of TBBPA may compete with estrogen metabolism, resulting in decreased excretion of E2 and increased E2 in the blood15. The second possibility is an in situ increase in aromatase caused by chemicals. While there is no clear evidence of this pathway in rodents, aromatase expression in endometrial adenocarcinomas has been reported in humans16. Aromatase is considered a key factor for mammary carcinogenesis. Therefore, in situ increases in aromatase may be involved in uterine carcinogenesis in rodents as well.
Pesticides as well as medical drugs are well-regulated chemicals; toxicological evaluations of these chemicals are required using a number of toxicity studies defined by the test guidelines of the responsible regulatory authorities. To protect the safety of consumers, all pesticides are required to be no genotoxic for humans by the authorities. The importance of MOA studies in toxicological evaluation of risk assessment of these chemicals is becoming apparent; indeed, if MOAs in nonclinical or toxicity/carcinogenicity studies, which are generally performed in rodents, are similar to those in humans, the toxicities/carcinogenicities of the chemicals could be evaluated as plausible/likely risks to human health. If any clear evidence showing differences between humans and experimental data can be provided, the MOA is not considered relevant to humans. On the other hand, as effective mechanistic or methodological studies to clarify MOAs have not been conducted, toxicological evaluations must have been determined based on conservative scenario. Therefore, the purpose of the present study was to predict MOAs in rodent uterine adenocarcinoma using published data from toxicological profiles of pesticides in rodents. We also proposed a flowchart showing a decision tree for predictive MOAs in uterine carcinogenesis.
Materials and Methods
Sources of published pesticide assessment reports
The data used in the present analysis were obtained from the risk assessment reports of approximately 300 pesticides evaluated by the Food Safety Commission (FSC) of Japan from 2003 to 2013 (published on their webpage)17. In these reports, we chose pesticides that increased the incidence of treatment-related endometrial adenocarcinomas in rodent carcinogenicity and/or chronic toxicity and carcinogenicity combined studies. After selecting the risk assessment reports from the FSC, available monographs of toxicological evaluations of these chemicals published by the Joint Meeting of Pesticides and Residues (JMPR) were also obtained using the JMPR database18.
Analysis of pesticides showing uterine carcinogenicity
For pesticides showing uterine carcinogenicity, chemical name (common name), pesticides classification (insecticide/fungicide/herbicide), chemical structure, mechanisms in target, toxicological profiles (toxicological targets, features of toxicity, acute toxicity), acceptable daily intake (ADI), lowest no-observed-adverse-effect-level (NOAEL), lowest low-observed-adverse-effect level (LOAEL), endpoints and corresponding study for setting ADI, and NOAEL/LOAEL for uterine carcinogenicity are listed.
Classification of MOAs of uterine carcinogenicity in rodents
The known or potential MOAs of rodent uterine carcinogenicity described in the introduction were classified into the following 5 types:
Type A: Estrogens/chemicals with estrogenic activity (Fig. 1). Any chemicals positive in a uterotrophic assay at the carcinogenic level are clearly recognized as chemicals with estrogenic activity. Estrogens have strong proliferative activity in estrogen-dependent tissues/organs. Uterotrophic assays are a highly sensitive method for detection of in vivo estrogenic activity. If the ovaries are not removed, the sensitivity for detection of activity becomes weaker. If the treatment level is enough to disrupt estrous cyclicity, vaginal cytology and morphologic features of estrogens/chemicals with estrogenic activity show persistent estrus (PE) and cornification, respectively. After disruption of cycling or anovulation occur, the morphological changes in the ovary are similar to those observed in the type B MOA (see below).
Fig. 1.
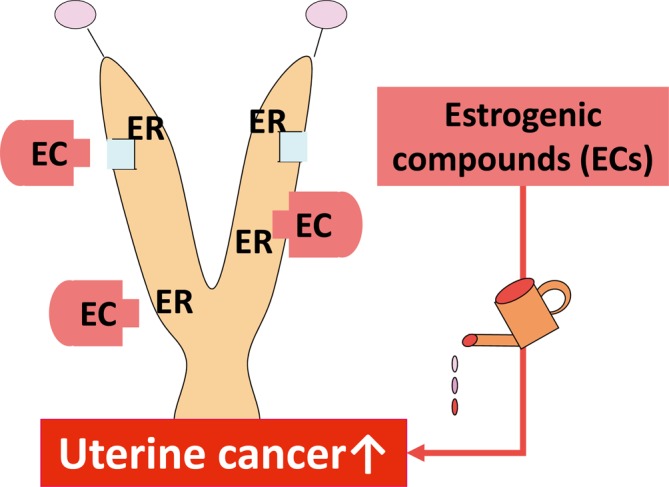
Predictive pathway of type A uterine tumor development. Estrogens/chemicals with estrogenic activity. ER, estrogen receptor.
Type B: Continuous increase of the E2 to P4 ratio (Fig. 2). Senescence or chemically induced imbalance in sex steroid hormones in the ovary leads to decreases in both E2 and P4 in the blood. When the decrease in P4 is predominant compared with that in E2, the E2 to P4 ratio is increased, resulting in a status similar to that of high E2. This status is manifested by PE in vaginal cytology, an atrophic ovary with cystic atretic follicles, lack of/few corpora lutea, cornification of the cervix/vaginal mucosa, and/or squamous metaplasia of endometrial epithelial cells. Atypical endometrial hyperplasia, a type of precancerous lesion, may be increased. An increased E2 to P4 ratio may be difficult to observe during short-term treatment. Estrous cycle-aware hormone measurement at middle age is useful to clarify.
Fig. 2.
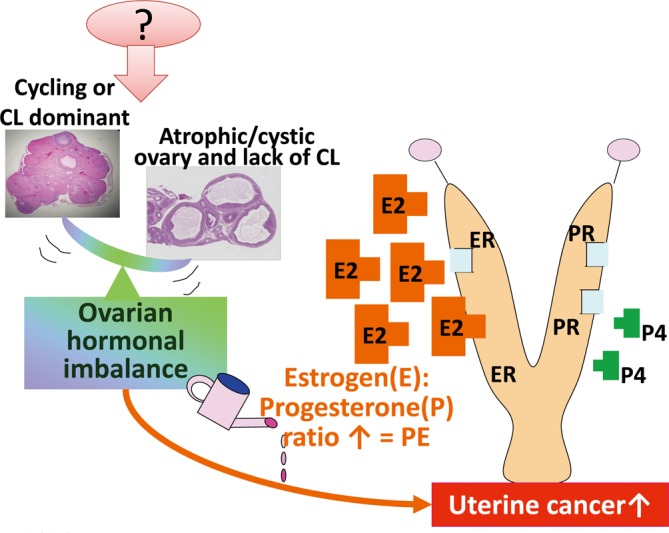
Predictive pathway of type B uterine tumor development. Increased ratio of E2 to P4. PE, persistent estrus; CL, corpus luteum; ER, estrogen receptor; PR, progesterone receptor.
Type C: Modulation of estrogen metabolism via induction of CYPs (Fig. 3). In type C, several key events should be checked. Analysis of increased 4HE in the blood is the most direct evidence for MOAs. Measurement of metabolic enzymes related to 4HE or 2HE in the liver or other tissues could be helpful. In addition, evaluation of the mRNA expression levels of genes related to the modulation of estrogen metabolism, such as CYP1B, would be useful. Liver hypertrophy may indicate induction of drug enzymes, including enzymes related to hydroxylation; however, this change is most commonly observed following induction by chemical treatment in rodents, and there are a number of drug metabolism-related enzymes in the liver. Therefore, while liver hypertrophy cannot be excluded, it is not an adequate indicator of a type C MOA. Moreover, in type C MOAs, no typical changes similar to those observed for types A and B, such as early occurrence of PE or morphological changes indicating increased E2 levels in the reproductive tract, are found in the female reproductive tract.
Fig. 3.
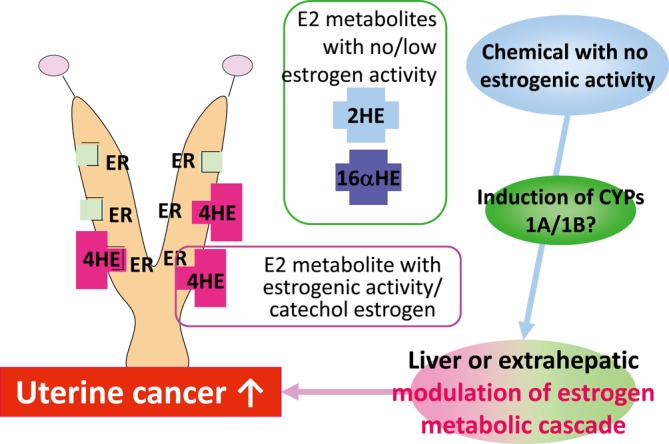
Predictive pathway of type C uterine tumor development. Modulation of estrogen metabolism via CYP induction. ER, estrogen receptor; 2HE, 2 hydroxyestradiol; 4HE, 4 hydroxyestradiol; 16α-E, 16α-hydroxyestradiol.
Type D: Decreased E2 excretion and increased E2 levels in the blood (Fig. 4). A little evidence has supported this MOA. In the case of TBBPA, absorption/distribution/metabolism/excretion (ADME) studies have been shown to be important13. High E2 levels in the blood provide strong evidence, but measurement of estrous cycle-dependent hormones is also necessary. If steroid hormones are measured in animals during the estrous cycle, E2 should be measured at baseline (i.e., estrus or metestrus) but not at the increased stages (i.e., proestrus or the later period of diestrus). Induction of phase 2 drug metabolism enzymes related to sulfoxylation is related to the MOA12; however, careful and adequate consideration should be needed to be interpreted because many chemicals induce expression of these enzymes. While continuously high E2 levels are predicted to result in morphological changes similar to those observed for type B MOAs, morphological changes and vaginal smears have not provided evidence yet for this MOA.
Fig. 4.
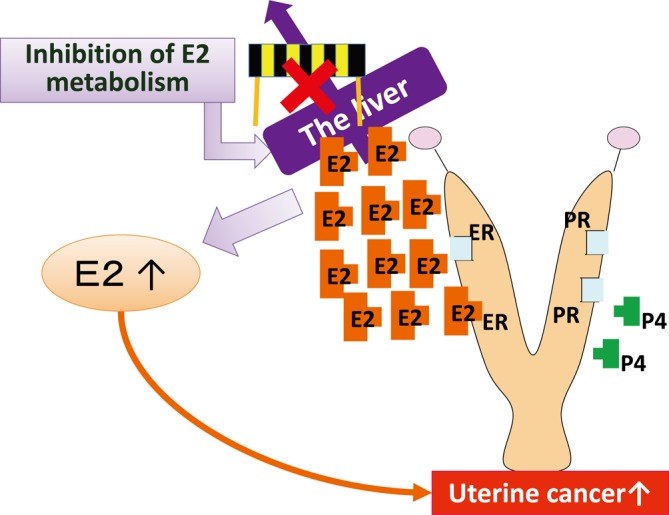
Predictive pathway of type D uterine tumor development. Inhibition of E2 excretion in the liver resulting in increased serum E2. PE, persistent estrus; CL, corpus luteum; ER, estrogen receptor; PR, progesterone receptor.
Type E: Increased in situ aromatase (Fig. 5). No studies have supported this MOA in rodents. In humans, increased protein and mRNA expression of aromatase has been demonstrated in epithelial stromal cells in endometrial carinomas14. While type E MOAs could potentially occur in rodents, more clear evidence is needed to show the mechanism of increased aromatase in situ.
Fig. 5.
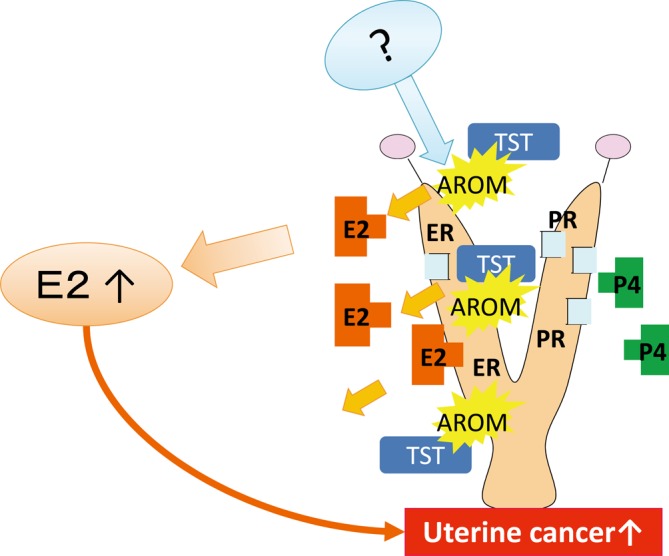
Predictive pathway of type E uterine tumor development. Increased aromatase in the uterus enhances the metabolism of testosterone (TST) to E2. PE, persistent estrus; CL, corpus luteum; ER, estrogen receptor; PR, progesterone receptor; AROM, aromatase.
Selection of parameters to be identified and MOA prediction
Parameters in mechanistic and/or toxicity studies related to mechanisms or MOAs of uterine carcinogenesis were selected, as follows: liver hypertrophy; induction of drug metabolism enzymes, including phases I and II; measurement of estrogen metabolism enzymes and/or catechol estrogens in the liver and/or other tissues; estrogenic activity measured using uterotrophic assays, estrogen receptor binding assays, or morphological evidence in the female reproductive tract indicating a high estrogen status; in situ expression of aromatase mRNA and/or protein by immunohistochemistry; hormone measurement (E2, P4 and possibly gonadotropins) and calculation of the E2 to P4 ratio; and metabolism studies or other related parameters, such as the timing of vaginal opening or estrous cycling in rat reproductive studies.
All changes listed above were reported in mechanistic studies of uterine carcinogenicity published in risk assessment reports or available references for the pesticides. After selection, we classified each pesticide having uterine carcinogenicity into the most appropriate predictive MOA based on the parameters identified for each pesticide.
Proposal of a flowchart diagram showing a decision tree for predictive MOAs in uterine carcinogenesis
Based on the analysis of pesticides with uterine carcinogenicity and classification for prediction of MOAs, we created a flowchart diagram showing a decision tree for the prediction of MOAs in uterine carcinogenesis. If possible, plausibility to human relevance was added to the chart.
Results
Seven pesticides showed treatment-related increases in endometrial adenocarcinomas. The increases were detected in combined chronic toxicity and carcinogenicity studies in rats. The strains in these studies were Wistar rats, with the exception of two studies of pyriminobac-methyl and benthiavalicarb-isopropyl, where Fischer rats were used. No increases were observed in mouse carcinogenicity studies. Of the 7 pesticides, 2, 3 and 2 were insecticides, fungicides, and herbicides, respectively (Table 1). The targets of each pesticide were as follows: pyriminobac-methyl and metazosulfuron, both herbicides, inhibited plant-specific acetolactate synthase (ALS); isopyrazam, sedaxane (both fungicides), and cyenopyrafen, an insecticide, inhibited mitochondrial electron transport, prevented cellular respiration and energy metabolism, and blocked ATP synthesis; and benthiavalicarb-isopropyl, a fungicide, and spirodiclofen, an insecticide, inhibited fatty acid biosynthesis. The chemical structures of these 7 chemicals were different, with the exception of isopyrazam and sedaxane, both of which had common chemical structures and functions as fungicides. The toxicological profiles of these 2 chemicals were similar; both chemicals were shown to induce liver hypertrophy and have secondary effects in the thyroid due to induction of hepatic drug enzymes induction, although there were discrepancies in some toxicological features, such as the effects on Leydig cells and reproductive toxicity. The toxicity profiles of the other chemicals varied.
Table 1. Pesticides Promoting Uterine Adenocarcinoma, their Toxicological Profiles and Comparisons of Doses for Setting ADIs as Uterine Carcinogens.
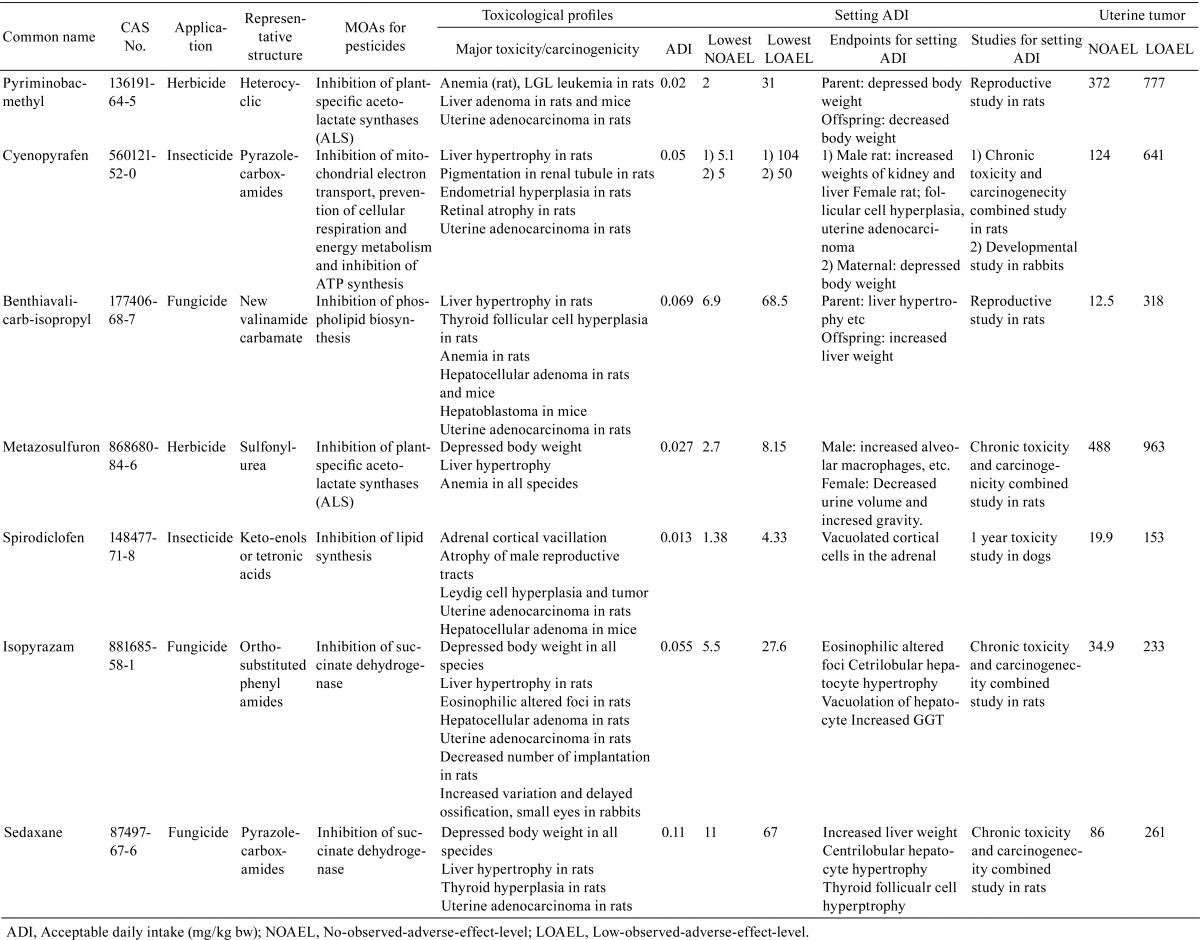
The doses showing uterine carcinogenicity were the highest doses tested in the chronic toxicity/carcinogenicity combined studies of 6 chemicals. In comparison with NOAELs/LOAELs for setting the ADI, there were no compounds where uterine carcinogenicity was an endpoint for setting the ADI. Benthiavalicarb-isopropyl had the smallest difference between the NOAEL for uterine carcinogenicity and the NOAEL for setting the ADI (approximately 2-fold difference). However the LOAEL for uterine carcinogenicity of this compound was relatively high at over 300 mg/kg body weight. For pyriminobac-methyl, a large difference between the NOAEL for uterine carcinogenicity and the NOAEL for setting the ADI (i.e., over 180-fold difference) was observed.
Possible endpoints involved in MOAs for uterine carcinogenicity were selected from the mechanistic and toxicity studies published in the risk assessment reports of the FSC (Table 2). Various types of mechanistic studies were conducted for 6 chemicals, i.e., pyriminobac-methyl, cyenopyrafen, benthiavalicarb-isopropyl, isopyrazam, metazosulfuron, and spirodiclofen. No mechanistic studies, except analysis of hepatic drug metabolism enzymes, were reported for sedaxane.
Table 2. Comparison of Parameters for Prediction of MOAs for Uterine Carcinogenicity.
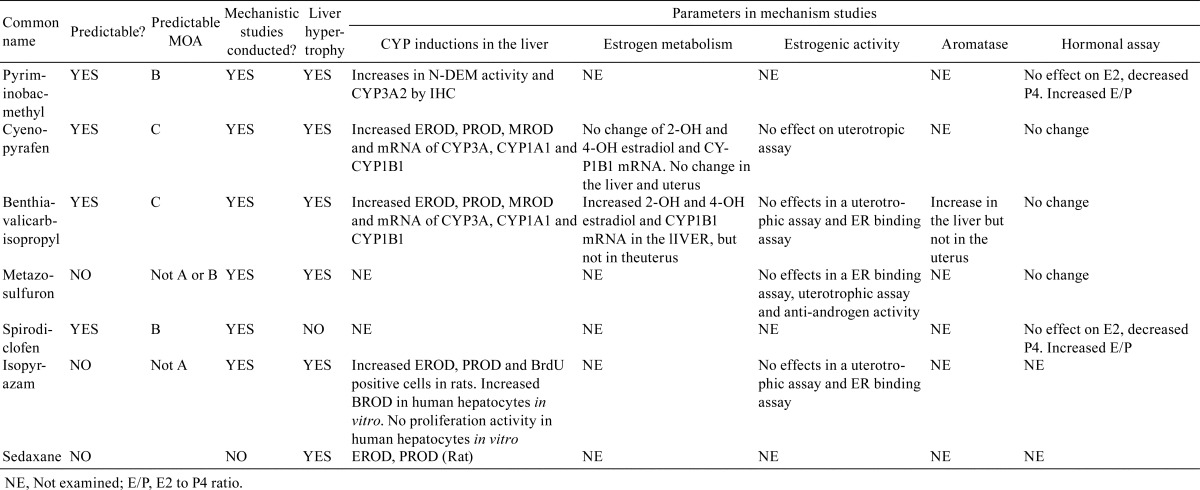
In the classification of pesticides into 5 different types of predictive MOAs, pyriminobac-methyl and spirodiclofen were classified as having type B MOAs because neither chemical elicited changes in E2 but both caused decreases in P4 in the blood, resulting in an increased E2 to P4 ratio. A number of mechanistic studies have been conducted for cyenopyrafen and benthiavalicarb-isopropyl. Cyenopyrafen was classified as having a type C MOA because of the following observations: induction of CYP1B1 protein and mRNA, increased 2- and 4-hydroxyestradiol in the liver, no estrogenic activity, and no hormonal changes; these alterations indicated that estrogen metabolism was shifted to increase 4HE production by CYP1B induction. Benthiavalicarb-isopropyl had effects similar to cyenopyrafen, except for the observation that aromatase activity was increased in the liver but not in the uterus. In mechanistic studies of metazosulfuron, no estrogenic activity in vivo or in vitro, no androgenic activity, and no changes in E2, P4, FSH/LH, and prolactin in the blood were observed. Isopyrazam also had no estrogenic activity in vivo or in vitro, as shown by the lack of reaction in uterotrophic assays and estrogen-receptor binding assays. No additional information was found. From these data, we could exclude types A and B MOAs and type A MOAs as MOAs of metazosulfuron and isopyrazam, respectively.
Next, we attempted to identify additional information to link to the 5 predictive MOAs described above in toxicological studies of the 7 pesticides. An increased incidence of cystic ovary was reported in the carcinogenicity study of metazosulfuron. Additionally, spirodiclofen increased the incidence of Leydig cell tumors at a dose that also induced uterine carcinogenicity. However, detailed information describing cell/tissue morphology or hormonal imbalances to support any of the predictive MOAs could not be obtained. All compounds except spirodiclofen induced hepatocellular hypertrophy.
Discussion
In the present study, we selected 7 pesticides exhibiting uterine carcinogenicity from approximately 300 pesticides evaluated in Japan from 2003 to 2013. We then predicted MOAs for induction of uterine carcinogenicity by these drugs and classified the MOAs into 5 types based on the evidence observed in mechanistic studies involving estrogen metabolism in the liver or uterus, induction of drug metabolism enzyme in the liver, hormone assay, or analysis of estrogenic activity. The reported mechanistic studies varied, and the types of studies were different depending on the specific compound. From our analysis, we were able to easily predict the MOAs of 4 compounds based on the endpoints of mechanistic studies, as follows: cyenopyrafen and benthiavalicarb-isopropyl had MOAs related to modulation of estrogen metabolism, and pyriminobac-methyl and spirodiclofen had MOAs related to increased E2 to P4 ratio. However, the MOAs of metazosulfuron and isopyrazam could not be defined, despite the fact that several mechanistic studies have been performed for these chemicals. If additional studies, such as analysis of estrogen metabolism by both compounds or hormonal assays for isopyrazam, had been reported, we would have been able to predict the MOAs for these compounds more definitively. Our results indicated that appropriate mechanistic studies were important for prediction of the MOAs of these chemicals with respect to uterine carcinogenicity.
In this study, we also identified parameters that were useful for prediction of different types of MOAs, as listed in Table 2; these parameters are expected to be applicable to prediction of the MOAs of chemicals during safety assessments of medical drugs and pesticides. Interestingly, centrilobular hepatocellular hypertrophy was observed in repeated oral toxicity studies of 6 of 7 pesticides This suggests that estrogen metabolism may be associated with induction of related CYP enzymes in the liver, but it also highlights that hepatocellular hypertrophy is the most common chemically induced alteration. Therefore, this information may provide additional insights, but it should not be considered definitive information for prediction19. Additional informative parameters for robust prediction of MOAs are needed.
We proposed 5 pathways as predictive MOAs for uterine adenocarcinoma development in rodents, and two pathways were found to be predictable in the present study. Regarding the predictability of other pathways, previous studies have clearly indicated that estrogens and/or chemicals with estrogenic activity increase the risk of uterine cancer in humans1 and rodents2, 20. Recent mechanistic studies of TBBPA have indicated the possibility that chemically induced inhibition of E2 excretion from the body results in increased serum E2, thereby promoting the development of uterine adenocarcinoma in rats. The relevance of this pathway in humans has not yet been established, and clarification of species differences in the toxicokinetics of these chemicals is crucial for confirmation of these results. However, it is possible that continuous increases in serum E2 levels through inhibition of E2 excretion may increase the risk of uterine cancer and may be a common MOA in humans. Thus, the predicted MOAs for these 4 compounds in rodents are considered to be feasible in humans. Although no evidence has shown that the risk of uterine adenocarcinoma increases due to in situ increases in aromatase expression in rodents, future studies should examine this possibility.
With regard to other possible factors affecting uterine carcinogenesis in rodents, the relationship between prolactin and uterine carcinogenesis should be discussed because the long-term effects of prolactin on uterine carcinogenicity in rats are controversial21,22,23 and the relevance of prolactin in the development of endometrial adenocarcinoma in humans has not yet been established. The function of prolactin in rats is known to be different from that in women, particularly in its role in the formation and regression of the corpus luteum. Rodents have a very short functional luteal phase; in cycling rats, granulosa cells in large growing follicles are the main sources of P4 production, while luteal cells produce small amounts of P4 at metestrus only. Dopamine blocking/inhibitory chemicals modulate prolactin production in rodents24. Recently, a P2Y12 receptor antagonist possessing dopamine agonistic activity was shown to increase the incidence of rat uterine adenocarcinomas in carcinogenicity studies21. Decreased pituitary and mammary tumors and approximately 20% lower body weight were also observed. The author speculated that the MOA of this chemical was a decreased P4 level compared with the level of E2, resulting in an increased E2 to P4 ratio, based on the inhibitory effects of single-dose exposure to the compound on prolactin in young ovariectomized rats with single E2 treatment. Moreover, prolactin has also been shown to promote or inhibit the development of spontaneous mammary or uterine tumors, respectively, in Wistar rats but not in Sprague-Dawley rats22. In contrast, bromocriptine, a dopamine agonist known to inhibit prolactin, did not affect estrous cycling during short-term treatment of young cycling rats24 and showed inhibitory effects on uterine adenocarcinoma development during long-term treatment of rats, with no effects on estrous cyclicity of E2, P4 and prolactin levels, except a decreased E2 to P4 ratio at 15 months of age, when the rats were sacrificed23. The specific mechanism of this has not yet been determined; however, the primary MOA was thought to be a decreased E2 to P4 ratio due to the modulation of P4 production by prolactin. Our current work provided interesting evidence demonstrating that short- and long-term treatment with sulpiride, a typical dopamine antagonist, clearly increased serum prolactin25 and inhibited uterine tumor development26. In addition, long-term treatment with sulpiride also decreased the E2 to P4 ratio, indicating that prolactin has various complex functions. Thus the possibility of secondary effects of prolactin change modulating the E2 to P4 ratio could not be excluded; however, we did not attempt to determine whether any other prolactin-driven pathways of rodent uterine carcinogenicity exist in the present study. If there may be an additional clear parameter, they should be added for the prediction of MOAs of uterine carcinogenicity. Therefore, to clarify the possibility of extrapolation to humans, more clear evidence is necessary.
Based on the analysis of predictive MOAs of uterine carcinogenicity using published toxicological data for pesticides, we proposed a flowchart diagram showing a decision tree for predictive MOAs in uterine carcinogenesis (Fig. 6). In the flowchart, the parameters used to predict different types of MOAs, as described above, are key events for classification. Due to the lack of evidence in rodent studies, the MOA related to increased in situ aromatase was excluded from the flowchart. The relevance of these MOAs in humans is also indicated in the chart with balloons. Estrogenic activity considered to be likely MOAs in humans. Without clear evidence indicating species differences between humans and rodents, increased E2 to P4 ratio and increased 4HE production via CYP induction are considered to be likely MOAs in humans, while increased E2 due to inhibition of E2 excretion was proposed as a possible MOA, with additional data required for confirmation.
Fig. 6.

Flowchart diagram showing a proposed decision tree for predictive MOAs in uterine carcinogenesis. Red and orange rectangles indicate predicted MOAs. Red rectangle indicates clear evidence for the predicted MOAs. The orange rectangle indicates that more evidence is required to support the predicted pathway of inhibition of E2 excretion. Balloons indicate plausibility of predictions in humans.
In conclusion, the present study indicates that MOAs for uterine carcinogenicity of 4 of the 7 pesticides were predictable using parameters detected during mechanistic studies. Thus, appropriate mechanistic studies are useful for MOA prediction in risk assessment. The flowchart proposed here is the first decision three for MOAs in uterine carcinogenesis and it might be useful for the hazard characterization in chemical risk assessment.
Acknowledgments
This study was partly supported by Health and Labor Sciences Research Grants, Research on Risks of Chemical Substances from the Ministry of Health, Labour and Welfare, Japan (H24-Toxicol-Japan-H010).
Footnotes
Disclosure of Potential Conflicts of Interest: This study was partly supported by Health and Labor Sciences Research Grants, Research on Risk of Chemical Substances, from the Ministry of Health, Labour and Welfare, Japan (H24-Toxicol-Japan-H010). All authors declare that they have no conflicts of interest to disclose.
References
- 1.Sherman ME. Theories of endometrial carcinogenesis: a multidisciplinary approach. Mod Pathol. 13: 295–308. 2000. [DOI] [PubMed] [Google Scholar]
- 2.Dixon D, Alison R, Bach U, Colman K, Foley GL, Harleman JH, Haworth R, Herbert R, Heuser A, Long G, Mirsky M, Regan K, Van Esch E, Westwood FR, Vidal J, and Yoshida M. Nonproliferative and proliferative lesions of the rat and mouse female reproductive system. J Toxicol Pathol. 27(Suppl): 1S–107S. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maekawa A, Takahashi M, Ando J, and Yoshida M. Uterine carcinogenesis by chemicals/hormones in rodents. J Toxicol Pathol. 12: 1–11. 1999. [Google Scholar]
- 4.Yoshida M, Katsuda S, and Maekawa A. Involvements of estrogen receptor, proliferating cell nuclear antigen and p53 in endometrial adenocarcinoma development in Donryu rats. J Toxicol Pathol. 25: 241–247. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fanta M. Is polycystic ovary syndrome, a state of relative estrogen excess, a real risk factor for estrogen-dependant malignancies? Gynecol Endocrinol. 29: 145–147. 2013. [DOI] [PubMed] [Google Scholar]
- 6.Gottschau M, Kjaer SK, Jensen A, Munk C, and Mellemkjaer L. Risk of cancer among women with polycystic ovary syndrome: a Danish cohort study. Gynecol Oncol. 136: 99–103. 2015. [DOI] [PubMed] [Google Scholar]
- 7.Yoshida M. A new hypothesis for uterine carcinogenesis: a pathway driven by modulation of estrogen metabolism through cytochrome P450 induction in the rat liver. J Toxicol Pathol. 19: 57–67. 2006. [Google Scholar]
- 8.Yoshida M, Katashima S, Ando J, Tanaka T, Uematsu F, Nakae D, and Maekawa A. Dietary indole-3-carbinol promotes endometrial adenocarcinoma development in rats initiated with N-ethyl-N’-nitro-N-nitrosoguanidine, with induction of cytochrome P450s in the liver and consequent modulation of estrogen metabolism. Carcinogenesis. 25: 2257–2264. 2004. [DOI] [PubMed] [Google Scholar]
- 9.Liehr JG, Ricci MJ, Jefcoate CR, Hannigan EV, Hokanson JA, and Zhu BT. 4-Hydroxylation of estradiol by human uterine myometrium and myoma microsomes: implications for the mechanism of uterine tumorigenesis. Proc Natl Acad Sci USA. 92: 9220–9224. 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Badawi AF, Cavalieri EL, and Rogan EG. Effect of chlorinated hydrocarbons on expression of cytochrome P450 1A1, 1A2 and 1B1 and 2- and 4-hydroxylation of 17beta-estradiol in female Sprague-Dawley rats. Carcinogenesis. 21: 1593–1599. 2000. [PubMed] [Google Scholar]
- 11.Tsuchiya Y, Nakajima M, Kyo S, Kanaya T, Inoue M, and Yokoi T. Human CYP1B1 is regulated by estradiol via estrogen receptor. Cancer Res. 64: 3119–3125. 2004. [DOI] [PubMed] [Google Scholar]
- 12.Doherty JA, Weiss NS, Freeman RJ, Dightman DA, Thornton PJ, Houck JR, Voigt LF, Rossing MA, Schwartz SM, and Chen C. Genetic factors in catechol estrogen metabolism in relation to the risk of endometrial cancer. Cancer Epidemiol Biomarkers Prev. 14: 357–366. 2005. [DOI] [PubMed] [Google Scholar]
- 13.Rylander-Rudqvist T, Wedrén S, Jonasdottir G, Ahlberg S, Weiderpass E, Persson I, and Ingelman-Sundberg M. Cytochrome P450 1B1 gene polymorphisms and postmenopausal endometrial cancer risk. Cancer Epidemiol Biomarkers Prev. 13: 1515–1520. 2004. [PubMed] [Google Scholar]
- 14.NTP Technical Report on the toxicology studies of tetrabromobisphenol A (Cas No.79–94–7) in F344/NTac rats and B6C3F1/N mice and toxicology and carcinogenesis study of tetrabromobisphenol A in Wistar Han [Crl:WI(Han)] rats and B6C3F1/N mice (gavage studies). NTP TR 58 7. National Toxicology Program, Research Triangle Rark, NC. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knudsen GA, Sanders JM, Sadik AM, and Birnbaum LS. Disposition and kinetics of tetrabromobisphenol A in female Wistar Han rats. Toxicol Rev. 1: 214–223. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sasano H, Kaga K, Sato S, Yajima A, Nagura H, and Harada N. Aromatase cytochrome P450 gene expression in endometrial carcinoma. Br J Cancer. 74: 1541–1544. 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Risk Assessment Reports of pesticides. Food Safety Commission website: http://www.fsc.go.jp/fsciis/evaluationDocument/list?itemCategory=001.
- 18.Inventory of evaluations performed by the Joint Meeting on Pesticide Residues (JMPR). WHO website: http://apps.who.int/pesticide-residues-jmpr-database.
- 19.Hall AP, Elcombe CR, Foster JR, Harada T, Kaufmann W, Knippel A, Küttler K, Malarkey DE, Maronpot RR, Nishikawa A, Nolte T, Schulte A, Strauss V, and York MJ. Liver hypertrophy: a review of adaptive (adverse and non-adverse) changes—conclusions from the 3rd International ESTP Expert Workshop. Toxicol Pathol. 40: 971–994. 2012. [DOI] [PubMed] [Google Scholar]
- 20.Katsuda S, Yoshida M, Kuroda H, Ando J, Takahashi M, Kurokawa Y, Watanabe G, Taya K, and Maekawa A. Uterine adenocarcinoma in N-ethyl-N’-nitro-N-nitrosoguanidine-treated rats with high-dose exposure to p-tert-octylphenol during adulthood. Jpn J Cancer Res. 93: 117–124. 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brott DA, Anersson HAS, Stewart J, Ewart L, Christoph G, Larlemen J, Armstrong D, and Kinter LB. A peripherally restricted P2Y12 receptor antagonist altered rat tumor incidences with no human relevance: Mode of action consistent with dopamine agonism. Toxicol Rev. 1: 1201–1212. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harleman JH, Hargreaves A, Andersson H, and Kirk S. A review of the incidence and coincidence of uterine and mammary tumors in Wistar and Sprague-Dawley rats based on the RITA database and the role of prolactin. Toxicol Pathol. 40: 926–930. 2012. [DOI] [PubMed] [Google Scholar]
- 23.Yoshida M, Watanabe G, Suzuki T, Inoue K, Takahashi M, Maekawa A, Taya K, and Nishikawa A. Long-term treatment with bromocriptine inhibits endometrial adenocarcinoma development in rats. J Reprod Dev. 55: 105–109. 2009. [DOI] [PubMed] [Google Scholar]
- 24.Kumazawa T, Nakajima A, Ishiguro T, Jiuxin Z, Tanaharu T, Nishitani H, Inoue Y, Harada S, Hayasaka I, and Tagawa Y. Collaborative work on evaluation of ovarian toxicity. 15) Two- or four-week repeated-dose studies and fertility study of bromocriptine in female rats. J Toxicol Sci. 34(Suppl 1): SP157–SP165. 2009. [DOI] [PubMed] [Google Scholar]
- 25.Taketa Y, Yoshida M, Inoue K, Takahashi M, Sakamoto Y, Watanabe G, Taya K, Yamate J, and Nishikawa A. Differential stimulation pathways of progesterone secretion from newly formed corpora lutea in rats treated with ethylene glycol monomethyl ether, sulpiride, or atrazine. Toxicol Sci. 121: 267–278. 2011. [DOI] [PubMed] [Google Scholar]
- 26.Taketa Y, Inoue K, Takahashi M, Sakamoto Y, Watanabe G, Taya K, and Yoshida M. Effects of sulpiride and ethylene glycol monomethyl ether on endometrial carcinogenicity in Donryu rats. J Appl Toxicol. 2015; (In press). [DOI] [PubMed] [Google Scholar]