

Abstract
The mammalian target of rapamycin (mTOR) is a central regulator of a diverse array of cellular processes, including cell growth, proliferation, autophagy, translation, and actin polymerization. Components of the mTOR cascade are present at synapses and influence synaptic plasticity and spine morphogenesis. A prevailing view is that the study of mTOR and its role in autism spectrum disorders (ASDs) will elucidate the molecular mechanisms by which mTOR regulates neuronal function under physiological and pathological conditions. Although many ASDs arise as a result of mutations in genes with multiple molecular functions, they appear to converge on common biological pathways that give rise to autism-relevant behaviors. Dysregulation of mTOR signaling has been identified as a phenotypic feature common to fragile X syndrome, tuberous sclerosis complex 1 and 2, neurofibromatosis 1, phosphatase and tensin homolog, and potentially Rett syndrome. Below are a summary of topics covered in a symposium that presents dysregulation of mTOR as a unifying theme in a subset of ASDs.
Keywords: mTOR signaling, Fragile X syndromes, protein synthesis pathway, mouse models of autism
Autism is a widespread disorder characterized by deficits in social interactions, impairments in communication, and repetitive and stereotypic behaviors. Identification of genetic markers has proved difficult, because of the highly complex and variable nature of the disease. Although autism spectrum disorders (ASDs) arise as a consequence of mutations in genes with multiple molecular functions, they appear to converge on common biological pathways to give rise to autism-relevant behaviors (Abrahams and Geschwind, 2008). One such pathway is the phosphatidylinositol 3-kinase (PI3K)–mammalian target of rapamycin (mTOR) signaling cascade. The mTOR pathway is a central regulator of a diverse array of cellular processes, including growth, proliferation, survival, and protein translation that requires a modified guanosine, termed a cap, at the 5′ end of the RNA. In the brain, components of the mTOR pathway are present at synapses, in which they regulate dendritic spine morphology and are essential for synaptogenesis. Growing evidence indicates that dysregulation of mTOR is involved in human diseases, including cancer, diabetes, and autism. Mutations in tuberous sclerosis complex 1 (TSC1), TSC2, neurofibromatosis 1 (NF1), and phosphatase and tensin homolog (PTEN) lead to an overactivated PI3K–mTOR pathway, autism-relevant behaviors, and tuberous sclerosis, neurofibromatosis, or macrocephaly (Kwon et al., 2006). Recent findings that PI3K–mTOR signaling is overactivated at synapses of fragile X syndrome (FXS) mice (Sharma et al., 2010) and in humans with FXS (Hoeffer et al., 2012) provide the first evidence that genetic mutation not only of components within the mTOR signaling cascade, but also distant regulatory proteins, can lead to autism-related phenotypes. mTOR signaling and protein synthesis are also impaired in a mouse model of Rett syndrome (Ricciardi et al., 2011; Jiang et al., 2013). In this symposium, we focus on four signaling mechanisms upstream and downstream of mTOR, including the following: (1) the translation initiation machinery; (2) metabotropic glutamate receptor 5 (mGluR5)–Homer scaffolds; (3) mTOR-dependent synaptic plasticity; and (4) actin polymerization. These four components of the mTOR signaling pathway, although separate and distinct under physiological conditions, lead to a common pathological phenotype that is most closely associated with ASDs and ASD-associated syndromes.
Exaggerated cap-dependent translation causes synaptic and behavioral aberrations associated with ASDs
Excessive translation is posited to be a common molecular anomaly that contributes to aberrant synaptic plasticity and behaviors associated with a number of neurodevelopmental disorders (Kelleher and Bear, 2008; Hoeffer and Klann, 2010; Auerbach et al., 2011). This idea was based primarily on the observation that FXS, the most commonly inherited form of mental retardation, has a high incidence of autism and is caused by lack of fragile X mental retardation protein (FMRP), usually a translational repressor (Darnell and Klann, 2013). FXS model mice exhibit excessive protein synthesis, exaggerated mGluR-dependent synaptic plasticity, and multiple autism-related behaviors. In addition, it has been shown that FXS mice exhibit upregulated mTOR complex 1 (mTORC1) signaling at hippocampal synapses (Sharma et al., 2010; Ronesi et al., 2012), which result in enhanced assembly of the cap-dependent translation initiation complex as measured by interactions of the eukaryotic initiation factor 4 (eIF4E; the cap-binding translation factor) with eIF4G (Sharma et al., 2010; Ronesi et al., 2012) and enhanced activation of p70 S6 kinase 1 (S6K1) (Bhattacharya et al., 2012). Moreover, direct evidence that upregulation of mTORC1 signaling plays a role in FXS pathophysiology was provided in studies in which Fmr1 knock-out (KO) mice were crossed with mice that lack both copies of the gene that encodes S6K1 (Bhattacharya et al., 2012). The genetic reduction of S6K1 prevented molecular, morphological, synaptic, and behavioral phenotypes displayed by Fmr1 KO mice (Bhattacharya et al., 2012). Notably, S6K1 has a wide range of substrates, many of which are not translational control molecules (Magnuson et al., 2012). Thus, direct evidence that excessive protein synthesis via S6K1 is a causative factor for exaggerated plasticity and ASD-like behaviors is still lacking in FXS mice (Darnell and Klann, 2013).
A direct genetic link between ASD and the cap-binding translation factor eIF4E was reported in 2009. It was shown that a boy with classic autism had a de novo chromosome translocation between 4q and 5q, and the breakpoint site was mapped to an alternative transcript of EIF4E (Neves-Pereira et al., 2009). Mutation screening identified two additional unrelated autism families that harbored the same single nucleotide insertion in the EIF4E promoter. In vitro studies demonstrated that this mutation enhances the binding of a nuclear factor and EIF4E promoter activity, suggesting that overexpression of eIF4E may be causative for ASD. Moreover, because eIF4E–eIF4G interactions are elevated in FXS model mice (Sharma et al., 2010), a similar aberrant translational control mechanism may be involved in behavioral abnormalities in FXS (Darnell and Klann, 2013).
The first direct evidence demonstrating that excessive cap-dependent translation can result in ASD-like behaviors was provided by studies of transgenic mice that overexpress eIF4E (Santini et al., 2013). It was observed that eIF4E transgenic mice exhibit a 25–50% increase in eIF4E expression throughout the brain (Santini et al., 2013). Increased expression of eIF4E resulted in increased eIF4E–eIF4G interactions in the striatum and hippocampus, which were blocked with intracerebroventricular injections of 4EGI-1 (Santini et al., 2013), which blocks eIF4E–eIF4G interactions (Moerke et al., 2007; Hoeffer et al., 2011). The increase in eIF4E–eIF4G interactions resulted in exaggerated protein synthesis, which also was blocked by 4EGI-1 (Santini et al., 2013). Behavioral analysis demonstrated that the eIF4E transgenic mice display enhanced repetitive behaviors as measured by self-grooming and marble burying. In addition, the eIF4E transgenic mice exhibited impaired arm choice reversal in the water-based Y maze. These behavioral abnormalities were correlated with exaggerated long-term depression (LTD) in both corticostriatal and hippocampal slices from the eIF4E transgenic mice. The behavioral and synaptic plasticity abnormalities displayed by the eIF4E transgenic mice could be reversed with 4EGI-1 treatments (Santini et al., 2013). Consistent with these findings, it was shown that mice that lack 4E-binding protein 2 (4EBP2), a repressor of eIF4E, exhibit synaptic plasticity and behavioral phenotypes similar to those displayed by the eIF4E transgenic mice (Banko et al., 2005; Gkogkas et al., 2013), which are reversed by 4EGI-1 (Gkogkas et al., 2013). In addition, it has been shown that altered eIF4E-dependent translation plays a role in FXS. Either genetic reduction or pharmacological inhibition of eIF4E phosphorylation prevented molecular, morphological, synaptic, and behavioral phenotypes exhibited by Fmr1 KO mice (Gkogkas et al., 2014). Notably, excessive translation of matrix metalloproteinase-9, which plays an important role in FXS pathophysiology (Sidhu et al., 2014), was normalized in FXS mice with reduced eIF4E phosphorylation (Gkogkas et al., 2014). Finally, crossing eIF4E transgenic mice with Fmr1 KO mice causes cognitive dysfunction in hippocampus-dependent memory tasks in the double mutant mice that are not exhibited by either the eIF4E transgenic mice or the Fmr1 KO mice (Huynh et al., 2015). Together, these findings indicate that increased eIF4E-dependent translation can cause synaptic dysfunction and behavioral aberrations associated with intellectual disabilities and ASD.
mGluR5–Homer scaffolds regulate signaling to PI3K and mTOR: implications for FXS and ASDs
Activation of the PI3K–mTORC1 pathway and signaling to translation control is necessary for rapid translation of new proteins and induction of translation-dependent long-term synaptic plasticity in response to Gq-coupled neurotransmitter receptors, including the mGluR5 and M1 muscarinic acetylcholine receptors (Hou and Klann, 2004; Volk et al., 2007). Furthermore, alterations in mGluR5 signaling to mTORC1 and translation-dependent synaptic plasticity occur in many mouse models of autism, such as FXS (Hou et al., 2006; Nosyreva and Huber, 2006; Sharma et al., 2010), tuberous sclerosis (Auerbach et al., 2011), Angelman syndrome, 16p11.2 microdeletion (Tian et al., 2015), and may contribute to disease-relevant behaviors. Recent work has provided a better understanding of the molecular mechanisms that couple mGluR5 signaling to mTORC1 and translational activation in both healthy and diseased neurons.
Pharmacological activation of acute hippocampal slices from wild-type mice with the group 1 mGluR agonist, (RS)-3,5-dihydroxyphenylglycine, results in a rapid activation of the PI3K–mTORC1 pathway as measured with phospho-specific antibodies to downstream effectors, such as p70 ribosomal S6K and 4EBP, which promotes mRNA translation initiation (Hou and Klann, 2004; Ronesi and Huber, 2008). Importantly, activation of PI3K–mTORC1 is necessary for mGluR5-stimulated LTD and synapse elimination, both of which rely on translation of dendritic mRNAs (Hou and Klann, 2004; Wilkerson et al., 2014). In Fmr1 KO mice, a mouse model of FXS, mGluR5-induced activation of PI3K and mTORC1 is deficient (Ronesi and Huber, 2008), but basal levels of PI3K and mTORC1 activity, as well as protein translation rates, are elevated in Fmr1 KO cortical regions. Basal or constitutive activity of mGluR5 may drive these changes, because enhanced PI3K activity and translation rates in Fmr1 KO tissues are sensitive to the mGluR5 antagonist MPEP (Gross et al., 2010; Osterweil et al., 2010; Sharma et al., 2010; Ronesi et al., 2012). These results suggest that there is constitutive signaling of mGluR5 to the PI3K–mTORC1 pathway in FXS that may reduce or occlude agonist-induced activation of the pathway.
Recent work by Huber and colleagues suggests that dissociation of mGluR5 with its postsynaptic scaffolding protein Homer mediates the abnormal mGluR5 signaling to PI3K–mTORC1 in the Fmr1 KO mouse (Giuffrida et al., 2005; Ronesi et al., 2012). The Homer family of proteins binds to the intracellular C-terminal tail of group 1 mGluRs and function to scaffold mGluRs to signaling complexes within the postsynaptic density through other Homer binding proteins (Shiraishi-Yamaguchi and Furuichi, 2007). Homers (Homer 1–Homer 3) share a common EVH1 domain at the N terminus, which binds to mGluR1α, mGluR5, and other scaffolding proteins (e.g., SHANK), ion channels (e.g., TrpC, CaV1.2), and signaling pathways [e.g., PI3K enhancer (PIKE), IP3 receptor]. Homers multimerize through their coiled-coil domains to form large signaling complexes at the postsynaptic density (Shiraishi-Yamaguchi and Furuichi, 2007). An activity-dependent short variant of Homer (Homer1a) lacks a coiled-coil domain and disrupts Homer scaffolds by competing with long Homers for mGluR5 and other Homer interacting proteins (Xiao et al., 1998).
A group 1 mGluR agonist stimulates association of Homer with PIKE, a GTPase that enhances PI3K activity by binding to the p85 subunit of PI3K. Homer and PIKE are both necessary for mGluR5 to stimulate PI3K activity in neurons (Rong et al., 2003). Consistent with this finding, acute disruption of mGluR5–Homer interactions in hippocampal slices, using a peptide that mimics the proline-rich Homer binding motif in the C terminus of mGluR5, blocks agonist-induced activation of PI3K–mTOR, mGluR–LTD, and the rapid translation of proteins required for LTD, such as Arc (Ronesi and Huber, 2008; Ronesi et al., 2012). In Fmr1 KO forebrain, mGluR5 is less associated with long Homer isoforms and more associated with Homer1a, suggesting that the disrupted mGluR5–Homer mediates the deficit in mGluR5 signaling to PI3K–mTOR and translation machinery in Fmr1 KO neurons (Giuffrida et al., 2005; Ronesi et al., 2012). In support of this idea, genetic deletion of Homer1a restores mGluR5–Homer scaffolds and mGluR5-induced activation of mTORC1 pathway, as well as mGluR5-induced activation of translation initiation (Ronesi et al., 2012). Homer is not necessary for all mGluR5 signaling because peptide disruption of mGluR5–Homer or Fmr1 KO has no effect on agonist-induced ERK activation in hippocampal slices (Ronesi and Huber, 2008; Ronesi et al., 2012), suggesting a specific role for Homer in coupling mGluR5 to PI3K–mTORC1 via PIKE.
Homer1a binding results in constitutive, agonist-independent activity of mGluR5 (Ango et al., 2001), suggesting that the Homer1a-bound mGluR5 in Fmr1 KO neurons may constitutively signal and contribute to the enhanced, mGluR5-dependent PI3K, mTORC1, translation initiation complex formation, and basal translation rates. In support of this idea, acute peptide-mediated disruption of mGluR5–Homer results in elevated protein synthesis rates in wild-type slices but not those from the Fmr1 KO slices. Homer1a deletion in the Fmr1 KO rescues elevated protein synthesis rates and enhanced translation initiation complex formation (Ronesi et al., 2012). The role of Homer or constitutive mGluR5 activation in driving elevated mTORC1 in Fmr1 KO is currently unknown. There may also be some regional differences in the contribution of PI3K–mTORC1 to enhanced basal translation rates in Fmr1 KOs. However, in both regions, protein synthesis rates are sensitive to mGluR5 antagonism, suggesting the constitutive mGluR5 signaling, as a result of Homer1a interactions, is driving signaling to enhance protein synthesis rates. What is unclear at this time is whether the enhanced constitutive signaling of mGluR5 saturates the PI3K–mTORC1 pathway, which prevents subsequent agonist-induced activation of the pathway, or whether Homer coupling is selectively required for agonist-induced, but not constitutive, activation of PI3K–mTORC1.
The blockade of agonist-induced activation of PI3K–mTORC1 by disruption of Homer interactions may have additional consequences on mGluR5-dependent translational activation (Fig. 1). mGluRs stimulate phosphorylation of elongation factor 2 (EF2) through activation of EF2 kinase (EF2K). Although P-EF2 inhibits elongation rate, EF2K and moderate inhibition of elongation are necessary for mGluR-induced synthesis of Arc, as well as mGluR–LTD (Park et al., 2008). Subthreshold inhibition of global elongation is thought to promote rate-limiting translation factors, which translate mRNAs that are poorly initiated and cannot compete effectively for these factors (Scheetz et al., 2000). EF2K is a Homer binding protein and also binds directly to mGluR5 (Park et al., 2008). Homer appears not to be required to couple mGluR5 activation to EF2K. On the contrary, peptide-mediated disruption of Homer–mGluR5 enhances agonist-induced activation of P-EF2, suggesting that Homer inhibits activation of EF2K (Ronesi et al., 2012). Consistent with this idea, agonist-induced activation of P-EF2 is enhanced in hippocampus of Fmr1 KO mice and rescued by deletion of Homer1a (Ronesi et al., 2012). Although as yet untested, the bidirectional effects of Homer on mGluR5 stimulation of mTORC1 and EF2K may be explained by the known inhibitory effects of S6K phosphorylation of EF2K. Homer disruption, via peptide or in Fmr1 KOs, prevents mGluR5 activation of mTORC1–S6K, which would also be expected to block the S6K phosphorylation and inhibition of EF2K, thus resulting in an overall increase in mGluR stimulation of P-EF2. Thus, the Homer–mGluR5 scaffold functions to balance mGluR5 signaling pathways for optimal translational activation. Homer is necessary for stimulation of translation initiation through activation of the PI3K–mTOR pathway (Ronesi and Huber, 2008) and limiting activation of EF2K and subsequent inhibition of elongation, which is necessary for rapid translation of new proteins required for synaptic plasticity.
Figure 1.
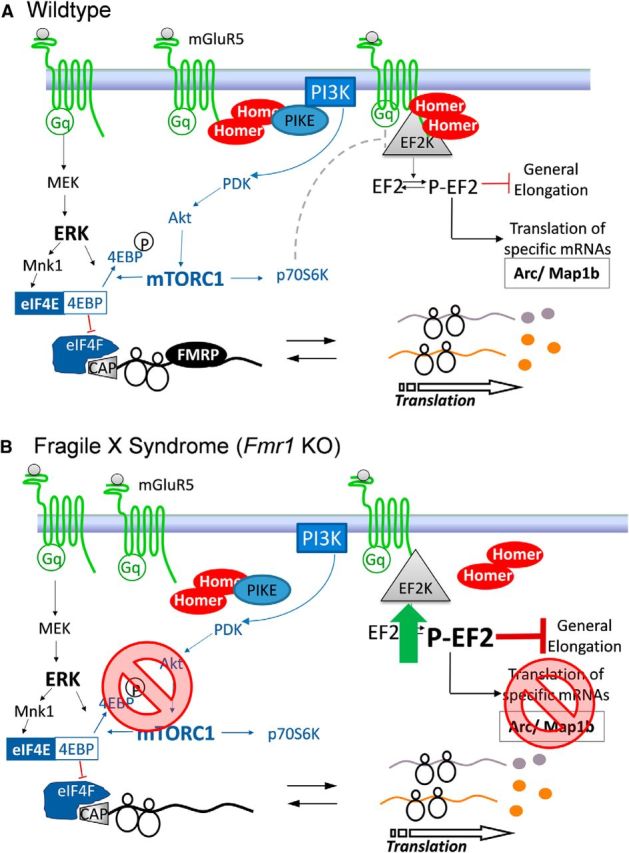
Working model of Homer scaffolds in agonist-stimulated mGluR5 signaling to translation machinery in wild-type (A) and FXS model (B) cortical neurons.
Abnormal mGluR5–Homer scaffolds may be a common deficiency across different genetic causes of autism. Recent work indicates that mGluR5–Homer interactions are enhanced in the Angelman syndrome mouse model (Ube3A maternal deletion), which may contribute to impairments in mGluR-induced LTD (Pignatelli et al., 2014). More work is needed to know whether and how the enhanced mGluR5–Homer interactions regulate Angelman syndrome pathophysiology.
Dysregulation of mTOR signaling is critical to impaired synaptic plasticity and aberrant spine morphology in a mouse model of FXS
FXS is the most common heritable form of intellectual disabilities and a leading genetic cause of autism. Whereas other syndromic ASDs arise from mutations in components of the PI3K–mTOR pathway, FXS arises from silencing of the gene encoding FMRP (Fmr1). FMRP is an RNA-binding protein that represses translation of a large array of RNAs, including PIKE. PIKE, an upstream activator of mTOR and identified target of FMRP (Darnell et al., 2011), is elevated at the synapses of Fmr1 KO mice (Gross et al., 2010; Sharma et al., 2010), humans (Hoeffer et al., 2012), and flies (McBride et al., 2005), providing a functional link between loss of FMRP and overactivated mTOR signaling (Fig. 2).
Figure 2.

Scheme showing the link between deficits in FMRP and overactivated mTOR signaling in the dendritic spine. Our findings support a model whereby FMRP represses PIKE, thereby inhibiting mTOR signaling, in wild-type mice. Under basal conditions, mTOR regulates LIM kinase and cofilin through Rac1, a member of the Rho family of GTPases. In FXS mice, we propose that dysregulation of mTORC2 leads to impaired actin polymerization and spine structure. DHPG, (RS)-3,5-dihydroxyphenylglycine.
Recent work from Sharma et al. (2010) has demonstrated that, in hippocampal neurons of Fmr1-deficient mice, upregulation of the PI3K-enhancer PIKE results in overactivation of PI3K/Akt and mTOR signaling in the hippocampus, as assessed by several functional readouts, including mTOR phosphorylation, binding to the regulatory protein Raptor, and formation of the eIF4F translation initiation complex. Moreover, synaptic plasticity in the form of mGluR–LTD is exaggerated and insensitive to the mTOR inhibitor rapamycin at hippocampal CA3–CA1 synapses of Fmr1-deficient mice (Sharma et al., 2010). These findings are consistent with a model in which, in the FXS mouse, the protein(s) required for mGluR-dependent AMPA receptor internalization and mGluR–LTD are already accumulated at CA1 synapses under basal conditions (Bear et al., 2004). These observations provide an important functional link between overactivated mGluR signaling, aberrant protein synthesis, and exaggerated mGluR–LTD in Fmr1-deficient mice (Sawicka and Zukin, 2012).
A hallmark feature of FXS that is also observed in Fmr1-deficient mice is synaptic spine abnormalities (Bagni and Greenough, 2005). Spines on hippocampal and cortical neurons of Fmr1 KO mice exhibit enhanced density and are thinner and longer than those of age-matched wild-type mice, resembling an immature morphology. These observations indicate that FMRP might normally repress local translation of proteins that inhibit synapse maturation, stabilization, and elimination (Bagni and Greenough, 2005). Cofilin is an actin depolymerizing factor and central regulator of apoptosis, activity-dependent synaptic plasticity, and spine morphology. Emerging evidence indicates that cofilin signaling is downstream of the mTORC2 pathway, an mTOR signaling complex insensitive to rapamycin and a critical regulator of embryonic development, actin cytoskeleton dynamics, synaptic efficacy, and the consolidation of long-term memory. Dysregulation of cofilin signaling has been linked to aberrant spine morphology in Tsc1+/− and Tsc2+/− mice (Tavazoie et al., 2005). Because mTOR is overactivated in FXS and is an upstream regulator of the postulated cofilin signaling pathway, an intriguing possibility currently under exploration is that aberrant cofilin signaling may be causally related to the spine abnormalities that are a neuroanatomical hallmark of FXS (Pyronneau et al., 2015) reminiscent of the morphological abnormalities and synaptic deficits observed in PTEN mutant mice (Kwon et al., 2006; Takeuchi et al., 2013).
mTORC2 and its role in ASDs
The evolutionarily conserved mTOR forms two functionally distinct complexes. mTORC1, which consists of mTOR, Raptor, and mLST8 (GβL), is sensitive to rapamycin and is thought to regulate mRNA translation rates. mTORC2 is mostly insensitive to rapamycin and contains the core components mTOR, mSIN1, mLST8, and Rictor. Rictor is highly expressed in the brain, in which it associates with membranes and regulates the actin cytoskeleton. Dysregulation of mTORC1 and mTORC2 signaling appears to have a crucial role in memory disorders, such as the cognitive deficits associated with ASDs. Notably, the activity of mTORC2 is altered in the brain of ASD patients harboring mutations in PTEN and/or TSC1 and TSC2 (two upstream negative regulators of mTORC1; Costa-Mattioli and Monteggia, 2013; Buffington et al., 2014). These findings raise the intriguing possibility that the neurological dysfunction in ASD is caused by dysregulation of mTORC2 rather than by mTORC1 signaling. Indeed, mTORC2 activity is altered in several cognitive disorders, including Huntington's disease, parkinsonism, Alzheimer-type dementia, and ASDs. Recent work from Huang et al. (2013) found that mTORC2 is an essential component of memory consolidation via its regulation of actin polymerization. Conditional deletion of Rictor in the postnatal murine forebrain greatly reduces mTORC2 activity and selectively impaired both long-term memory and the late phase of hippocampal long-term potentiation (L-LTP). A comparable impairment of long-term memory is also discovered in dTORC2-deficient Drosophila, highlighting the evolutionary conservation of long-term memory in the mTORC2 signaling pathway. Basal levels of actin polymerization are reduced in the hippocampus of mTORC2-deficient mice, and treatment with jasplakinolide restores actin polymerization to wild-type levels and rescues both L-LTP and long-term memory. Moreover, a small-molecule activator of mTORC2 activity converts early LTP into L-LTP and enhanced long-term memory. These results identify both the molecular and synaptic events occurring upstream and downstream of mTORC2 in the brain and suggest that mTORC2 could be a therapeutic target for the treatment of cognitive dysfunction.
The topics highlighted in this symposium support the central hypothesis that dysregulation of mTOR-dependent signaling, both upstream and downstream of its kinase activity, contributes to the pathophysiology associated with multiple ASDs. Mutations in components of the mTOR signaling cascade, including TSC1, TSC2, NF1, and PTEN, as well as the more distant regulatory proteins FMRP and MeCP2, present similar phenotypes in both mice and humans. Although these ASD-associated syndromes have their own distinct clinical features, the unifying theme of mTOR dysregulation underscores the significance of synaptic deficits and its relevance to autism-relevant behaviors, such as social deficits and cognitive impairment. On the basis of observations that mTOR signaling, synaptic plasticity, and protein translation are overactivated in many of these disorders, compounds that can potentially inhibit the mTOR pathway represent promising therapeutic candidates for their treatment. Indeed, the mTORC1 inhibitor rapamycin has shown promising results in PTEN KO mice (Zhou et al., 2009) and TSC2+/− mice (Ehninger et al., 2008). Thus, interventions that target mTOR signaling should be at the leading edge of future translational research in the autism field.
References
- Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9:341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ango F, Prézeau L, Muller T, Tu JC, Xiao B, Worley PF, Pin JP, Bockaert J, Fagni L. Agonist-independent activation of metabotropic glutamate receptors by the intracellular protein Homer. Nature. 2001;411:962–965. doi: 10.1038/35082096. [DOI] [PubMed] [Google Scholar]
- Auerbach BD, Osterweil EK, Bear MF. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature. 2011;480:63–68. doi: 10.1038/nature10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagni C, Greenough WT. From mRNP trafficking to spine dysmorphogenesis: the roots of fragile X syndrome. Nat Rev Neurosci. 2005;6:376–387. doi: 10.1038/nrn1667. [DOI] [PubMed] [Google Scholar]
- Banko JL, Poulin F, Hou L, DeMaria CT, Sonenberg N, Klann E. The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J Neurosci. 2005;25:9581–9590. doi: 10.1523/JNEUROSCI.2423-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, Murphy JP, Pierre P, Klann E. Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron. 2012;76:325–337. doi: 10.1016/j.neuron.2012.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffington SA, Huang W, Costa-Mattioli M. Translational control in synaptic plasticity and cognitive dysfunction. Annu Rev Neurosci. 2014;37:17–38. doi: 10.1146/annurev-neuro-071013-014100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Monteggia LM. mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat Neurosci. 2013;16:1537–1543. doi: 10.1038/nn.3546. [DOI] [PubMed] [Google Scholar]
- Darnell JC, Klann E. The translation of translational control by FMRP: therapeutic targets for FXS. Nat Neurosci. 2013;16:1530–1536. doi: 10.1038/nn.3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD, Darnell RB. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med. 2008;14:843–848. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffrida R, Musumeci S, D'Antoni S, Bonaccorso CM, Giuffrida-Stella AM, Oostra BA, Catania MV. A reduced number of metabotropic glutamate subtype 5 receptors are associated with constitutive homer proteins in a mouse model of fragile X syndrome. J Neurosci. 2005;25:8908–8916. doi: 10.1523/JNEUROSCI.0932-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB, Vasuta C, Yee S, Truitt M, Dallaire P, Major F, Lasko P, Ruggero D, Nader K, Lacaille JC, Sonenberg N. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature. 2013;493:371–377. doi: 10.1038/nature11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkogkas CG, Khoutorsky A, Cao R, Jafarnejad SM, Prager-Khoutorsky M, Giannakas N, Kaminari A, Fragkouli A, Nader K, Price TJ, Konicek BW, Graff JR, Tzinia AK, Lacaille JC, Sonenberg N. Pharmacogenetic inhibition of eIF4E-dependent Mmp9 mRNA translation reverses fragile X syndrome-like phenotypes. Cell Rep. 2014;9:1742–1755. doi: 10.1016/j.celrep.2014.10.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Nakamoto M, Yao X, Chan CB, Yim SY, Ye K, Warren ST, Bassell GJ. Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J Neurosci. 2010;30:10624–10638. doi: 10.1523/JNEUROSCI.0402-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffer CA, Cowansage KK, Arnold EC, Banko JL, Moerke NJ, Rodriguez R, Schmidt EK, Klosi E, Chorev M, Lloyd RE, Pierre P, Wagner G, LeDoux JE, Klann E. Inhibition of the interactions between eukaryotic initiation factors 4E and 4G impairs long-term associative memory consolidation but not reconsolidation. Proc Natl Acad Sci U S A. 2011;108:3383–3388. doi: 10.1073/pnas.1013063108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffer CA, Sanchez E, Hagerman RJ, Mu Y, Nguyen DV, Wong H, Whelan AM, Zukin RS, Klann E, Tassone F. Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav. 2012;11:332–341. doi: 10.1111/j.1601-183X.2012.00768.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2004;24:6352–6361. doi: 10.1523/JNEUROSCI.0995-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51:441–454. doi: 10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Huang W, Zhu PJ, Zhang S, Zhou H, Stoica L, Galiano M, Krnjević K, Roman G, Costa-Mattioli M. mTORC2 controls actin polymerization required for consolidation of long-term memory. Nat Neurosci. 2013;16:441–448. doi: 10.1038/nn.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh A, DuPage M, Priyadharshini B, Sage PT, Quiros J, Borges CM, Townamchai N, Gerriets VA, Rathmell JC, Sharpe AH, Bluestone JA, Turka LA. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat Immunol. 2015;16:188–196. doi: 10.1038/ni.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Ash RT, Baker SA, Suter B, Ferguson A, Park J, Rudy J, Torsky SP, Chao HT, Zoghbi HY, Smirnakis SM. Dendritic arborization and spine dynamics are abnormal in the mouse model of MECP2 duplication syndrome. J Neurosci. 2013;33:19518–19533. doi: 10.1523/JNEUROSCI.1745-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Bear MF. The autistic neuron: troubled translation? Cell. 2008;135:401–406. doi: 10.1016/j.cell.2008.10.017. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnuson B, Ekim B, Fingar DC. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem J. 2012;441:1–21. doi: 10.1042/BJ20110892. [DOI] [PubMed] [Google Scholar]
- McBride SM, Choi CH, Wang Y, Liebelt D, Braunstein E, Ferreiro D, Sehgal A, Siwicki KK, Dockendorff TC, Nguyen HT, McDonald TV, Jongens TA. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron. 2005;45:753–764. doi: 10.1016/j.neuron.2005.01.038. [DOI] [PubMed] [Google Scholar]
- Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, Gross JD, Degterev A, Yuan J, Chorev M, Halperin JA, Wagner G. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell. 2007;128:257–267. doi: 10.1016/j.cell.2006.11.046. [DOI] [PubMed] [Google Scholar]
- Neves-Pereira M, Müller B, Massie D, Williams JH, O'Brien PC, Hughes A, Shen SB, Clair DS, Miedzybrodzka Z. Deregulation of EIF4E: a novel mechanism for autism. J Med Genet. 2009;46:759–765. doi: 10.1136/jmg.2009.066852. [DOI] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM. Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol. 2006;95:3291–3295. doi: 10.1152/jn.01316.2005. [DOI] [PubMed] [Google Scholar]
- Osterweil EK, Krueger DD, Reinhold K, Bear MF. Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J Neurosci. 2010;30:15616–15627. doi: 10.1523/JNEUROSCI.3888-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Park JM, Kim S, Kim JA, Shepherd JD, Smith-Hicks CL, Chowdhury S, Kaufmann W, Kuhl D, Ryazanov AG, Huganir RL, Linden DJ, Worley PF. Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron. 2008;59:70–83. doi: 10.1016/j.neuron.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignatelli M, Piccinin S, Molinaro G, Di Menna L, Riozzi B, Cannella M, Motolese M, Vetere G, Catania MV, Battaglia G, Nicoletti F, Nisticò R, Bruno V. Changes in mGlu5 receptor-dependent synaptic plasticity and coupling to homer proteins in the hippocampus of Ube3A hemizygous mice modeling angelman syndrome. J Neurosci. 2014;34:4558–4566. doi: 10.1523/JNEUROSCI.1846-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyronneau AP, Porch MP, Hwang JY, Zukin RS. The actin depolymerizing factor, cofilin, is critical to aberrant spine morphology in fragile X syndrome. Soc Neurosci Abstr. 2015;41:491.25. [Google Scholar]
- Ricciardi S, Boggio EM, Grosso S, Lonetti G, Forlani G, Stefanelli G, Calcagno E, Morello N, Landsberger N, Biffo S, Pizzorusso T, Giustetto M, Broccoli V. Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum Mol Genet. 2011;20:1182–1196. doi: 10.1093/hmg/ddq563. [DOI] [PubMed] [Google Scholar]
- Ronesi JA, Huber KM. Homer interactions are necessary for metabotropic glutamate receptor-induced long-term depression and translational activation. J Neurosci. 2008;28:543–547. doi: 10.1523/JNEUROSCI.5019-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi JA, Collins KA, Hays SA, Tsai NP, Guo W, Birnbaum SG, Hu JH, Worley PF, Gibson JR, Huber KM. Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat Neurosci. 2012;15:431–440. S1. doi: 10.1038/nn.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong R, Ahn JY, Huang H, Nagata E, Kalman D, Kapp JA, Tu J, Worley PF, Snyder SH, Ye K. PI3 kinase enhancer-Homer complex couples mGluRI to PI3 kinase, preventing neuronal apoptosis. Nat Neurosci. 2003;6:1153–1161. doi: 10.1038/nn1134. [DOI] [PubMed] [Google Scholar]
- Santini E, Huynh TN, MacAskill AF, Carter AG, Pierre P, Ruggero D, Kaphzan H, Klann E. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature. 2013;493:411–415. doi: 10.1038/nature11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicka K, Zukin RS. Dysregulation of mTOR signaling in neuropsychiatric disorders: therapeutic implications. Neuropsychopharmacology. 2012;37:305–306. doi: 10.1038/npp.2011.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheetz AJ, Nairn AC, Constantine-Paton M. NMDA receptor-mediated control of protein synthesis at developing synapses. Nat Neurosci. 2000;3:211–216. doi: 10.1038/72915. [DOI] [PubMed] [Google Scholar]
- Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, Zukin RS. Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci. 2010;30:694–702. doi: 10.1523/JNEUROSCI.3696-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi-Yamaguchi Y, Furuichi T. The Homer family proteins. Genome Biol. 2007;8:206. doi: 10.1186/gb-2007-8-2-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhu H, Dansie LE, Hickmott PW, Ethell DW, Ethell IM. Genetic removal of matrix metalloproteinase 9 rescues the symptoms of fragile X syndrome in a mouse model. J Neurosci. 2014;34:9867–9879. doi: 10.1523/JNEUROSCI.1162-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, Gertner MJ, Zhou J, Parada LF, Bennett MV, Zukin RS. Dysregulation of synaptic plasticity precedes appearance of morphological defects in a Pten conditional knockout mouse model of autism. Proc Natl Acad Sci U S A. 2013;110:4738–4743. doi: 10.1073/pnas.1222803110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8:1727–1734. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- Tian D, Stoppel LJ, Heynen AJ, Lindemann L, Jaeschke G, Mills AA, Bear MF. Contribution of mGluR5 to pathophysiology in a mouse model of human chromosome 16p11.2 microdeletion. Nat Neurosci. 2015;18:182–184. doi: 10.1038/nn.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk LJ, Pfeiffer BE, Gibson JR, Huber KM. Multiple Gq-coupled receptors converge on a common protein synthesis-dependent long-term depression that is affected in fragile X syndrome mental retardation. J Neurosci. 2007;27:11624–11634. doi: 10.1523/JNEUROSCI.2266-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson JR, Tsai NP, Maksimova MA, Wu H, Cabalo NP, Loerwald KW, Dictenberg JB, Gibson JR, Huber KM. A role for dendritic mGluR5-mediated local translation of Arc/Arg3.1 in MEF2-dependent synapse elimination. Cell Rep. 2014;7:1589–1600. doi: 10.1016/j.celrep.2014.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Tu JC, Petralia RS, Yuan JP, Doan A, Breder CD, Ruggiero A, Lanahan AA, Wenthold RJ, Worley PF. Homer regulates the association of group 1 metabotropic glutamate receptors with multivalent complexes of homer-related, synaptic proteins. Neuron. 1998;21:707–716. doi: 10.1016/S0896-6273(00)80588-7. [DOI] [PubMed] [Google Scholar]
- Zhou J, Blundell J, Ogawa S, Kwon CH, Zhang W, Sinton C, Powell CM, Parada LF. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci. 2009;29:1773–1783. doi: 10.1523/JNEUROSCI.5685-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]