

Abstract
Transient changes in striatal dopamine (DA) concentration are considered to encode a reward prediction error (RPE) in reinforcement learning tasks. Often, a phasic DA change occurs concomitantly with a dip in striatal acetylcholine (ACh), whereas other neuromodulators, such as adenosine (Adn), change slowly. There are abundant adenylyl cyclase (AC) coupled GPCRs for these neuromodulators in striatal medium spiny neurons (MSNs), which play important roles in plasticity. However, little is known about the interaction between these neuromodulators via GPCRs. The interaction between these transient neuromodulator changes and the effect on cAMP/PKA signaling via Golf- and Gi/o-coupled GPCR are studied here using quantitative kinetic modeling. The simulations suggest that, under basal conditions, cAMP/PKA signaling could be significantly inhibited in D1R+ MSNs via ACh/M4R/Gi/o and an ACh dip is required to gate a subset of D1R/Golf-dependent PKA activation. Furthermore, the interaction between ACh dip and DA peak, via D1R and M4R, is synergistic. In a similar fashion, PKA signaling in D2+ MSNs is under basal inhibition via D2R/Gi/o and a DA dip leads to a PKA increase by disinhibiting A2aR/Golf, but D2+ MSNs could also respond to the DA peak via other intracellular pathways. This study highlights the similarity between the two types of MSNs in terms of high basal AC inhibition by Gi/o and the importance of interactions between Gi/o and Golf signaling, but at the same time predicts differences between them with regard to the sign of RPE responsible for PKA activation.
SIGNIFICANCE STATEMENT Dopamine transients are considered to carry reward-related signal in reinforcement learning. An increase in dopamine concentration is associated with an unexpected reward or salient stimuli, whereas a decrease is produced by omission of an expected reward. Often dopamine transients are accompanied by other neuromodulatory signals, such as acetylcholine and adenosine. We highlight the importance of interaction between acetylcholine, dopamine, and adenosine signals via adenylyl-cyclase coupled GPCRs in shaping the dopamine-dependent cAMP/PKA signaling in striatal neurons. Specifically, a dopamine peak and an acetylcholine dip must interact, via D1 and M4 receptor, and a dopamine dip must interact with adenosine tone, via D2 and A2a receptor, in direct and indirect pathway neurons, respectively, to have any significant downstream PKA activation.
Keywords: acetylcholine, D1R/M4R, D2R/A2AR, dopamine, reward learning, striatal plasticity
Introduction
Reinforcement learning is a kind of association building where an organism, based on its prior experiences, associates an environmental stimulus to either a reward-producing response or to the reward itself by updating the salience of the stimulus. Basal ganglia play a crucial role in reinforcement learning, and the input nucleus of basal ganglia, striatum, receives sensorimotor and reward-related signals (Liljeholm and O'Doherty, 2012). The reward-related signal, encoded as a reward prediction error (RPE), which is the difference between the expected and the obtained reward, is typically represented by the activity of midbrain dopaminergic neurons (DAN) projecting onto striatum (Schultz, 1998; Steinberg et al., 2013). A salient cue or an unexpected reward produces burst activity in DANs (positive RPE), whereas omission of an expected reward leads to a depression in DAN activity (negative RPE) (Schultz, 1998). The positive RPE signal increases the striatal dopamine (DA) concentration, whereas the negative RPE signal decreases it (Hart et al., 2014). The increase in DAN activity is often accompanied by a simultaneous pause in the activity of striatal cholinergic interneurons, which largely constitute a population of tonically active neurons (TANs), resulting in a dip in the striatal acetylcholine (ACh) level (Aosaki et al., 1995; Goldberg and Reynolds, 2011). These striatal DA and ACh signals are fast, but other neuromodulators, such as adenosine (Adn), seem to change more slowly (Cechova and Venton, 2008; Aosaki et al., 2010; Pajski and Venton, 2010). There are several adenylyl cyclase (AC5)-coupled GPCRs for these neuromodulators in striatal neurons (Lovinger, 2010). However, little is known, quantitatively, about the way in which the aforementioned transient neuromodulatory signals might interact and affect the AC activity via the Golf and Gi/o type G-proteins.
Striatum is mainly populated by the medium sized spiny projection neurons (MSNs). There are two types of MSNs, striatonigral (D1+ MSNs) and striatopallidal (D2+ MSNs), each of which expresses a unique dopamine receptor class; D1 receptors (D1R) coupled to Golf in D1+ MSNs and D2 receptors (D2R) coupled to Gi/o in D2+ MSNs. Striatal D1R is known to be expressed with the muscarinic M4 acetylcholine receptor (M4R), which is coupled to Gi/o, whereas D2R with the adenosine A2a receptors (A2aR), which are coupled to Golf (Fig. 1) (Fink et al., 1992; Ferré et al., 1993; Hersch et al., 1994, 1995; Ince et al., 1997). These receptors show functional interaction and regulate the AC5/cAMP/PKA signaling, which in turn is important for the long-term potentiation of the corticostriatal synapses and motor learning (Brandon et al., 1998; Price et al., 1999; Svenningsson et al., 2000; Onali and Olianas, 2002; Augustin et al., 2014). In this study, we explored interactions between the aforementioned transient neuromodulatory signals via Golf- and Gi/o-coupled receptors in MSNs and how they affect the downstream cAMP/PKA signaling using quantitative kinetic modeling. Our results suggest that, in D1+ MSNs, AC5 is tonically inhibited by the ACh/M4R/Gi/o pathway under the basal condition; and a DA peak, in response to a positive RPE, is not able to activate the AC5/cAMP/PKA signaling significantly unless it is accompanied by an ACh dip. The ACh dip leads to a reduction in the M4R-mediated inhibition of AC5 and thus acts as a gate for the PKA activation by the DA peak. On the other hand, in D2+ MSNs, a DA dip stimulates cAMP/PKA by disinhibiting the A2aR signaling, which is tonically inhibited by D2R under basal conditions. These results highlight the important role of M4R and A2aR interaction with DA receptors in MSNs along with the common theme that a dip in Gi/o activity is required to gate Golf-dependent PKA responses in these neurons. The basal inhibition of AC by Gi/o and gating of Golf signaling by a decrease in Gi/o could be viewed as a general mechanism to filter out spurious Golf signals.
Figure 1.

An overview of the biochemical network modeled in this study. A, The biochemical network for the D1+ MSN. B, The biochemical network for the D2+ MSN. The signaling core is similar, but the receptors coupled to the Gi (Gi/o) and Golf (Golf) proteins are different, and this opens up for alternative processing of changes in neurotransmitter signaling in the two neuron types.
Materials and Methods
Biochemical networks.
Here, we study the functional interactions between the following receptor pairs: (1) interaction between D1R and M4R in the striatal direct pathways, D1+ MSNs; and (2) interaction between D2R and A2aR in striatal indirect pathways, D2+ MSNs.
To this end, we developed two related model variants: one for D1+ MSN and the second for D2+ MSN. The biochemical networks modeled for both the cell types focus on the intracellular signaling events related to the cAMP/PKA cascade (Fig. 1). The biochemical reactions in each network are modeled as ordinary differential equations according to the mass-action kinetics with a well-mixed approximation. The resulting system of ordinary differential equations is solved using ode15s solver in the Simbiology toolbox of MATLAB (The MathWorks). The models are deposited in BioModels database (Chelliah et al., 2014) in SBML format along with tabular description (Model id: MODEL1502200000 for D1+ MSN; Model id: MODEL1502200001 for D2+ MSN).
D1+ MSN model.
The reaction network modeled for the D1+ MSN takes into account a subset of intracellular signaling triggered by the activation of the D1R and M4R in these neurons. The signaling cascades triggered by the two receptors converge on AC5, which is the predominant adenylate cyclase in MSNs (Glatt and Snyder, 1993), where M4R-activated Gi/o inhibits the D1R/Golf-induced AC5 activation. The downstream signaling module is a reparameterized version of the AC5/cAMP/PKA axis of previously developed signaling models (Lindskog et al., 2006; Oliveira et al., 2012; Gutierrez-Arenas et al., 2014). The M4R/Gi/o/AC5 signaling branch is a crucial addition in the current model. Figure 1 shows the major interactions involved in this signaling cascade. The parameter values for the AC5/cAMP/PKA signaling module were further improved on the basis of additional recent experimental evidences related to the cAMP and the PKA dynamics (Castro et al., 2013). Castro et al. (2013) have used AKAR3 to measure the PKA dynamics. Thus, we also included AKAR3 into this model to fit the experimental data.
In this model, the AC5 is assumed to be under a high basal inhibition due to a high activity of M4R resulting from the tonically active TANs. Previously published experiments show that the M4R activation at saturating concentration of agonist is sufficient to fully inhibit the D1R-induced AC5 activity in the striatal membrane preparation (Onali and Olianas, 2002; Jeon et al., 2010). This suggests a high concentration of M4R and associated Gi/o protein assembly. There are reports of nanomolar affinity (25–50 nm) of striatal M4R receptors for ACh (Quirion et al., 1989). The basal level of extracellular ACh in striatum is estimated to be ∼20–30 nm by microdialysis (Rada et al., 2010; Farrar et al., 2011), and it may even be higher as suggested by the large underestimation recently disclosed through electrochemical measurements in prefrontal cortex (Mattinson et al., 2011). Thus, the combination of high concentration of M4R and a basal concentration of ACh around its half-occupancy level suggest a high basal activity of these receptors, which would lead to a high basal inhibition of AC5.
The inputs to the D1+ MSN model were the DA and ACh concentrations. DA could either be represented as a low tonic basal level input or a transient increase in the DA concentration due to burst firing in DANs (e.g., in response to a salient reward-producing stimulus). The possible patterns for ACh release are either a tonic basal level or else a dip in ACh concentration assumed to result from the pause in TANs (e.g., concomitant with DAN bursting as seen in response to a salient stimulus) (Aosaki et al., 2010). The values for the basal level of ACh and DA used in the model are 100 and 10 nm, respectively (Heien et al., 2005). A value of 1500 nm is used for the peak amplitude of the DA transient, and ACh is assumed to be cleared out completely during a TAN pause unless stated otherwise.
D2+ MSN model.
The biochemical network modeled for the D2+ MSN also considers the AC5/cAMP/PKA signaling, but in this case the downstream signaling events are triggered by the D2R and A2aR. The signaling by D2R and A2aR also converges onto the AC5 where the D2R, via Gi/o, inhibits the A2aR/Golf triggered signaling. The signaling downstream of D2R and A2aR is similar to the D1+ MSN case and is modeled by the same core model (Fig. 1). However, few of the parameter values have been modified to fit the previously published experimental results specific to D2+ MSNs neurons (Nishi et al., 1997, 2000; Bibb et al., 1999; Lindskog et al., 2002; Bateup et al., 2008). AC5 is considered to be under a high basal inhibition in this model, too, but here due to the activity of D2R. Traditionally, it has been thought that a considerable fraction of the D2R population in these neurons is present in the high-affinity state (Cumming, 2011). However, the reported percentage of the high-affinity fraction has a high variability because the receptor affinity state critically depends on the experimental conditions (Skinbjerg et al., 2012). However, in vivo experiments using PET provide indirect evidence that ∼20%-30% of the D2R population are occupied under the basal condition (Laruelle et al., 1997), which could be approximated as the fraction of high-affinity receptors because the basal DA level is reported to be in the nanomolar range (Abi-Dargham et al., 2000; Venton et al., 2003; Dreyer et al., 2010; Spanos et al., 2013). Moreover, several FRET measurements have demonstrated that the Gi/o protein has a higher tendency to precouple with its receptors, including D2R (Bünemann et al., 2003; Nobles et al., 2005). Such kind of precoupling increases the affinity between the receptor and its ligand (Hulme and Trevethick, 2010). In our D2+ MSN model, the fraction corresponding to the high-affinity D2R is assumed to be 30% of the total D2R population, which produces a good fit to experimental data as indicated in the following sections, and this number also falls into the range of the high affinity fraction estimated in vivo (Laruelle et al., 1997). The basal concentration of DA and Adn were set at 10 and 150 nm, respectively (Ballarín et al., 1991).
The inputs to the D2+ MSN model are DA and Adn. Two possible patterns for the DA input are used. First, DA input pattern is an increase in the extracellular DA concentration due to a burst firing in DANs (e.g., in response to an unexpected reward). The other input pattern is a dip in the extracellular DA concentration due to a depression in the DAN activity (e.g., in response to the omission of an expected reward) (Schultz, 1998). The duration of the DA dip is assumed to be of similar duration as the DA peak (Roitman et al., 2008; Hart et al., 2014). These DA inputs are combined with the Adn inputs. We have taken two different types of Adn inputs into consideration: the tonic level of basal Adn (Håkansson et al., 2006) concentration, 150 nm, and an activity-dependent elevation in the Adn concentration produced in response to a high neuronal activity (Sciotti et al., 1993). The activity-dependent Adn elevation reaches its maximum ∼10 s after the stimulus and then slowly returns to the basal level (Cechova and Venton, 2008; Pajski and Venton, 2010; Sims and Dale, 2014).
Molecular phenotypes for model constraining.
The models were constrained using quantitative molecular/subcellular phenotypes collected from previously published results (Table 1). In the context of this study, the word “phenotype” is used in a broader sense to refer a set of markers/observables measured in an experimental sample, such as striatal slices or heterologous expression system, in response to a specific stimulus, such as receptor agonist or ligand uncaging (Gutierrez-Arenas et al., 2014). We used three target phenotypes (refer to “Phenotype Name” in Table 1) specific for the D1+ MSNs. The DARPP-32 phenotype, “DAsliceD32D1,” is the same as that used in a previous version of the D1+ MSN model (Nishi et al., 1997, 2000; Bibb et al., 1999; Bateup et al., 2008; Gutierrez-Arenas et al., 2014). The new phenotypes include the time-series data for receptor-G-protein and PKA activity. The time-series data were parameterized, and these parameters are considered as the phenotypic variables for the time-series data. For example, the data from “ReceptorGprotein” phenotype, which corresponds to the activation of G-proteins, were parameterized in terms of time constants obtained from fitting a monoexponential (Gutierrez-Arenas et al., 2014). Similarly, the PKA activation dynamics (in terms of AKAR3 signal) upon addition of D1 agonist from “DAsliceAKAR3D1” was also parameterized in terms of time constant and amplitude obtained from fitting a monoexponential (Eq. 1) as follows:
The PKA dynamics produced by transient D1R agonist application and DA uncaging were parameterized using the difference of two decaying exponentials (Eq. 2), resulting in three quantities: amplitude of the total response, Bmax, and two decay rates for each exponential, k1 and k2. This equation represents a quantity that has a finite source and a sink (Gutierrez-Arenas et al., 2014).
 |
The data for “ReceptorGprotein” were inferred from the activation of G-protein-dependent processes in neurons. For example, the activation of D2R could be in between 10 ms in cholinergic interneurons (Chuhma et al., 2014) to a little >100 ms in D2+ MSN GIRK currents (Ford et al., 2009; Marcott et al., 2014); and similarly, PKA activation in D1+ MSN and hippocampal neurons is also significantly faster than other cellular systems (Nikolaev et al., 2004; Castro et al., 2013). These G-protein activation data are considerably faster than the activation reported for heterologous expression system (Bünemann et al., 2003; Hein et al., 2005, 2006; Lohse et al., 2008; Vilardaga, 2012). We assume that the G-protein activation time constant range is between 10 and 100 ms. The “ReceptorGprotein” phenotype is used for constraining both D1+ MSN and D2+ MSN model. Apart from “ReceptorGprotein,” the D2+ MSN model is also constrained by D2+ MSN specific data, listed in Table 1.
Table 1.
Quantitative target phenotypes used to constrain the D1+ MSN and D2+ MSN models
| Neuron type | Phenotype name | Treatment | Marker | Phenotypic variable and values | References |
|---|---|---|---|---|---|
| D1+ MSN, D2+ MSN | ReceptorGprotein | Receptor + saturating concentration of ligand | G-protein activation (range of activation time constant = 10–100 ms; parameterized with Eq. 1) | Bmax = 100, c = 1.0, k = 10–100 | Chuhma et al., 2014; Marcott et al., 2014 |
| D1+ MSN | BasalD1 | — | cAMP | 30–100 nm | Bacskai et al., 1993; Mironov et al., 2009 |
| D32p34 | 0.4 μm | Nishi et al., 1997; Bateup et al., 2008 | |||
| D32p75 | 12 μm | Bibb et al., 1999; Bateup et al., 2008 | |||
| DAsliceD32D1 | Striatal slices + DA ≥ 10 μm; sampled at 5 min | D32p34 | 11 × basal | Nishi et al., 2000; Bateup et al., 2008 | |
| D32p75 | 0.5 × basal | ||||
| DAsliceAKAR3D1 | Striatal slices + high D1R agonist (equivalent to 10 μm DA) | cAMP | 2.5 μm | Castro et al., 2013 | |
| AKAR3 phosphorylation (parameterized with Eq. 1) | Bmax = 100, c = 1.04, k = 0.118 | ||||
| Striatal slices + high D1R agonist (equivalent to 10 μm DA) for 10 s | AKAR3 phosphorylation and dephosphylation (parameterized with Eq. 2) | Bmax = 94.73, k1 = 0.172, k2 = 2.317 | |||
| Striatal slices + transient DA uncaging (for 1.0 s) | AKAR3 phosphorylation and dephosphylation (parameterized with Eq. 2) | Bmax = 92.34, k1 = 0.736, k2 = 2.726 | |||
| Striatal slices + transient DA uncaging (for 0.3 s) | AKAR3 phosphorylation and dephosphylation (parameterized with Eq. 2) | Bmax = 95.23, k1 = 0.698, k2 = 3.006 | |||
| Striatal slices + transient DA uncaging (for 0.18 s) | AKAR3 phosphorylation and dephosphylation (parameterized with Eq. 2) | Bmax = 45.111, k1 = 0.731, k2 = 6.1 | |||
| D2+ MSN | BasalD2 | — | D32p34 | 0.7 μm | Nishi et al., 1997; Bateup et al., 2008 |
| D32p75 | 13 μm | Bibb et al., 1999; Bateup et al., 2008 | |||
| DAsliceD32D2 | Striatal slices + DA ≥ 10 μm; sampled at 5 min | D32p34 | 0.4 × basal | Bateup et al., 2008 | |
| D32p75 | 1.5 × basal | ||||
| D2AntagonistSliceD32 | Striatal slices + D2R antagonist; sampled at 15 min | D32p34 | 2.0 × basal | Bateup et al., 2008 | |
| D32p75 | 0.9 × basal | ||||
| A2aAntagonistSliceD32 | Striatal slices + A2aR antagonist; sampled at 15 min | D32p75 | 2 × basal | Lindskog et al., 2002 |
The models also take into account that the G-proteins, both Gi/o and Golf, are acted upon by GTPase-activating proteins (GAPs). GAPs, for instance, RGS (Regulator of G-protein signaling), increase the GTPase activity of heterotrimeric G-protein 20–40 times resulting in their fast deactivation (Doupnik et al., 1997). RGS9-2 is an RGS enriched in striatum and is known to produce faster deactivation of Gi/o proteins (Xie et al., 2012). AC5 also acts as a GAP for the Gs proteins increasing the Gs GTPase activity at least fivefold (Scholich et al., 1999); and by following the argument of homology between Gs and Golf, we assume that it acts as a GAP for Golf.
Computing the interaction between two input signals.
The strength of interaction between two different input signals, Signal 1 and Signal 2, affecting the same marker (e.g., active PKA) at each time step of a simulation is quantified with synergy(t) which is defined as follows:
 |
where synergy(t) is the synergy between Signal 1 and Signal 2 at the level of PKA calculated at time t, PKAstimulated (Signal 1, Signal 2, t) is the PKA activation level in the presence of both Signal 1 and Signal 2 at time t, PKAstimulated (Signal 1,t) and PKAstimulated (Signal 2,t) are the PKA activation level in response to either Signal 1 or Signal 2 at time t, and PKAbasal is the basal PKA level at t = 0. A similar synergy(t) calculation is done for another effector whenever required, like the ratio of PKA and PP1.
We also use mean synergy (μ[synergy]) as a single quantity to represent the overall synergy of interaction for a simulation. μ[synergy] is the mean of synergy values calculated every time-step (0.05 s) between two time points t1 and t2. We calculate the μ[synergy] for the first hundred seconds of the simulation. Thus, in our case, t1 = 0 and t2 = 100.
According to this phenomenological metric, the interaction is additive if μ[synergy] = 1, synergistic for μ[synergy] > 1, and antagonistic for μ[synergy] < 1.
Results
The constrained models reproduce a range of experimental data
In this study, we developed quantitative models for interaction between neuromodulatory signals and their effects on AC5/cAMP/PKA signaling for the two MSN types, D1+ MSN and D2+ MSN (Fig. 1). The D1+ MSN model is an updated version of previously published models (Lindskog et al., 2006; Oliveira et al., 2012; Gutierrez-Arenas et al., 2014). The current D1+ MSN model includes an additional signaling branch for M4R activation by ACh and the further downstream inhibition of AC5. Similar to the D1+ MSN model, the D2+ MSN model also features the AC5/cAMP/PKA signaling. However, the receptors involved in the D2+ MSN model are D2R and A2aR, which converge onto AC5. Both models have been constrained to data from the respective MSN type and reproduce various kinetic and steady-state experimental data listed in Table 1. The receptor and the G-protein reaction parameters in each of the models are constrained within a range of previously published observable activation kinetics in neurons. Both the models closely follow the experimentally observed G-protein activation range (Table 1, “ReceptorGprotein”) (Fig. 2A1,B1,A2,B2).
Figure 2.
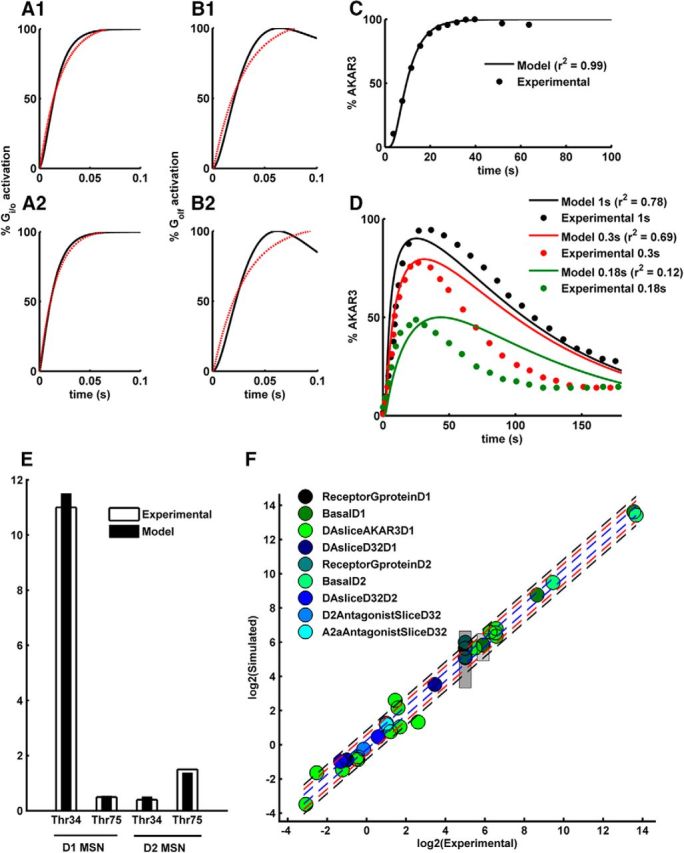
Model fitting to experimental data mentioned in Table 1. A, Gi/o activation rate for (A1) M4R and (A2) D2R in the presence of saturating concentration of their respective ligands (phenotype “ReceptorGprotein”). Black curve represents the simulated activation. Red dotted curve represents a best fitted monoexponential with time constant in the range of neuronal G-protein activation mentioned in ReceptorGprotein. B, Golf activation rate for (B1) D1R and (B2) A2aR in the presence of saturating concentration of their respective ligands (phenotype “ReceptorGprotein”). Black curve represents the simulated activation. Red dotted curve represents a best fitted monoexponential with time constant in the range of neuronal G-protein activation mentioned in ReceptorGprotein. C, Percentage of AKAR3 phosphorylation in response to 10 μm tonic DA (phenotype “DASliceAKAR3D1”). D, Percentage of AKAR3 phosphorylation in response to different duration of DA pulse (phenotype “DASliceAKAR3D1”). y-axis is normalized with respect to the maximum phosphorylation produced by 10 μm tonic DA. E, Changes in the DARPP32 Thr-34 and Thr-75 phosphorylation level measured 5′ after the application of 10 μm DA in the D1+ MSN and D2+ MSN (phenotype “DAsliceD32D1” and “DAsliceD32D2”). F, Experimental versus Simulated values for all the phenotypic variables. Blue lines indicate 20% divergence from experimental values. Red lines indicate 50% divergence from experimental values. Black lines indicate 80% divergence from experimental values. Patches represent the range for the phenotypic variables cAMP basal level and G-protein decay constants. The legends represent various phenotype names listed in Table 1. Each phenotype contains several points corresponding to different phenotypic variables. The “ReceptorGproteinD1” and “ReceptorGproteinD2” correspond to the “ReceptorGprotein” phenotype for the D1+ MSN and D2+ MSN model, respectively.
The models are furthermore constrained to match the basal and activated levels of a number of different signaling molecules. In the case of D1+ MSNs, the basal activity of the D1R is low. Thus, the basal level of cAMP is also expected to be low. There is no basal cAMP measurement for MSNs. However, the cAMP level has been estimated for other neuron types having low basal adenylyl cyclase activity (Bacskai et al., 1993; Mironov et al., 2009), and the current D1+ MSN model is tuned to reproduce a similar cAMP level. The steady-state level of cAMP following a high D1R specific agonist (SKF38393) has been previously reported to be >2.4 μm for MSNs using Epac1-camp biosensor (Castro et al., 2013), and this is captured by the D1+ MSN model presented here. The model also reproduces the previously reported fast PKA dynamics measured in striatal slices using the AKAR3 biosensor in response to sustained application of SKF38393 or a brief (1.0, 0.3, and 0.1–0.2 s) DA uncaging (Fig. 2C,D) (Castro et al., 2013). The basal and the stimulated levels of the phosphorylated forms of DARPP-32 have been used to constrain both the D1+ MSN and D2+ MSN models (Table 1). Figure 2E shows an example of how both models match the DARPP-32 phosphorylation changes after 5 min incubation of striatal slices with a D1R agonist in D1+ MSNs and a D2R agonist in D2+ MSNs. In general, the D1+ MSN and the D2+ MSN model versions have been constrained with respective neuron-specific data, and there is good quality of fit (i.e., <20% divergence) for most of the phenotypic variables for both models (Fig. 2F). However, there are few points that diverge 80%, which are mostly the dephosphorylation phase of the AKAR3 (Fig. 2D).
A DA peak together with an ACh dip in the D1+ MSNs synergistically activate PKA
We used the D1+ MSN model to study the potential integration of the transient DA peak with the concomitant ACh dip via the D1R and M4R. A transient increase in DA results in an increased activation of D1R in D1+ MSNs leading to an increased level of cAMP and PKA activation. Simulation experiments with the D1+ MSN model suggest that the level of PKA activation by a DA peak (1 s in our simulations) depends on the extracellular concentration of striatal ACh (Fig. 3A). It is known that the striatal ACh level could vary depending upon the activity of other brain regions (Rada et al., 2010). The DA peak produces low PKA activation if accompanied by high tonic ACh levels. This is due to the activation of M4R, which inhibits the cAMP/PKA pathway.
Figure 3.

PKA response in D1+ MSNs following ACh and DA transients. A, PKA activation produced by a 1 s DA 1.5 μm elevation as a function of varying basal level of tonic ACh; 100% ACh represents the assumed basal level, which is 100 nm. y-axis is normalized to steady-state level of active PKA produced by saturating D1R agonist in the presence of basal ACh. B, Active PKA levels in response to the different combinations of inputs. The DA and ACh signals have the durations of 1 s and 0.4 s, respectively. y-axis is normalized to steady-state level of active PKA produced by saturating D1R agonist in the presence of basal ACh. ↑, Peak; ↓, dip. C, Synergy at the level of PKA activation following a DA peak and ACh dip starting at the same time. Synergy is calculated at every time-step of the simulation using Equation 3. Mean synergy, μ[synergy], is 2.4. D, μ[synergy] (top) and maximum PKA level (bottom) as a function of the ACh dip amplitude. ACh dip amplitude is represented in percentage of maximum dip amplitude: 100% corresponds to full extracellular ACh depletion, and 0% means no dip. E, PKA increase for varying DA amplitude in the presence and absence of an ACh dip. Vertical line indicates the assumed basal concentration of DA, 10 nm. Amplitudes >10 nm represent a DA peak, and <10 nm represent a DA dip. Transient DA dips are not detected by D1+ MSNs. F, PKA fold increase (maximum/basal) as a function of DA peak amplitude and DA peak duration in the presence of (F1) a 0.4-s-long ACh dip and (F2) a 0.5-s-long ACh dip. Zero percentage in y-axis represents no peak, and 100% represents the 1500 nm amplitude of the peak. x-axis and y-axis labels are shared for both the plots.
Often a DA increase resulting from a salient stimulus is accompanied with pausing activity in TANs and thus an ACh dip. The ACh dip is known to be sensed by various receptors, such as nAChRs (Drenan et al., 2010). However, the capability of adenylyl cyclase-coupled M4R in sensing this ACh dip is not known. Using the D1+ MSN model, we therefore explored whether, and under which circumstances, the ACh dip could be sensed by M4R, and how the ACh signaling is integrated with the DA transients. As mentioned previously, a DA transient alone, in the presence of tonic ACh, is not able to produce high levels of PKA activation. However, the same DA transient is able to produce high PKA activation when accompanied with an ACh dip (Fig. 3B). Thus, the simulations suggest that D1+ MSNs are able to sense the ACh dip via M4R, and the TAN pause thus would gate the D1R-induced PKA activation. However, the ACh dip itself is not able to produce any significant PKA activation (Fig. 3B). Even though the ACh dip itself leads to the disinhibition of AC5, a further D1R-dependent AC5 activation is required to produce downstream cAMP/PKA because the basal AC5 activity in D1+ MSN is not high enough to produce significant cAMP/PKA activation by itself for such as small duration. Because the basal DA level is not enough to produce any significant D1R activation, a DA peak, therefore, with an ACh dip is required for significant PKA activation.
The synergy of interaction between a DA peak and an ACh dip for every time step computed according to Equation 3 is shown in Figure 3C. The mean synergy value, μ[synergy], for the interaction is 2.4 (see Materials and Methods). This highly synergistic response is the way in which a brief DA peak above a low basal level and a brief ACh dip from a high basal level could interact at the AC5 signaling axis for colocalized D1R and M4R. A high synergy for the interaction between a DA peak and an ACh dip implies that the system acts as an AND gate for brief DA “on” and ACh “off” signals. Thus, a dip in ACh gates the effect of DA transients in D1+ MSNs. As the synergy increases, the ACh gate thus becomes more important. One consequence is that the basal ACh tone produced by TAN activity has a potential to filter out a brief DA pulse that is not accompanied by an ACh dip. The synergy of interaction and the PKA activation produced by the concomitant DA peak and ACh dip critically depend on the amplitude of the ACh dip (Fig. 3D). As the amplitude of the ACh dip reduces, the synergy and the PKA level decrease.
The pause in TAN activity is at times preceded or followed by a burst (Morris et al., 2004). Even though the physiological role of the associated TAN burst is not clear, this phenomenon seems to be region-dependent (Chuhma et al., 2014). We also looked at how an ACh peak could affect the DA-dependent cAMP/PKA activation. The PKA activation produced by a DA peak is slightly reduced in the presence of an ACh peak compared with the DA peak alone, but the difference is small (Fig. 3B). The effect of an ACh peak is considerably low because AC5 is already under high inhibition even at the basal level of ACh and there is little room for further inhibition with increasing ACh. Thus, an ACh dip rather than an ACh peak is sensed via M4R in D1+ MSNs, and there are no significant differences in PKA response between the multiphasic ACh dip and a normal dip.
DA signals encode the error in the reward prediction. A higher RPE leads to a higher DAN activity (Fiorillo et al., 2003). The ACh tone keeps a strong inhibitory control of the PKA activation produced even at high DA levels and the ACh dip is important to produce high PKA changes (Fig. 3E). There is no effect on PKA level in D1+ MSNs in response to negative DA amplitudes (i.e., amplitudes less than the basal DA level of 10 nm) (Fig. 3E). Such negative DA amplitudes are thought to represent omission of an expected reward. Thus, the cAMP/PKA signaling axis in D1+ MSN is responsible for sensing only the positive RPE signals. However, we wondered whether there is any role of the peak duration in coding the magnitude of positive RPE. Thus, we looked at how the PKA activation is affected by the duration of the DA peak compared with the DA amplitude. The simulation results suggest that the PKA activation is mostly affected by the DA peak amplitude rather than the peak duration (Fig. 3F1). There is certainly a variation in the PKA activation produced by varying the peak duration, but it is significantly lower than the change produced by varying the peak amplitude. Thus, the amplitude of DA peak rather than its duration seems to be more relevant to encode the graded level of PKA activation. However, whether a DA peak of given amplitude produces a significant PKA activation depends on the ACh dip (e.g., some values of amplitude for the DA peak may not produce PKA activation in the presence of a shorter ACh dip, but the same peak could produce PKA when coupled to a slightly longer dip) (Fig. 3F1,F2). This is especially true for lower values of the amplitude. Thus, whether a D1-dependent PKA signaling occurs in response to a DA peak is flagged by the ACh dip, which goes along with the previously suggested idea that the ACh dip sets the temporal frame for DA peak response (Morris et al., 2004).
Synergy between a DA peak and an ACh dip depends on signal duration
The pause in the TANs in response to a reward signal mirrors the burst in the DANs (Aosaki et al., 1995; Morris et al., 2004). A similar relation may hold for the duration of the peak and the dip in extracellular DA and ACh, respectively. As there are not direct experimental measurements of the duration of the dip in ACh concentration, and the duration of the DA peak also varies according to the mismatch between the expected and the delivered reward, we explored the dependence of the μ[synergy] of PKA activation on the DA peak and the ACh dip duration. The results show that the gating potential of the ACh dip depends critically on its duration as a dip longer than 250 ms is necessary to generate significant synergy values (Fig. 4A). A DA peak in the presence of a very brief (<100 ms) ACh dip does not produce any significant synergy because such a dip does not lead to sufficient disinhibition of AC5 to gate the DA peak-induced AC5·Golf complex formation. This threshold-like relation between synergy and dip duration has the potential to act as a filter for DA noise if it is not accompanied by an ACh dip longer than a certain duration. In addition to synergy, the actual PKA activation too follows such a threshold-like behavior (Fig. 4B).
Figure 4.

Temporal dependence of synergy in D1+ MSNs. A, μ[synergy] for different combinations of the duration of the DA peak and the ACh dip. In these calculations, the start of the peak and dip coincide with each other. B, Maximum activated PKA level for different combinations of the duration of the DA peak and the ACh dip. The PKA activation values are normalized to steady-state level of active PKA produced by saturating D1R agonist in the presence of basal ACh. C1, μ[synergy] as a function of delay between the ACh dip and the DA peak. The delay in the x-axis represents the time of onset of the ACh dip minus the time of DA peak (tAch dip − tDA peak). A negative value means that the ACh dip is starting before the DA peak and vice-versa. C2, Maximum activated PKA level as a function of delay between the ACh dip and the DA peak. The activated PKA level is normalized to steady-state level of active PKA produced by saturating level of D1R agonist in the presence of basal ACh. D, Maximum activation level of PKA in the absence of an ACh dip and with varying DA signal duration. y-axis is normalized to steady-state level of active PKA produced by saturating level of D1R agonist (10 μm).
The D1+ MSN model also suggests that the delay between the DA peak and ACh dip affects the synergy produced (Fig. 4C1). If the two signals arrive simultaneously, then the synergy is high. However, subsecond delay between these signals still produces synergy, suggesting that slight variations in the timing of the arrival of DA peak and ACh dip may not impair PKA activation (Fig. 4C2).
Interestingly, the relative gating potential of the ACh dip decreases at longer DA peaks of 5–10 s, which could produce significant PKA activation even in the absence of an ACh dip (Fig. 4D). Recently, such long-duration DA elevations are shown to be physiologically relevant and are involved in providing a drive toward distant goals (Howe et al., 2013). In this work, however, we restrict the analysis to transients no longer than 1 s.
DA dip, and not DA peak, affects the PKA level in D2+ MSNs
Similar to the D1+ MSNs, the D2+ MSNs also detect extracellular DA changes. In this case, the response to DA is mediated via the D2R, which is coupled to Gi/o, inhibiting the cAMP production. However, the D2R-mediated PKA changes produced by transient DA signal remain largely unknown. Here, we used the D2+ MSN model to explore the PKA activation produced by DA transients in D2+ MSNs. In our simulations, we find that the effect of a DA peak on the PKA activation is very little in the D2+ MSNs (Fig. 5A). The fraction of high-affinity D2R in the model is ∼30% (see Materials and Methods), and the activity of basal level of DA on this fraction is sufficient to keep the basal PKA activation at a low level despite the tonic A2aR activity. Thus, a further increase in the D2R activation by a transient DA peak does not lead to a further decrease in the PKA level. Because the DA peak had no significant effect on the PKA level in D2+ MSNs, we further tested the possible effect of a DA dip on these neurons. DA dips are often associated with the omission of an expected reward (Schultz, 1998). The D2+ MSN model detects a DA dip (1 s in our simulations); and in response to this DA dip, there is a significant increase in the PKA activation (Fig. 5A). In the basal state of the D2+ MSN, the inhibition posed by the basal D2R activity keeps the PKA level low despite the basal A2a activity (Fig. 5B), and a DA dip removes the D2R inhibition, resulting in an increased PKA activation. This PKA activation trend produced by the D2+ MSN model (i.e., a DA dip is sensed by D2+ MSNs) is in accordance with some of the hypotheses in field (Frank, 2005; Dreyer et al., 2010).
Figure 5.

Response of D2+ MSNs following changes in DA and Adn stimulation. A, PKA activation in response to a DA peak (1500 nm) and a DA dip (0 nm) of 1 s duration. The y-axis is normalized to steady-state level of active PKA produced by saturating A2aR agonist in the presence of basal DA. ↑, Peak; ↓, dip. B, PKA activation produced by the basal Adn as a function of varying DA level. The y-axis is normalized to steady-state level of active PKA produced by saturating A2aR agonist in the presence of basal DA. The x-axis represents the basal level of DA as a factor of assumed basal level of 10 nm. C, PKA activation in response to a DA dip, Adn elevation, and DA dip in the presence of Adn elevation. The y-axis is normalized to steady-state level of active PKA produced by saturating A2aR agonist in the presence of basal DA. ↑, Increase; ↓, dip. D, The activation half-time for PKA response in response to DA dip, Adn elevation, and DA dip in the presence of Adn elevation. E, PKA fold change (maximum/basal) as a function of DA dip duration and DA dip amplitude. DA dip amplitude is represented in percentage DA dip amplitude: 100% corresponds to full DA depletion upon a dip, and 0% corresponds to no dip at all. F, Free Gi/o-βγ level in response to a DA peak and a DA dip (1 s). The y-axis is normalized to steady-state level produced by high A2aR agonist. ↑, Peak; ↓, dip.
It has previously been observed that dopaminergic transmission is often accompanied with Adn release in the striatum (Cechova and Venton, 2008). This Adn elevation has been attributed to the level of neuronal activity such that high activity leads to increased Adn elevation (Pajski and Venton, 2010). This increase in the Adn concentration is rather slow and reaches its maximum in ∼10 s and then slowly decays to the basal level (Cechova and Venton, 2008; Pajski and Venton, 2010). We looked at how this Adn change interacts with the DA dip and find that the DA dip-induced PKA activation in D2+ MSNs becomes more pronounced in the presence of the activity-dependent Adn increase (Fig. 5C). Thus, in these neurons, similar to the D1+ MSN, the change in the activity of the colocalized receptors potentiates the DA-dependent PKA response. However, unlike the D1+ MSNs where the interaction between the DA peak and ACh dip is synergistic, the interaction between the DA dip and the Adn increase in D2+ MSN is not strongly synergistic because DA dip could produce significant PKA activation alone (Fig. 5C). The Adn elevation alone could also produce PKA activation even though the level of PKA is much lower than the level produced by a DA dip. However, the PKA activation onset kinetics is significantly different between a DA dip response and an Adn elevation response (Fig. 5C,D). The PKA response produced by the Adn elevation alone is considerably slower than the response produced by the DA dip. The difference between response time is not due to any receptor-specific kinetics because the kinetics of all the receptors in this study is constrained by the same set of data (Table 1). Rather, this difference emerges at the level of AC5. In the case of “Adn elevation alone,” there is a higher competition between D2R-activated Gi/o and A2aR-activated Golf, which slows down the rate of AC5 activation and thus PKA activation.
Whether the amplitude of a dip is sufficient enough to encode the negative RPE information is still not clear, and there exist observations either way (Bayer and Glimcher, 2005; Hart et al., 2014). There have also been suggestions that the magnitude information could be encoded with the duration of the dip (Bayer et al., 2007). Thus, we looked at how the PKA activation is affected by the dip duration and amplitude. The PKA activation in response to a DA dip depends on both amplitude and duration of the dip (Fig. 5E) unlike the activation produced by DA peak in D1+ MSNs where the peak amplitude is much more relevant than the duration. It required a 70%–100% reduction in the DA concentration to produce any significant PKA change (Fig. 5E). The dip amplitude has only a very small dynamic range (70%–100% of dip amplitude) to encode the whole range of PKA activation. In terms of actual DA concentration, assuming 10 nm as basal level, this would translate into a very small range (0–3 nm). In such a steep system, a concentration noise of even 1 nm could significantly affect the PKA activation, thus making the system less robust. On the other hand, the dip duration (for deeper dips) shows a more graded range to encode the whole PKA activation (e.g., PKA changes in response to different dip durations for a 100% dip amplitude in Figure 5E). Thus, it could be inferred that a role of dip duration in coding the magnitude information of negative RPE could make the downstream signaling, at least at the level of PKA activation, more robust compared with when it is coded only by DA amplitude.
The PKA activation produced by the DA dip in D2+ MSNs suggests that the cAMP/PKA signaling axis in this neuronal type mainly detects the negative RPE signals, associated with reward omission, depending upon basal state of the system. This is in contrast with the D1+ MSNs where the cAMP/PKA cascade seems to be responsible for sensing the positive RPE signals. cAMP/PKA signaling is not the only downstream effects of D2R activity. D2R activation modulates L-type calcium channels and GIRK channels via the G-βγ subunit (Hernandez-Lopez et al., 2000; Ford et al., 2009). In our model, because the occupancy of D2R is not saturated at basal DA level, a DA peak could lead to an increase in the Gi/o activation; thus, free G- βγ is produced (Fig. 5F). This increase in free G-βγ could lead to the activation of respective effector, like GIRK2. G-βγ has a low affinity to GIRK2 (Wang et al., 2014); thus, GIRK2 modulation might require a higher level of G-βγ, which is produced by a DA peak. This aligns with the known observations about D2-dependent modulation of GIRK in response to a DA peak (Marcott et al., 2014). The DA peak also increases the α subunit of Gi/o, but this increase does not affect the cAMP/PKA levels significantly because the cAMP/PKA signaling axis is already highly inhibited by the basal level of DA and a further increase in Gi/o-α has a little effect as previously mentioned in this section. Thus, the effects of DA transients on the D2+ MSNs are effector specific (i.e., D2R is able to sense both a DA peak and a dip, but whether a peak or dip produces a downstream effect could significantly depend on the effector).
DARPP-32 can significantly amplify the effect of neuromodulators on the PKA to PP1 activity ratio (PKA:PP1)
PP1 is an important phosphatase in MSNs, which dephosphorylates several PKA targets, such as GluR1. Thus, the PKA-to-PP1 ratio, , is an important variable affecting the phosphorylation level of downstream signaling proteins, which are substrates to both PKA and PP1. Thus, we looked at the changes in PKA:PP1 in response to the DA transients in D1+ MSNs and D2+ MSNs. In both D1+ and D2+ MSNs, the PKA:PP1 follows the same trend as the PKA activation. The PKA:PP1 increase produced by a concomitant DA peak and an ACh dip in D1+ MSNs is significantly higher than the PKA:PP1 produced by the DA peak or ACh dip alone (Fig. 6A1). Similarly, in the D2+ MSNs, there is an increase in PKA:PP1 in response to a DA dip; and in the presence of an Adn elevation, the effect produced by the DA dip becomes even more pronounced. Like PKA activation, PKA:PP1 increases in response to DA transients, but the amplitude of PKA:PP1 change is significantly higher than PKA (Figs. 3B, 5C, 6A1,A2). The higher level of PKA:PP1 compared with the PKA activation is produced by DARPP-32 via the PKA-dependent inhibition of PP1 and via the phosphorylation of the Thr-34 residue. Simulations of the D1+ MSN and D2+ MSN models with the mutation of Thr-34, by switching off the phosphorylation of this residue, indeed confirm this. The Thr-34 mutant version of the D1+ MSN and D2+ MSN models produces PKA:PP1 similar to the PKA activation level (Fig. 6B1,B2,C1,C2). Thus, the upstream cAMP/PKA signals in both MSNs are amplified at the PKA:PP1 level via DARPP-32 Thr-34 phosphorylation. In D1+ MSNs, the synergy between the DA peak and ACh dip is also enhanced at the level of PKA:PP1 compared with the synergy at the level of PKA (Fig. 6D shows synergy(t) calculated for every time step using Eq. 3), suggesting that the downstream effect of the DA peak and ACh dip integration stays on for a longer duration. This illustrates the importance of DARPP-32 in translating a transient neuromodulator signal into a longer-lasting effector response. As mentioned above, the DA amplitude correlates to the error in the reward prediction. In the case of D1+ MSN, the PKA:PP1 increased with an increase in DA amplitude. At higher amplitudes (>1000 nm), the PKA:PP1 change is significantly higher than the PKA change. The difference between PKA:PP1 and PKA is abolished in the Thr-34 mutant, thus indicating that DARPP-32 is critical in the process of amplifying the positive RPE signal encoded by DA amplitude further downstream (Fig. 6E1). In contrast, the PKA:PP1 in D2+ MSN increased in response to a dip in DA level (Fig. 6E2). The basal DA amplitude for the model is 10 nm, and the DA amplitudes below this value in Figure 6E2 represent a decrease in the DA level, which is often associated with omission of an expected reward.
Figure 6.
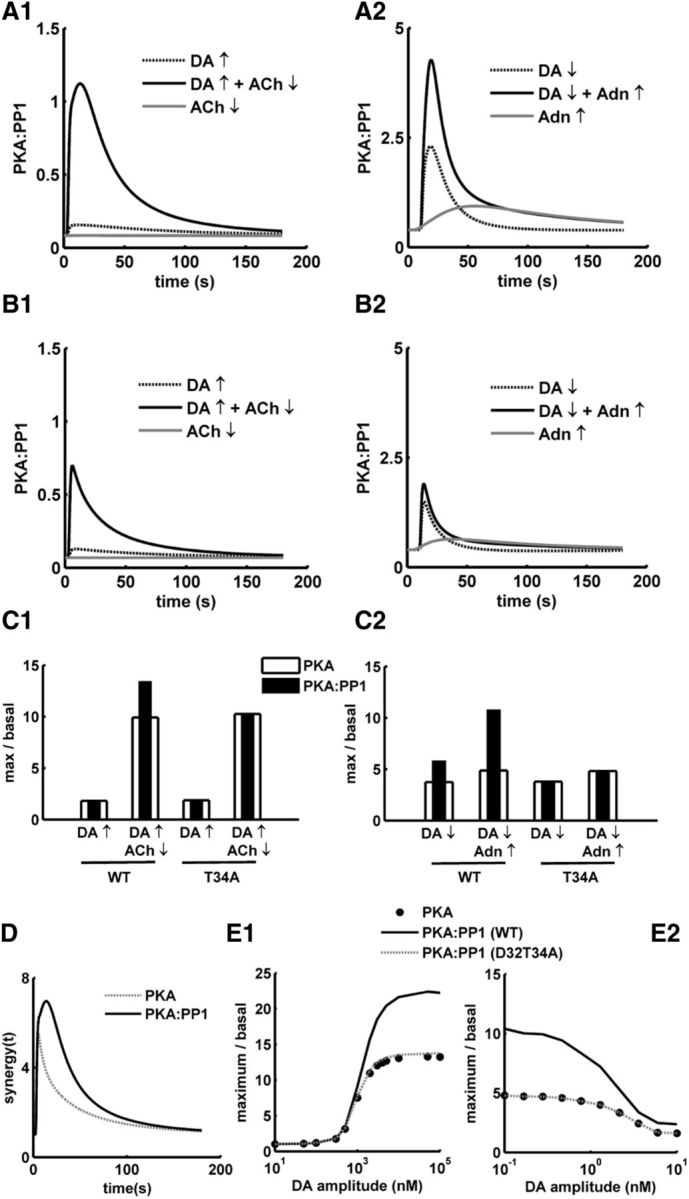
Amplification of signal downstream by DARPP-32. A, PKA:PP1 ratio in response to various input combinations in (A1) D1+ MSN and (A2) D2+ MSN models. B, PKA:PP1 ratio in response to various input combinations for T34A mutant in (B1) D1+ MSN and (B2) D2+ MSN models. C, Comparison of the change in PKA:PP1 ratio and PKA in response to various input for the wild-type (WT) and T34A mutant in (C1) D1+ MSN and (C2) D2+ MSN. D, Comparison of the synergy(t) at PKA:PP1 level and the synergy(t) produced at PKA level in D1+ MSN. E, PKA and PKA:PP1 in both wild-type (WT) and D32T34A (T34A) as a function of varying dopamine amplitude. E1, D1+ MSN model. DA amplitude in x-axis represents the amplitude of a DA peak. E2, D2+ MSN model. The basal level of DA is 10 nm, and the amplitude in x-axis represents the decreased level of DA for a dip.
Discussion
PKA activation in MSNs is crucial for long-term plasticity events in corticostriatal synapses (Shen et al., 2008a; Wickens, 2009; Lerner and Kreitzer, 2012). In this study, we investigated, using kinetic ordinary differential equation modeling, the interaction between the Golf- and Gi/o-coupled GPCRs (M4R and D1R in D1+ MSNs and D2R and A2aR in D2+ MSNs) on the AC5/cAMP/PKA signaling for detecting brief reward-related neuromodulator transients and how these interactions could shape the downstream PKA signaling.
AND gate in D1+ MSNs: PKA activation upon convergent ACh dip AND DA peak
Mice knock-out of M4R in D1+ MSNs shows a hyperlocomotion phenotype characteristic of sustained D1R stimulation (Jeon et al., 2010). M2/M4 antagonists were among the earliest anti-parkinsonian medication (Olianas and Onali, 1996) possibly because they activate the direct pathway, and M4 allosteric activators are also under study for the treatment of schizophrenia (Leach et al., 2010) as they shift the balance of activity toward the indirect pathway, just as D2R antagonists, such as haloperidol. However, despite these facts and high expression levels of M4R in D1+ MSNs, very little is known about its role in processing striatal ACh reward-related transients, such as an ACh dip, which co-occurs with a peak in DA. In this study, we advance the prediction that M4R could sense this reward-related ACh dip through a synergistic interaction with the concomitant DA peak acting through D1R, resulting in a significant PKA activation not achievable with any of the transients independently. This coincidence detection capability may seem redundant if the ACh dip and DA peak are tightly correlated (Graybiel et al., 1994; Threlfell et al., 2012); however, the picture is more diverse. In addition to dopaminergic control, the interruption of the autonomous tonic firing of TANs by brief pauses and bursts is also controlled by cortical and thalamic innervations (Doig et al., 2014), GABAergic innervation from VTA (Brown et al., 2012), and possibly other circuits (Lim et al., 2014). Furthermore, in striatal slices, the induction of a pause in TANs by a DAN burst is highly region-dependent (Chuhma et al., 2014). In the face of these various relations between ACh and DA transients, the synergistic interaction between M4R and D1R on the AC5/cAMP/PKA could be a signaling module specialized in detecting a subset of them. In addition, the coincidence detection capability predicted here would act as a filter of very brief local changes in DA and ACh concentrations arising either from random fluctuations (Faisal et al., 2008) in the mechanisms regulating these transients or from the pace-making activity of these neurons (Grace and Bunney, 1984; Zhang et al., 2009).
A requirement for this synergistic interaction to occur is the high and low occupancies in basal conditions of M4R and D1R, respectively. If the basal activation of M4R decreases due to local and stable changes in the level of ACh, this would open up the possibility for an isolated DA peak to produce PKA even without an ACh dip and thus reducing the potential for synergy. However, there seem to be adaptations in M4R density to counter the changes in ACh level (Bernard et al., 1999), which could be interpreted as the basal inhibition is maintained even on the face of long-term changes in the ACh level. This could mean that the M4R-dependent basal inhibition, and thus synergy, is less variable and might be physiologically important. The second important requirement for synergy is the colocalization of these receptors. The M4R and D1R are both found in the soma and distal dendritic spines (Hersch et al., 1994, 1995); albeit little is known about the colocalization of these receptors with regard to nanoscale resolution.
ACh transients could still be directly detected by D1+ MSNs by other mechanisms (e.g., isolated ACh pauses), such as the one arising on conditioning stimulus presentation in overtrained animals (Aosaki et al., 1994), and could be processed by M1R; and indeed, pauses in M1R activation have been suggested to promote LTD in both MSN types (Wang et al., 2006).
Gate opening to tonic Adn: PKA activation upon DA dip on D2+ MSNs
Unlike D1+ MSNs, the results from the D2+ MSN model suggest that the DA peak has no significant effect on the active PKA levels of D2+ MSNs. However, a DA dip, which represents an omission of expected reward, produced a significant increase in the D2+ MSN PKA level. Systemic administration of the antipsychotic haloperidol, a D2R antagonist, activates PKA in D2+ MSNs of mice (Bateup et al., 2008). The PKA activation in response to the DA dip is due to the disinhibition of tonic A2aR activity in the D2+ MSNs, which, under the basal condition, is effectively opposed by a basal D2R activation. However, this may not mean that D2R sites are saturated under the basal condition, as previously thought (Dreyer et al., 2010), rather a small fraction of the high affinity D2R sufficiently inhibit cAMP production. The PKA triggered by a DA dip might facilitate LTP induction, which aligns with the effect of A2aR agonists and D2R antagonist on D2+ MSNs (Shen et al., 2008b; Lerner and Kreitzer, 2012). A hypothesis about such an effect of a DA dip already exists (Frank, 2005; Cox et al., 2015), and our results support them with subcellular details and highlight the importance of receptor and neuromodulator interaction in the process. The PKA response resulting from a DA dip is even more pronounced in the presence of striatal Adn elevation. There are several reasons for the Adn elevation, but the physiological importance of such elevation is currently not clear (Cechova and Venton, 2008; Pajski and Venton, 2010; Sims and Dale, 2014). A hypothesis could be that, in a series of repeated trials, if the adenosine level increases with each passing trial due to increased striatal activity, then this could make the system more prone to extinction. This, of course, assumes that the intertrial duration is less than the Adn decay duration and the extinction process is related to the cAMP/PKA signaling in D2+ MSN.
Even though the cAMP/PKA signaling in D2+ MSNs is largely insensitive to a DA peak, this does not necessarily mean that a peak does not hold any information for this neuron type (e.g., a DA peak significantly increases the level of Gi/o-βγ in these neurons) (Fig. 5F), and such an increase might have physiological roles considering the Gβγ-mediated regulation of ion channels (Hernandez-Lopez et al., 2000). Such a Gβγ-dependent GIRK activation in response to a peak is observed for D2+ MSNs (Ford et al., 2009; Marcott et al., 2014). Thus, D2+ MSNs could sense both dips and peaks of DA at the receptor level, but the downstream effects of these signals are effector specific.
The Golf/AC5/Gi/o core, a common signaling motif for opposing functions
The cAMP/PKA signaling is highly inhibited by the basal activity of Gi/o in both types of MSNs even though the neuromodulators responsible for this basal activity are different. Such a basal inhibition appears due to a combination of tonic neuromodulator level. The parameters, which are most critical for the current models, are related to Gi/o-coupled GPCR activation–deactivation cycle. These are responsible for maintaining the basal level of AC5 inhibition. To a lesser extent, the model results could also be affected by the Golf-coupled GPCR activation kinetic parameters and the AC5-related parameters. These parameters affect the efficacy of receptor activation on AC5. Apart from the kinetic parameters, the total amounts of chemical species, which control the tonic inhibition of AC5, are also important. A transient dip in the levels of the neuromodulator coupled to Gi/o operates as a highly dynamic gate for the Golf-dependent activation of AC5/cAMP/PKA signaling in these neurons. Fast inactivation of the Gi/o protein is a critical requirement for the transient dip to act as an effective gate-opener, which suggests an important role of GAP proteins, which are known to be expressed in MSNs (Gold et al., 1997; Xie et al., 2012).
Even though the configuration of the G-protein system is similar between the two neuronal types (i.e., high basal activity of Gi/o and a gating of Golf signaling by a dip), they serve opposite function in the context of reward learning. In D1+ MSN, the PKA activation is produced by a DA peak; whereas in D2+ MSNs, a DA dip affects PKA (Figure 7), which aligns with the notion that positive RPE signals lead to the potentiation of corticostriatal synapses onto D1+ MSNs, whereas negative signals rather potentiate D2+ MSN (Frank, 2005). It is thought that the DA amplitude is sufficient to encode both positive and negative RPE (Hart et al., 2014). However, the PKA activation suggests that amplitude might be sufficient in the case of positive RPE; whereas for negative RPE, the duration might also play an important role (Figs. 3F1, 5E) (Bayer et al., 2007). Outside the context of reinforcement learning, the basal inhibition of AC by Gi/o and gating of Golf (Gs) signaling by a decrease in Gi/o could be viewed as a general mechanism to filter out spurious Golf (Gs) signals and should be further explored and extended for cell types other than MSNs.
Figure 7.

Overview of the interaction between ACh dip and DA peak in D1+ MSN and DA dip and Adn in D2+ MSN.
Footnotes
This work was supported by European Union Seventh Framework Programme (FP7/2007–2013) under Grant agreement no. 604102 (HBP), the Swedish Research Council, National Institute on Alcohol Abuse and Alcoholism Grant 2R01AA016022, Swedish e-Science Research Center, and EuroSPIN, an Erasmus Mundus Joint Doctorate program.
The authors declare no competing financial interests.
References
- Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce Y, Gil R, Kegeles LS, Weiss R, Cooper TB, Mann JJ, Van Heertum RL, Gorman JM, Laruelle M. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci U S A. 2000;97:8104–8109. doi: 10.1073/pnas.97.14.8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aosaki T, Tsubokawa H, Ishida A, Watanabe K, Graybiel AM, Kimura M. Responses of tonically active neurons in the primate's striatum undergo systematic changes during behavioral sensorimotor conditioning. J Neurosci. 1994;14:3969–3984. doi: 10.1523/JNEUROSCI.14-06-03969.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aosaki T, Kimura M, Graybiel AM. Temporal and spatial characteristics of tonically active neurons of the primate's striatum. J Neurophysiol. 1995;73:1234–1252. doi: 10.1152/jn.1995.73.3.1234. [DOI] [PubMed] [Google Scholar]
- Aosaki T, Miura M, Suzuki T, Nishimura K, Masuda M. Acetylcholine-dopamine balance hypothesis in the striatum: an update. Geriatr Gerontol Int. 2010;10(Suppl 1):S148–S157. doi: 10.1111/j.1447-0594.2010.00588.x. [DOI] [PubMed] [Google Scholar]
- Augustin SM, Beeler JA, McGehee DS, Zhuang X. Cyclic AMP and afferent activity govern bidirectional synaptic plasticity in striatopallidal neurons. J Neurosci. 2014;34:6692–6699. doi: 10.1523/JNEUROSCI.3906-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacskai BJ, Hochner B, Mahaut-Smith M, Adams SR, Kaang BK, Kandel ER, Tsien RY. Spatially resolved dynamics of cAMP and protein kinase A subunits in Aplysia sensory neurons. Science. 1993;260:222–226. doi: 10.1126/science.7682336. [DOI] [PubMed] [Google Scholar]
- Ballarín M, Fredholm BB, Ambrosio S, Mahy N. Extracellular levels of adenosine and its metabolites in the striatum of awake rats: inhibition of uptake and metabolism. Acta Physiol Scand. 1991;142:97–103. doi: 10.1111/j.1748-1716.1991.tb09133.x. [DOI] [PubMed] [Google Scholar]
- Bateup HS, Svenningsson P, Kuroiwa M, Gong S, Nishi A, Heintz N, Greengard P. Cell type-specific regulation of DARPP-32 phosphorylation by psychostimulant and antipsychotic drugs. Nat Neurosci. 2008;11:932–939. doi: 10.1038/nn.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer HM, Glimcher PW. Midbrain dopamine neurons encode a quantitative reward prediction error signal. Neuron. 2005;47:129–141. doi: 10.1016/j.neuron.2005.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer HM, Lau B, Glimcher PW. Statistics of midbrain dopamine neuron spike trains in the awake primate. J Neurophysiol. 2007;98:1428–1439. doi: 10.1152/jn.01140.2006. [DOI] [PubMed] [Google Scholar]
- Bernard V, Levey AI, Bloch B. Regulation of the subcellular distribution of m4 muscarinic acetylcholine receptors in striatal neurons in vivo by the cholinergic environment: evidence for regulation of cell surface receptors by endogenous and exogenous stimulation. J Neurosci. 1999;19:10237–10249. doi: 10.1523/JNEUROSCI.19-23-10237.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibb JA, Snyder GL, Nishi A, Yan Z, Meijer L, Fienberg AA, Tsai L, Kwon YT, Girault J, Czernik AJ, Huganir RL, Hemmings HC, Jr, Nairn AC, Greengard P. Phosphorylation of DARPP-32 by Cdk5 modulates dopamine signalling in neurons. Nature. 1999;402:669–671. doi: 10.1038/45251. [DOI] [PubMed] [Google Scholar]
- Brandon EP, Logue SF, Adams MR, Qi M, Sullivan SP, Matsumoto AM, Dorsa DM, Wehner JM, McKnight GS, Idzerda RL. Defective motor behavior and neural gene expression in RIIbeta-protein kinase A mutant mice. J Neurosci. 1998;18:3639–3649. doi: 10.1523/JNEUROSCI.18-10-03639.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MT, Tan KR, O'Connor EC, Nikonenko I, Muller D, Lüscher C. Ventral tegmental area GABA projections pause accumbal cholinergic interneurons to enhance associative learning. Nature. 2012;492:452–456. doi: 10.1038/nature11657. [DOI] [PubMed] [Google Scholar]
- Bünemann M, Frank M, Lohse MJ. Gi protein activation in intact cells involves subunit rearrangement rather than dissociation. Proc Natl Acad Sci U S A. 2003;100:16077–16082. doi: 10.1073/pnas.2536719100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro LR, Brito M, Guiot E, Polito M, Korn CW, Hervé D, Girault JA, Paupardin-Tritsch D, Vincent P. Striatal neurones have a specific ability to respond to phasic dopamine release. J Physiol. 2013;591:3197–3214. doi: 10.1113/jphysiol.2013.252197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cechova S, Venton BJ. Transient adenosine efflux in the rat caudate-putamen. J Neurochem. 2008;105:1253–1263. doi: 10.1111/j.1471-4159.2008.05223.x. [DOI] [PubMed] [Google Scholar]
- Chelliah V, et al. BioModels: ten-year anniversary. Nucleic Acids Res. 2014;43:542–548. doi: 10.1093/nar/gku1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuhma N, Mingote S, Moore H, Rayport S. Dopamine neurons control striatal cholinergic neurons via regionally heterogeneous dopamine and glutamate signaling. Neuron. 2014;81:901–912. doi: 10.1016/j.neuron.2013.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox SM, Frank MJ, Larcher K, Fellows LK, Clark CA, Leyton M, Dagher A. Striatal D1 and D2 signaling differentially predict learning from positive and negative outcomes. Neuroimage. 2015;109:95–101. doi: 10.1016/j.neuroimage.2014.12.070. [DOI] [PubMed] [Google Scholar]
- Cumming P. Absolute abundances and affinity states of dopamine receptors in mammalian brain: a review. Synapse. 2011;65:892–909. doi: 10.1002/syn.20916. [DOI] [PubMed] [Google Scholar]
- Doig NM, Magill PJ, Apicella P, Bolam JP, Sharott A. Cortical and thalamic excitation mediate the multiphasic responses of striatal cholinergic interneurons to motivationally salient stimuli. J Neurosci. 2014;34:3101–3117. doi: 10.1523/JNEUROSCI.4627-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doupnik CA, Davidson N, Lester HA, Kofuji P. RGS proteins reconstitute the rapid gating kinetics of gbetagamma-activated inwardly rectifying K+ channels. Proc Natl Acad Sci U S A. 1997;94:10461–10466. doi: 10.1073/pnas.94.19.10461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenan RM, Grady SR, Steele AD, McKinney S, Patzlaff NE, McIntosh JM, Marks MJ, Miwa JM, Lester HA. Cholinergic modulation of locomotion and striatal dopamine release is mediated by alpha6alpha4* nicotinic acetylcholine receptors. J Neurosci. 2010;30:9877–9889. doi: 10.1523/JNEUROSCI.2056-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyer JK, Herrik KF, Berg RW, Hounsgaard JD. Influence of phasic and tonic dopamine release on receptor activation. J Neurosci. 2010;30:14273–14283. doi: 10.1523/JNEUROSCI.1894-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faisal AA, Selen LP, Wolpert DM. Noise in the nervous system. Nat Rev Neurosci. 2008;9:292–303. doi: 10.1038/nrn2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar AM, Callahan JW, Abercrombie ED. Reduced striatal acetylcholine efflux in the R6/2 mouse model of Huntington's disease: an examination of the role of altered inhibitory and excitatory mechanisms. Exp Neurol. 2011;232:119–125. doi: 10.1016/j.expneurol.2011.08.010. [DOI] [PubMed] [Google Scholar]
- Ferré S, O'Connor WT, Fuxe K, Ungerstedt U. The striopallidal neuron: a main locus for adenosine-dopamine interactions in the brain. J Neurosci. 1993;13:5402–5406. doi: 10.1523/JNEUROSCI.13-12-05402.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink JS, Weaver DR, Rivkees SA, Peterfreund RA, Pollack AE, Adler EM, Reppert SM. Molecular cloning of the rat A2 adenosine receptor: selective co-expression with D2 dopamine receptors in rat striatum. Brain Res Mol Brain Res. 1992;14:186–195. doi: 10.1016/0169-328X(92)90173-9. [DOI] [PubMed] [Google Scholar]
- Fiorillo CD, Tobler PN, Schultz W. Discrete coding of reward probability and uncertainty by dopamine neurons. Science. 2003;299:1898–1902. doi: 10.1126/science.1077349. [DOI] [PubMed] [Google Scholar]
- Ford CP, Phillips PE, Williams JT. The time course of dopamine transmission in the ventral tegmental area. J Neurosci. 2009;29:13344–13352. doi: 10.1523/JNEUROSCI.3546-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MJ. Dynamic dopamine modulation in the basal ganglia: a neurocomputational account of cognitive deficits in medicated and nonmedicated Parkinsonism. J Cogn Neurosci. 2005;17:51–72. doi: 10.1162/0898929052880093. [DOI] [PubMed] [Google Scholar]
- Glatt CE, Snyder SH. Cloning and expression of an adenylyl cyclase localized to corpus striatum. Nature. 1993;361:536–538. doi: 10.1038/361536a0. [DOI] [PubMed] [Google Scholar]
- Gold SJ, Ni YG, Dohlman HG, Nestler EJ. Regulators of G-protein signaling (RGS) proteins: region-specific expression of nine subtypes in rat brain. J Neurosci. 1997;17:8024–8037. doi: 10.1523/JNEUROSCI.17-20-08024.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JA, Reynolds JN. Spontaneous firing and evoked pauses in the tonically active cholinergic interneurons of the striatum. Neuroscience. 2011;198:27–43. doi: 10.1016/j.neuroscience.2011.08.067. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: single spike firing. J Neurosci. 1984;4:2866–2876. doi: 10.1523/JNEUROSCI.04-11-02866.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM, Aosaki T, Flaherty AW, Kimura M. The basal ganglia and adaptive motor control. Science. 1994;265:1826–1831. doi: 10.1126/science.8091209. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Arenas O, Eriksson O, Hellgren Kotaleski JH. Segregation and crosstalk of D1 receptor-mediated activation of ERK in striatal medium spiny neurons upon acute administration of psychostimulants. PLoS Comput Biol. 2014;10:e1003445. doi: 10.1371/journal.pcbi.1003445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Håkansson K, Galdi S, Hendrick J, Snyder G, Greengard P, Fisone G. Regulation of phosphorylation of the GluR1 AMPA receptor by dopamine D2 receptors. J Neurochem. 2006;96:482–488. doi: 10.1111/j.1471-4159.2005.03558.x. [DOI] [PubMed] [Google Scholar]
- Hart AS, Rutledge RB, Glimcher PW, Phillips PE. Phasic dopamine release in the rat nucleus accumbens symmetrically encodes a reward prediction error term. J Neurosci. 2014;34:698–704. doi: 10.1523/JNEUROSCI.2489-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heien ML, Khan AS, Ariansen JL, Cheer JF, Phillips PE, Wassum KM, Wightman RM. Real-time measurement of dopamine fluctuations after cocaine in the brain of behaving rats. Proc Natl Acad Sci U S A. 2005;102:10023–10028. doi: 10.1073/pnas.0504657102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein P, Frank M, Hoffmann C, Lohse MJ, Bünemann M. Dynamics of receptor/G protein coupling in living cells. EMBO J. 2005;24:4106–4114. doi: 10.1038/sj.emboj.7600870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein P, Rochais F, Hoffmann C, Dorsch S, Nikolaev VO, Engelhardt S, Berlot CH, Lohse MJ, Bünemann M. Gs activation is time-limiting in initiating receptor-mediated signaling. J Biol Chem. 2006;281:33345–33351. doi: 10.1074/jbc.M606713200. [DOI] [PubMed] [Google Scholar]
- Hernandez-Lopez S, Tkatch T, Perez-Garci E, Galarraga E, Bargas J, Hamm H, Surmeier DJ. D2 dopamine receptors in striatal medium spiny neurons reduce L-type Ca2+ currents and excitability via a novel PLC[beta]1-IP3-calcineurin-signaling cascade. J Neurosci. 2000;20:8987–8995. doi: 10.1523/JNEUROSCI.20-24-08987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersch SM, Gutekunst CA, Rees HD, Heilman CJ, Levey AI. Distribution of m1–m4 muscarinic receptor proteins in the rat striatum: light and electron microscopic immunocytochemistry using subtype-specific antibodies. J Neurosci. 1994;14:3351–3363. doi: 10.1523/JNEUROSCI.14-05-03351.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersch SM, Ciliax BJ, Gutekunst CA, Rees HD, Heilman CJ, Yung KK, Bolam JP, Ince E, Yi H, Levey AI. Electron microscopic analysis of D1 and D2 dopamine receptor proteins in the dorsal striatum and their synaptic relationships with motor corticostriatal afferents. J Neurosci. 1995;15:5222–5237. doi: 10.1523/JNEUROSCI.15-07-05222.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe MW, Tierney PL, Sandberg SG, Phillips PE, Graybiel AM. Prolonged dopamine signalling in striatum signals proximity and value of distant rewards. Nature. 2013;500:575–579. doi: 10.1038/nature12475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme EC, Trevethick MA. Ligand binding assays at equilibrium: validation and interpretation. Br J Pharmacol. 2010;161:1219–1237. doi: 10.1111/j.1476-5381.2009.00604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince E, Ciliax BJ, Levey AI. Differential expression of D1 and D2 dopamine and m4 muscarinic acetylcholine receptor proteins in identified striatonigral neurons. Synapse. 1997;27:357–366. doi: 10.1002/(SICI)1098-2396(199712)27:4<357::AID-SYN9>3.0.CO%3B2-B. [DOI] [PubMed] [Google Scholar]
- Jeon J, Dencker D, Wörtwein G, Woldbye DP, Cui Y, Davis AA, Levey AI, Schütz G, Sager TN, Mørk A, Li C, Deng CX, Fink-Jensen A, Wess J. A subpopulation of neuronal M4 muscarinic acetylcholine receptors plays a critical role in modulating dopamine-dependent behaviors. J Neurosci. 2010;30:2396–2405. doi: 10.1523/JNEUROSCI.3843-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laruelle M, D'Souza CD, Baldwin RM, Abi-Dargham A, Kanes SJ, Fingado CL, Seibyl JP, Zoghbi SS, Bowers MB, Jatlow P, Charney DS, Innis RB. Imaging D2 receptor occupancy by endogenous dopamine in humans. Neuropsychopharmacology. 1997;17:162–174. doi: 10.1016/S0893-133X(97)00043-2. [DOI] [PubMed] [Google Scholar]
- Leach K, Loiacono RE, Felder CC, McKinzie DL, Mogg A, Shaw DB, Sexton PM, Christopoulos A. Molecular mechanisms of action and in vivo validation of an M4 muscarinic acetylcholine receptor allosteric modulator with potential antipsychotic properties. Neuropsychopharmacology. 2010;35:855–869. doi: 10.1038/npp.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner TN, Kreitzer AC. RGS4 is required for dopaminergic control of striatal LTD and susceptibility to parkinsonian motor deficits. Neuron. 2012;73:347–359. doi: 10.1016/j.neuron.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liljeholm M, O'Doherty JP. Contributions of the striatum to learning, motivation, and performance: an associative account. Trends Cogn Sci. 2012;16:467–475. doi: 10.1016/j.tics.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SA, Kang UJ, McGehee DS. Striatal cholinergic interneuron regulation and circuit effects. Front Synaptic Neurosci. 2014;6:22. doi: 10.3389/fnsyn.2014.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindskog M, Svenningsson P, Pozzi L, Kim Y, Fienberg AA, Bibb JA, Fredholm BB, Nairn AC, Greengard P, Fisone G. Involvement of DARPP-32 phosphorylation in the stimulant action of caffeine. Nature. 2002;418:774–778. doi: 10.1038/nature00817. [DOI] [PubMed] [Google Scholar]
- Lindskog M, Kim M, Wikström MA, Blackwell KT, Kotaleski JH. Transient calcium and dopamine increase PKA activity and DARPP-32 phosphorylation. PLoS Comput Biol. 2006;2:e119. doi: 10.1371/journal.pcbi.0020119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse M, Hein P, Hoffmann C, Nikolaev V, Vilardaga JP, Bünemann M. Kinetics of G-protein-coupled receptor signals in intact cells. Br J Pharmacol. 2008;153:125–132. doi: 10.1038/sj.bjp.0707656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM. Neurotransmitter roles in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology. 2010;58:951–961. doi: 10.1016/j.neuropharm.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcott PF, Mamaligas AA, Ford CP. Phasic dopamine release drives rapid activation of striatal D2-receptors. Neuron. 2014;84:164–176. doi: 10.1016/j.neuron.2014.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattinson CE, Burmeister JJ, Quintero JE, Pomerleau F, Huettl P, Gerhardt GA. Tonic and phasic release of glutamate and acetylcholine neurotransmission in sub-regions of the rat prefrontal cortex using enzyme-based microelectrode arrays. J Neurosci Methods. 2011;202:199–208. doi: 10.1016/j.jneumeth.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov SL, Skorova E, Taschenberger G, Hartelt N, Nikolaev VO, Lohse MJ, Kügler S. Imaging cytoplasmic cAMP in mouse brainstem neurons. BMC Neurosci. 2009;10:29. doi: 10.1186/1471-2202-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G, Arkadir D, Nevet A, Vaadia E, Bergman H. Coincident but distinct messages of midbrain dopamine and striatal tonically active neurons. Neuron. 2004;43:133–143. doi: 10.1016/j.neuron.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Nikolaev VO, Bünemann M, Hein L, Hannawacker A, Lohse MJ. Novel single chain cAMP sensors for receptor-induced signal propagation. J Biol Chem. 2004;279:37215–37218. doi: 10.1074/jbc.C400302200. [DOI] [PubMed] [Google Scholar]
- Nishi A, Snyder GL, Greengard P. Bidirectional regulation of DARPP-32 phosphorylation by dopamine. J Neurosci. 1997;17:8147–8155. doi: 10.1523/JNEUROSCI.17-21-08147.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi A, Bibb JA, Snyder GL, Higashi H, Nairn AC, Greengard P. Amplification of dopaminergic signaling by a positive feedback loop. Proc Natl Acad Sci U S A. 2000;97:12840–12845. doi: 10.1073/pnas.220410397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobles M, Benians A, Tinker A. Heterotrimeric G proteins precouple with G protein-coupled receptors in living cells. Proc Natl Acad Sci U S A. 2005;102:18706–18711. doi: 10.1073/pnas.0504778102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olianas MC, Onali P. Antagonism of striatal muscarinic receptors inhibiting dopamine D1 receptor-stimulated adenylyl cyclase activity by cholinoceptor antagonists used to treat Parkinson's disease. Br J Pharmacol. 1996;118:827–828. doi: 10.1111/j.1476-5381.1996.tb15474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira RF, Kim M, Blackwell KT. Subcellular location of PKA controls striatal plasticity: stochastic simulations in spiny dendrites. PLoS Comput Biol. 2012;8:e1002383. doi: 10.1371/journal.pcbi.1002383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onali P, Olianas MC. Muscarinic M4 receptor inhibition of dopamine D1-like receptor signalling in rat nucleus accumbens. Eur J Pharmacol. 2002;448:105–111. doi: 10.1016/S0014-2999(02)01910-6. [DOI] [PubMed] [Google Scholar]
- Pajski ML, Venton BJ. Adenosine release evoked by short electrical stimulations in striatal brain slices is primarily activity dependent. ACS Chem Neurosci. 2010;1:775–787. doi: 10.1021/cn100037d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price CJ, Kim P, Raymond LA. D1 dopamine receptor-induced cyclic AMP-dependent protein kinase phosphorylation and potentiation of striatal glutamate receptors. J Neurochem. 1999;73:2441–2446. doi: 10.1046/j.1471-4159.1999.0732441.x. [DOI] [PubMed] [Google Scholar]
- Quirion R, Araujo D, Regenold W, Boksa P. Characterization and quantitative autoradiographic distribution of [3H]acetylcholine muscarinic receptors in mammalian brain: apparent labelling of an M2-like receptor subtype. Neuroscience. 1989;29:271–289. doi: 10.1016/0306-4522(89)90057-2. [DOI] [PubMed] [Google Scholar]
- Rada P, Barson JR, Leibowitz SF, Hoebel BG. Opioids in the hypothalamus control dopamine and acetylcholine levels in the nucleus accumbens. Brain Res. 2010;1312:1–9. doi: 10.1016/j.brainres.2009.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roitman MF, Wheeler RA, Wightman RM, Carelli RM. Real-time chemical responses in the nucleus accumbens differentiate rewarding and aversive stimuli. Nat Neurosci. 2008;11:1376–1377. doi: 10.1038/nn.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholich K, Mullenix JB, Wittpoth C, Poppleton HM, Pierre SC, Lindorfer MA, Garrison JC, Patel TB. Facilitation of signal onset and termination by adenylyl cyclase. Science. 1999;283:1328–1331. doi: 10.1126/science.283.5406.1328. [DOI] [PubMed] [Google Scholar]
- Schultz W. Predictive reward signal of dopamine neurons. J Neurophysiol. 1998;80:1–27. doi: 10.1152/jn.1998.80.1.1. [DOI] [PubMed] [Google Scholar]
- Sciotti VM, Park TS, Berne RM, Van Wylen DG. Changes in extracellular adenosine during chemical or electrical brain stimulation. Brain Res. 1993;613:16–20. doi: 10.1016/0006-8993(93)90448-V. [DOI] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ. Control of striatal synaptic plasticity. 2008a:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008b;321:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RE, Dale N. Activity-dependent adenosine release may be linked to activation of Na(+)-K(+) ATPase: an in vitro rat study. PLoS One. 2014;9:e87481. doi: 10.1371/journal.pone.0087481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinbjerg M, Sibley DR, Javitch JA, Abi-Dargham A. Imaging the high-affinity state of the dopamine D2 receptor in vivo: fact or fiction? Biochem Pharmacol. 2012;83:193–198. doi: 10.1016/j.bcp.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanos M, Gras-Najjar J, Letchworth JM, Sanford AL, Toups JV, Sombers LA. Quantitation of hydrogen peroxide fluctuations and their modulation of dopamine dynamics in the rat dorsal striatum using fast-scan cyclic voltammetry. ACS Chem Neurosci. 2013;4:782–789. doi: 10.1021/cn4000499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg EE, Keiflin R, Boivin JR, Witten IB, Deisseroth K, Janak PH. A causal link between prediction errors, dopamine neurons and learning. Nat Neurosci. 2013;16:966–973. doi: 10.1038/nn.3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsson P, Lindskog M, Ledent C, Parmentier M, Greengard P, Fredholm BB, Fisone G. Regulation of the phosphorylation of the dopamine- and cAMP-regulated phosphoprotein of 32 kDa in vivo by dopamine D1, dopamine D2, and adenosine A2A receptors. Proc Natl Acad Sci U S A. 2000;97:1856–1860. doi: 10.1073/pnas.97.4.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, Cragg SJ. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron. 2012;75:58–64. doi: 10.1016/j.neuron.2012.04.038. [DOI] [PubMed] [Google Scholar]
- Venton BJ, Zhang H, Garris PA, Phillips PE, Sulzer D, Wightman RM. Real-time decoding of dopamine concentration changes in the caudate-putamen during tonic and phasic firing. J Neurochem. 2003;87:1284–1295. doi: 10.1046/j.1471-4159.2003.02109.x. [DOI] [PubMed] [Google Scholar]
- Vilardaga JP. Theme and variations on kinetics of GPCR activation/deactication. J Recept Signal Transduct Res. 2012;30:304–312. doi: 10.3109/10799893.2010.509728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Whorton MR, MacKinnon R. Quantitative analysis of mammalian GIRK2 channel regulation by G proteins, the signaling lipid PIP 2 and Na + in a reconstituted system. Elife. 2014;3:1–17. doi: 10.7554/eLife.03671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Kai L, Day M, Ronesi J, Yin HH, Ding J, Tkatch T, Lovinger DM, Surmeier DJ. Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron. 2006;50:443–452. doi: 10.1016/j.neuron.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Wickens JR. Synaptic plasticity in the basal ganglia. Behav Brain Res. 2009;199:119–128. doi: 10.1016/j.bbr.2008.10.030. [DOI] [PubMed] [Google Scholar]
- Xie K, Masuho I, Brand C, Dessauer CW, Martemyanov KA. The complex of G protein regulator RGS9-2/Gβ5 controls sensitization and signaling kinetics of type 5 adenylyl cyclase. Sci Signal. 2012;5:ra63. doi: 10.1126/scisignal.2002922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Doyon WM, Clark JJ, Phillips PE, Dani JA. Controls of tonic and phasic dopamine transmission in the dorsal and ventral striatum. Mol Pharmacol. 2009;76:396–404. doi: 10.1124/mol.109.056317. [DOI] [PMC free article] [PubMed] [Google Scholar]