

Abstract
During mammalian development, myelin-forming oligodendrocytes are generated and axons ensheathed according to a tightly regulated sequence of events. Excess premyelinating oligodendrocytes are eliminated by apoptosis and the timing of the onset of myelination in any specific CNS region is highly reproducible. Although the developing CNS recovers more effectively than the adult CNS from similar insults, it is unknown whether early loss of oligodendrocyte lineage cells leads to long-term functional deficits. To directly assess whether the loss of oligodendrocytes during early postnatal spinal cord development impacted oligodendrogenesis, myelination, and remyelination, transgenic mouse lines were generated in which a modified caspase-9 molecule allowed spatial and temporal control of the apoptotic pathway specifically in mature, myelin basic protein expressing oligodendrocytes (MBP-iCP9). Activating apoptosis in MBP+ cells of the developing spinal cord during the first postnatal week inhibited myelination. This inhibition was transient, and the levels of myelination largely returned to normal after 2 weeks. Despite robust developmental plasticity, MBP-iCP9-induced oligodendrocyte apoptosis compromised the rate and extent of adult remyelination. Remyelination failure correlated with a truncated proliferative response of oligodendrocyte progenitor cells, suggesting that depleting the oligodendrocyte pool during critical developmental periods compromises the regenerative response to subsequent demyelinating lesions.
SIGNIFICANCE STATEMENT This manuscript demonstrates that early insults leading to oligodendrocyte apoptosis result in the impairment of recovery from demyelinating diseases in the adult. These studies begin to provide an initial understanding of the potential failure of recovery in insults, such as periventricular leukomalacia and multiple sclerosis.
Keywords: development, lysolecithin, myelination, oligodendrocyte
Introduction
During CNS development, oligodendrocyte precursor cells (OPCs) are generated in distinct regions of the CNS as a result of local signals (Miller, 2002; Cai et al., 2005; Vallstedt et al., 2005). These cells subsequently disperse throughout the CNS before differentiating into oligodendrocytes (Noble et al., 1988; Tsai et al., 2002) that then myelinate adjacent axons in a tightly regulated and highly reproducible pattern. OPCs are generated in excess during development, and unnecessary cells are eliminated by a regulated program of cell death called apoptosis (Barres et al., 1992; Trapp et al., 1997) to achieve a predictable number of myelinating oligodendrocytes. Even in experimental conditions where the number of OPCs is artificially increased, the number of oligodendrocytes and extent of myelination remain constant (Calver et al., 1998; Woodruff et al., 2004). Likewise, elimination of a major source of OPCs during development does not compromise myelination because their target axons are myelinated by oligodendrocytes from alternative sources (Kessaris et al., 2006). Despite robust short-term plasticity, perinatal white matter injury often triggers permanent neurological deficits. For example, periventricular leukomalacia, a neonatal condition characterized by necrosis of the periventricular white matter (Shang et al., 2015), perturbs the subsequent development and survival of oligodendrocyte lineage cells and is associated with a high risk of long-lasting clinical deficits (Back et al., 2001; Volpe, 2001, 2009; Marlow et al., 2005). It is therefore important to determine whether perturbations in oligodendrocyte development confer long-term functional consequences. The current study addresses the potential long-term consequences of developmental oligodendrocyte death on remyelination in the spinal cord.
In contrast to the limited repair seen in periventricular leukomalacia, many animal models of white matter insults repair rapidly and effectively (Matsushima and Morell, 2001). Local demyelination induced by the injection of gliotoxins, such as lysolecithin (LPC) repairs rapidly in young rodents, even after multiple lesions (Penderis et al., 2003), as a result of the recruitment and proliferation of adult OPCs adjacent to the lesion (Blakemore, 1978; Ruckh et al., 2012). How new myelinating oligodendrocytes are recruited in response to white matter perturbations and why this fails in certain situations are not completely understood. New approaches in which cells of the oligodendrocyte lineage are selectively ablated in the adult CNS demonstrate rapid demyelination after the focal induction of apoptosis in MBP+ cells (Caprariello et al., 2012) and widespread slow demyelination and repair following global oligodendrocyte depletion (Traka et al., 2010). Whether the developing CNS has similar responses to oligodendrocyte loss and whether there are long-term consequences of selectively perturbing oligodendrogenesis are unknown. To address these issues, we generated a novel transgenic mouse line in which a modified, inducible caspase-9 (iCP9) molecule (MacCorkle et al., 1998) allowed experimental control of the apoptotic pathway specifically in mature, MBP-expressing oligodendrocytes (MBP-iCP9). We previously used a similar lentiviral approach to induce oligodendrocyte apoptosis in the adult corpus callosum (Caprariello et al., 2012). In the current study, iCP9 was activated by daily intrathecal injection of the small molecule chemical inducer of dimerization (CID) from P4-P7, a period of intense gliogenesis. The subsequent development of myelination and the response of developmentally perturbed white matter to a subsequent demyelinating lesion were assessed. Early oligodendrocyte ablation delayed myelination but recovered over a 2 week period. Targeting subsequent demyelinating lesions to previously depleted regions revealed a lasting effect that compromised the rate and success of remyelination. The reduced remyelination capacity correlated with a reduction in the proliferative response of responding OPCs, suggesting that oligodendrocyte ablation during early CNS development compromises adult myelin repair. These results identify early oligodendrocyte death as a potential predisposing factor for failed remyelination in a number of pathological conditions.
Materials and Methods
Generation of MBP-iCP9 transgenic animals.
Transgenic animals expressing the iCP9 construct (MBP-iCP9) were generated in the CWRU Transgenic Core by pronuclear injection of a linearized 4.2 kb insert isolated from a commercially available plasmid (pIRES2 DsRedExpress2, Clontech). Both the 1.4 kb fragment of the myelin basic protein promoter (pMBP) as well as the 1.3 kb iCP9 sequence were identical to that used in our previous lentiviral experiments (Caprariello et al., 2012). The offspring from 69 founders were screened for expression of reporter DsRed. Two lines with different expression levels were established, and subsequent experiments were conducted in the high expressing line.
RT-PCR.
Spinal cord tissues from MBP-iCP9 animals were flash frozen and RNA isolated using Trizol/chloroform according to the manufacturer's protocol (Invitrogen). After DNase treatment for 15 min at room temperature, RNA was transcribed into cDNA using SuperScript first-Strand Synthesis Kit (Invitrogen). cDNA was run in a PCR together with primers that flank the insertion site of iCP9 into the DsRed Express2 plasmid backbone, as follows: forward, TGGGCTCACTCTGAAGACCT; reverse, AGGAACTGCTTCCTTCACGA. Transgenic MBP-iCP9 animals from both founder lines (3 total) were compared against plasmid DNA as a positive control and nontransgenic animals from both lines (3 total) as negative controls.
Western blot.
To verify iCP9 expression at the protein level, Western blots were performed per the manufacturer's instructions (Bio-Rad). Briefly, spinal cord samples from MBP-iCP9 animals and wild-type (WT) littermate controls were flash frozen and incubated in a solution of RIPA buffer/protease/EDTA, manually homogenized, and spun at 13,000 rpm. Samples were denatured in β-mercaptoethanol and run in a 15% gel, transferred to a PVDF membrane, and probed using a caspase-9 antibody (1:1000, Abcam 25758) that recognized an epitope common to both endogenous and transgenic caspase-9 species. GAPDH (1:10,000, Abcam 9484) was used as a loading control. Three segments from each of two MBP-iCP9 animals were assayed against three segments from WT littermate control. Significance was determined using an unpaired Student's t test.
Primary mixed glial and neurosphere cultures.
Primary spinal cord cell cultures were prepared from individual transgenic and WT littermates of either sex at P2. Following removal of the meninges, cells were dissociated with trypsin (0.025%), plated on PLL-coated coverslips at a density of 100,000 cells/well, and grown in media consisting of 1% FBS in DMEM/F12 (Invitrogen 21331-020), N2 supplement (1×; Invitrogen 17502-048), PDGF (10 ng/ml FC; Sigma P3076), Glutamax (Invitrogen 35050-061), and penicillin (50 U/ml) and streptomycin (50 μg/ml). Fresh media was provided every 2 d. Twenty-four hours after CID treatment, cells were fixed in 4% PFA and labeled with cell type-specific antibodies O1 (1:4; in-house hybridoma), GFAP (1:500, Dako #Z0334), and Tuj1 (1:500, Abcam ab14545). Labeled cells were counted in 10 different 40× fields. Data represent mean ± SEM of at least three coverslips in three separate experiments. Neurosphere cultures were grown as previously described (Moore et al., 2011). Briefly, neurospheres derived from spinal cords of P3 MBP-iCP9 transgenic mice were CID treated (10 μm) while in suspension after 5 d in vitro. Four days after CID treatment, spheres were plated onto PLL-coated coverslips to induce differentiation. Two days after plating, spheres were fixed in 4% PFA and labeled with cell type-specific antibodies CC1 (1:250, Millipore OP80) and anti-GFAP. Labeled cells were counted in 5 different 40× fields from at least three different neurosphere preparations. Data represent mean ± SEM of three independent experiments. Significance was determined using an unpaired Student's t test.
Intrathecal CID injection.
Under isoflurane anesthesia, male MBP-iCP9 or WT littermate mice at P4, P5, or P6 were positioned so that the animal's thoracic spine was elevated with respect to its anterior–posterior body axis. Using the caudal-most rib as a landmark for the lower thoracic spine, a 30 gauge needle attached to a 10 μl Hamilton syringe was carefully inserted, without a skin incision, between spinous processes into the fluid-filled space surrounding the spinal cord at a depth of ∼2 mm, including skin thickness. After withdrawing clear CSF to ensure intrathecal targeting, 2 μl of CID (0.25 mg/kg) was slowly dispensed. After injection, the needle was left in place for ∼1 min to prevent liquid reflux upon needle withdrawal. Animals were recovered on a heating pad before returning to their cage. Controls included CID injections into WT littermates as well as injection of 2 μl of saline into MBP-iCP9 animals.
LPC injection.
To determine the efficiency of adult myelin repair, LPC lesions were induced in thoracic spinal cord white matter of 6-week-old MBP-iCP9 or WT littermate controls. Under isoflurane anesthesia, a small incision was made in the skin to expose the spinal column. Access to the spinal cord between T12 and T13 was obtained by a combination of blunt and micro-scissor dissection. A 30 mm interior-diameter pulled-glass pipette fitted to a microinjector was used to administer 1 μl of a 1% solution of LPC in 0.9% saline at a depth of 0.3 mm into the dorsal column of the spinal cord. After LPC injection, the needle was left in place for 2 min to prevent liquid reflux. The skin was sutured closed, and the animals were recovered on a heating pad before returning to its cage. Controls included injection of the same volume of 0.9% saline into both MBP-iCP9 and nontransgenic animals (3 of each). The 5 d post-lesion (dpl) studies involved 3 MBP-iCP9 and 3 WT littermate controls. The 14 dpl studies included 5 MBP-iCP9 and 3 WT littermate controls.
Histology, tissue processing, and analysis.
For histological analyses, mice were killed at P7 and P21 via transcardial perfusion with 0.9% saline followed by 4% PFA. Spinal cords were removed, placed in 20% sucrose overnight, embedded in OCT, and sectioned at 20 μm on a cryostat (Leica CM3050S). The extent of transgene expression was assessed using antibodies to the DsRed reporter protein (1:100, Clontech #632496). Oligodendrocyte-specific markers included CC1 (1:250, Millipore OP80) or MBP (1:500, Covance, SMI99R). All antibodies were incubated overnight at 4°C. For DsRed and CC1, light antigen retrieval using a 10 min incubation in 10 mm sodium citrate buffer was performed before incubation in primary antibody. Other primary antibodies used were NeuN (1:250, Millipore MAB377), Neurofilament (1:500, Millipore MAB1592), cCP3 (1:100, Cell Signaling Technology 9661), Olig2 (1:250, Millipore AB9610), Ki67 (1:250, BD PharMingen 550609), and IBA1 (1:500, Wako 019-19741). An individual blinded to animal phenotype generated quantitative data from counts of immunolabeled cells in the dorsal column white matter of the spinal cord. The same area of tissue was analyzed in at least 3 sections from 3–5 animals per experiment. Statistical significance was assessed using an unpaired Student's t test. For experiments involving multiple time points and treatments, a two-way ANOVA was performed.
In situ hybridization.
A 969 bp probe for proteolipid protein was generated using PCR primers from Allen Brain Atlas flanked with T7/T3 (T3 forward, 5′-AATTAACCCTCACTAAAGGGGGGGATGCCTGAGAAGGT3′; and T7 reverse, 5′-GTAATACGACTCACTATAGGGCGTGTGATGCTTTCTGCCCA-3′). PCR products were generated using isolated RNA and the SuperScript One-Step RT-PCR kit (Invitrogen), reamplified, and confirmed by sequencing. Probes were then transcribed using the Ambion Maxiscript transcription kit (Invitrogen) and digoxigenin-11-UTP (Roche), using either T3 for the sense control probe or T7 for the antisense probe. Probes were precipitated overnight at −20°C, centrifuged, and the pellet rinsed twice with 70% ethanol, air dried, and resuspended in 50 μl of DEPC water and stored at −80°C until use. Alexandra Joyner's (Memorial Sloan-Kettering Cancer Center, New York, NY) nonradioactive in situ hybridization protocol was followed (www.mskcc.org/research/lab/alexandra-joyner/laboratory-p r o t o c o l s). Probes were diluted 1:50 in hybridization solution and hybridized overnight at 55°C–60°C.
EM processing and analysis.
For ultrastructure analyses, mice under terminal sedation were transcardially perfused with 1× PBS followed immediately by 4% PFA/2% gluteraldehyde in 0.1 m sodium cacodylate. Spinal cords were dissected and left in EM fixative solution at 4°C for at least 24 h. Samples were osmicated (1% in cacodylate buffer), treated with 1% uranyl acetate, dehydrated through gradated alcohols and propylene oxide, and embedded in Epon 812. Sections were cut on an ultramicrotome (Leica EM UC7). The 0.5–1 μm thin sections were stained with a solution of 1% toluidine blue/1% sodium borate for 2–3 min. Ultrathin sections were stained with lead citrate and uranyl acetate and imaged at 1500× and 3000× on a Joel 100X electron microscope at 80 kV. To quantify the level of myelination, the number of myelinated axons was counted in a fixed region of the spinal cord white matter in at least 4 animals per condition. The level of myelination was assessed by analysis of g-ratios in which the thickness of the myelin sheath was calculated relative to the diameter of the axon. At least 100 axons were measured in at least 3 mice per condition. Statistical significance of remyelination was assessed using an unpaired Student's t test.
Results
Inducible Caspase-9 transgene is expressed exclusively in oligodendrocytes
To induce apoptosis selectively in myelinating oligodendrocytes in the intact CNS, transgenic mouse lines were generated using a construct in which a cell-selective and experimentally iCP9 and reporter DsRed were under transcriptional control of a CNS-specific fragment of the pMBP (Fig. 1A). Sixty-nine potential founder mice (MBP-iCP9-IRES-DsRed) were screened for expression of the DsRed reporter from which two independent mouse lines, henceforth referred to as MBP-iCP9, were identified with differing levels of DsRed expression. In each founder line, transgene expression was validated at both the mRNA and protein levels. In spinal cord homogenates from P10 MBP-iCP9 animals, RT-PCR detected a single mRNA species of the appropriate size corresponding to the transcribed region of genomic DNA at which the cloned iCP9 construct was inserted into the DsRed plasmid. Present in MBP-iCP9 transgenic animals but not WT littermate controls (Fig. 1B), this mRNA species confirmed transcription of the transgene as well as the DsRed reporter gene.
Figure 1.
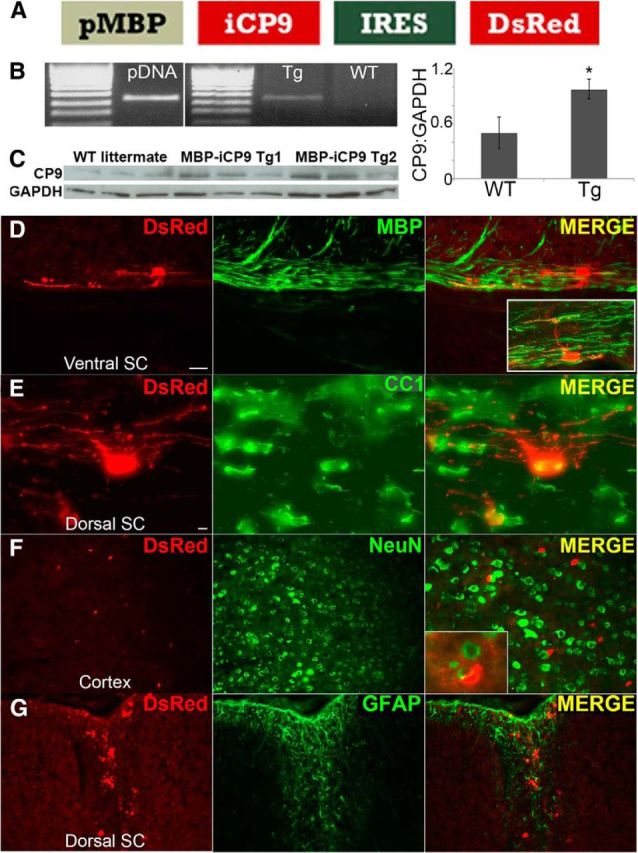
MBP-iCP9-IRES-DsRed transgenic mice (MPB-iCP9) express iCP9 mRNA and protein selectively in MBP+ oligodendrocytes. A, Schematic of the transgene demonstrates two key points: (1) iCP9 is transcriptionally regulated by an oligodendrocyte-specific promoter sequence (pMBP); and (2) an internal ribosome entry site (IRES) facilitates cotranscription of reporter DsRed fluorescent protein. B, RT-PCR gel image demonstrates mRNA corresponding to cotranscribed iCP9 and DsRed in homogenized MBP-iCP9 spinal cord tissue; n = 3 for both genotypes. *p < 0.05. Error bars indicate SEM. C, Western blot with an antibody common to both endogenous and transgenic iCP9 indicates increased protein expression in MBP-iCP9 spinal cords at P30 compared with age-matched WT littermates; n = 3 WT; n = 6 MBP-iCP9. D–G, DsRed immunohistochemistry confirmed robust and oligodendrocyte-specific expression of reporter DsRed in multiple adult CNS tissues; n = 5. D, DsRed was expressed in MBP+ cell processes in a longitudinal section of P14 MBP-iCP9 ventral spinal cord. Far-right inset, Single DsRed+ oligodendrocyte myelinating multiple nearby axons. E, Double labeling of DsRed and CC1 demonstrates strong expression of DsRed in CC1+ oligodendrocytes in longitudinal sections of P14 dorsal spinal cord. The vast majority of DsRed+ cells expressed CC1 (97 ± 2.5%, n = 3). Neither cortical NeuN+ neurons (F) nor GFAP+ astrocytes (G) express iCP9. Scale bars, 50 μm.
To detect iCP9 at the protein level, spinal cord homogenates from P30 MBP-iCP9 and WT mice were probed by Western blot with an antibody specific to an epitope of the caspase-9 protein common to both endogenous and inducible forms. A significant increase in the total levels of preactivated caspase-9 protein was observed in samples derived from MBP-iCP9 animals compared with WT littermates, suggesting that the transgene transcript identified by RT-PCR was indeed being translated into protein (Fig. 1C). To confirm the protein translation as well as the cell type specificity of the MBP-iCP9 transgene, frozen sections from various CNS tissues were assayed by immunohistochemistry. Double-labeling studies using antibodies to DsRed and to the major CNS cell types demonstrated robust DsRed expression in both the cell body and processes of MBP+ and CC1+ oligodendrocytes (Fig. 1D,E). By contrast, no coincident expression of DsRed was observed in NeuN+ neurons (Fig. 1F) or GFAP+ astrocytes (Fig. 1G). Not all oligodendrocytes expressed DsRed, however. In the high-expressing founder line, ∼65% of CC1+ oligodendrocytes in P14 thoracic spinal cords expressed DsRed, compared with ∼20% in the low-expressing line. In both lines, essentially 100% of DsRed+ cells were CC1+ (97% ± 2.5%, n = 3). The relative level of DsRed expression in different regions of the CNS was dependent on the region within the CNS. Highest levels of expression were detected in optic nerve, spinal cord, and cerebellum white matter, lower levels in brainstem and cortex, and minimal labeling was detected in hippocampus. Little or no DsRed expression was seen in peripheral tissues, including kidney, muscle, liver, spleen, and sciatic nerve, supporting the CNS specificity of the MBP promoter sequence as established previously (Foran et al., 1992; Gow et al., 1992; Goujet-Zalc et al., 1993; Asipu et al., 2001). Together, these data validate the transcription, translation, and cell specificity of the MBP-iCP9 transgene in multiple independent founder lines. All subsequent studies used the highest expressing line.
Experimental activation of iCP9 results in selective death of mature oligodendrocytes in MBP-iCP9 primary cultures
In previous studies using a viral delivery system, we have shown that experimental activation of an identical iCP9 construct driven by the same fragment of the MBP promoter is sufficient to stimulate oligodendrocyte apoptosis in vivo (Caprariello et al., 2012). To verify iCP9 activation in oligodendrocytes derived from MBP-iCP9 transgenic animals, mixed primary spinal cord cell cultures from neonatal MBP-iCP9 animals were treated with the CID after 4 days in vitro. Twenty-fours after CID treatment, classical signs of cell apoptosis, including nuclear condensation and membrane blebbing, were observed by bright-field microscopy (Fig. 2A,B). Quantitation of the relative proportion of O1+ mature oligodendrocytes demonstrated a reduction from 15 ± 1.8% in vehicle-treated control cultures to 3.5 ± 2.6% in CID-treated MBP-iCP9 cultures (Fig. 2C,D). By contrast, no reduction in the proportion of GFAP+ astrocytes (Fig. 2E,F) or Tuj1+ neurons (Fig. 2G,H) was seen following CID treatment of MBP-iCP9-derived cultures. Although the relative number of Tuj1+ neurons was not significantly different from controls, the relative number of GFAP+ astrocytes significantly increased from 29 ± 1.8% in control cultures to 41 ± 2.5% in CID-treated MBP-iCP9 cultures. An increase in GFAP+ astrocytes following CID was also observed in neurosphere preparations from MBP-iCP9 animals. Treatment of neurosphere cells in suspension for 24 hours with CID before plating and adhesion-dependent differentiation resulted in a significant increase in the proportion of GFAP+ cells following 2 d in vitro (Fig. 2I,J). The effects of CID treatment were specific to MBP-iCP9 cultures. In cultures derived from WT littermates, the addition of CID had no effect either on cell survival or the proportions of different cell populations in the cultures. For example, nontreated cultures had 6.7 ± 1.5 O4+ cells per field, whereas parallel CID-treated WT cultures had 8.3 ± 3.0 O4+ cells. Similarly, nontreated cultures had 64 ± 5.1 GFAP+ cells per field, whereas parallel CID-treated WT cultures had 63 ± 5.9 GFAP+ cells (n = 4) with no significant difference in total cell numbers between conditions for either cell type. Furthermore, the relative proportions of oligodendrocytes, astrocytes, and neurons in MBP-iCP9 cultures in vehicle-treated controls was similar to that of WT cultures, demonstrating that the induction of iCP9-mediated apoptosis was dependent on both the expression of the iCP9 transgene and its activation by CID.
Figure 2.

CID treatment of neonatal MBP-iCP9 cultures induces cell death selectively in O1+ oligodendrocytes. A, B, Bright-field microscopy indicates extensive cell death 24 hours after treatment with 10 μm CID. C, D, Labeling with the mAb O1 demonstrates significant and selective depletion of oligodendrocytes in CID-treated cultures. By contrast, neither GFAP+ astrocytes (E, F) nor Tuj1+ neurons (G, H) were reduced by CID treatment, demonstrating that CID is specific for oligodendrocyte death. The number of GFAP+ cells increased significantly in dissociated cultures as well as in CID-treated neurosphere preparations (I, J). K, Summary histogram of cell counts of oligodendrocytes (O1), astrocytes (GFAP), neurons (Tuj1), and neurosphere-derived astrocytes (GFAP-ns) from MBP-iCP9 cultures with vehicle or CID treatment. n = 3 coverslips per condition per cell marker repeated in cultures from 2 or 3 different mice. *p < 0.05. Error bars indicate SEM. Phase image scale bar, 100 μm; white scale bar, 30 μm, applies to all fluorescent images, except 50 μm scale bar in I.
Induction of oligodendrocyte apoptosis during development results in delayed myelination
To determine whether the induction of apoptosis in mature oligodendrocytes during the first postnatal week resulted in an inhibition of myelination, MBP-iCP9 and WT littermates were treated with CID on postnatal days 4, 5, and 6 (P4-P6). To avoid direct insult to spinal cord tissue, cell-permeable CID was administered intrathecally by microinjection into the CSF surrounding the thoracic spinal cord, a procedure validated using surrogate injections with Evans blue (Fig. 3A, middle-right panel). During all experiments, charcoal was applied to the tip of the needle to verify proper CID targeting (Fig. 3A, far-right panel). Analysis of harvested tissue at P7 indicated no significant differences in myelination in the spinal cords of nontreated or saline-treated MBP-iCP9 animals compared with identical regions in nontreated or CID-treated WT littermates. By contrast, in thoracic spinal cord regions of MBP-iCP9 animals treated with CID, there was a dramatic reduction in the level of myelination. In the dorsal columns of control animals, individual oligodendrocytes surrounded by multiple myelinated axons were apparent at both the light and electron microscopic level (Fig. 3B,D). These were largely absent in MBP-iCP9 CID-treated animals (Fig. 3C,E). The remaining oligodendrocytes displayed hallmark morphological characteristics of apoptosis, including process retraction, membrane blebbing, and nuclear condensation (Fig. 3C,E, arrows). As a result, there was a significant reduction in the number of myelinated axons in MBP-iCP9 animals following CID treatment from 7120 ± 1334 mm2 in CID-treated WT controls to 2428 ± 526.0 myelinated axons in MBP-iCP9 animals (Fig. 3F). Residual myelin was abnormally thin as revealed by g-ratio analysis and was largely absent from smaller-caliber axons (Fig. 3G). Despite intrathecal CID treatment, the loss of oligodendrocytes and myelin was not uniform throughout all regions of the spinal cord. In general, dorsal columns were more severely affected than ventral columns, although MBP-iCP9 ventral regions had clearly reduced myelination and contained apoptotic oligodendrocytes (data not shown). The extent of oligodendrocyte loss was quantified by in situ hybridization for proteolipid protein (Fig. 4A,B) as well as by labeling with oligodendrocyte-specific markers CC1 or MBP and axonal neurofilaments (Fig. 4C–F). The number of DsRed+ cells in CID-treated MBP-iCP9 regions was reduced by nearly 75% compared with CID-treated littermate controls (Fig. 4G). Similarly, there was a threefold reduction in CC1+ cells, whereas MBP+ cells were essentially eliminated in CID-treated MBP-iCP9 animals. The apoptotic nature of oligodendrocyte death was confirmed by labeling with cleaved caspase-3, the immediate downstream effector of caspase-9, as well as TUNEL, which identifies the DNA fragmentation of the organized cellular dismantling that occurs during programmed cell death (Fig. 4H–J). Similar results were obtained from parallel studies in independent MBP-iCP9 founder lines demonstrating that experimentally induced apoptosis in the initial population of mature oligodendrocytes leads to significant disruption of myelination in the neonatal thoracic spinal cord.
Figure 3.
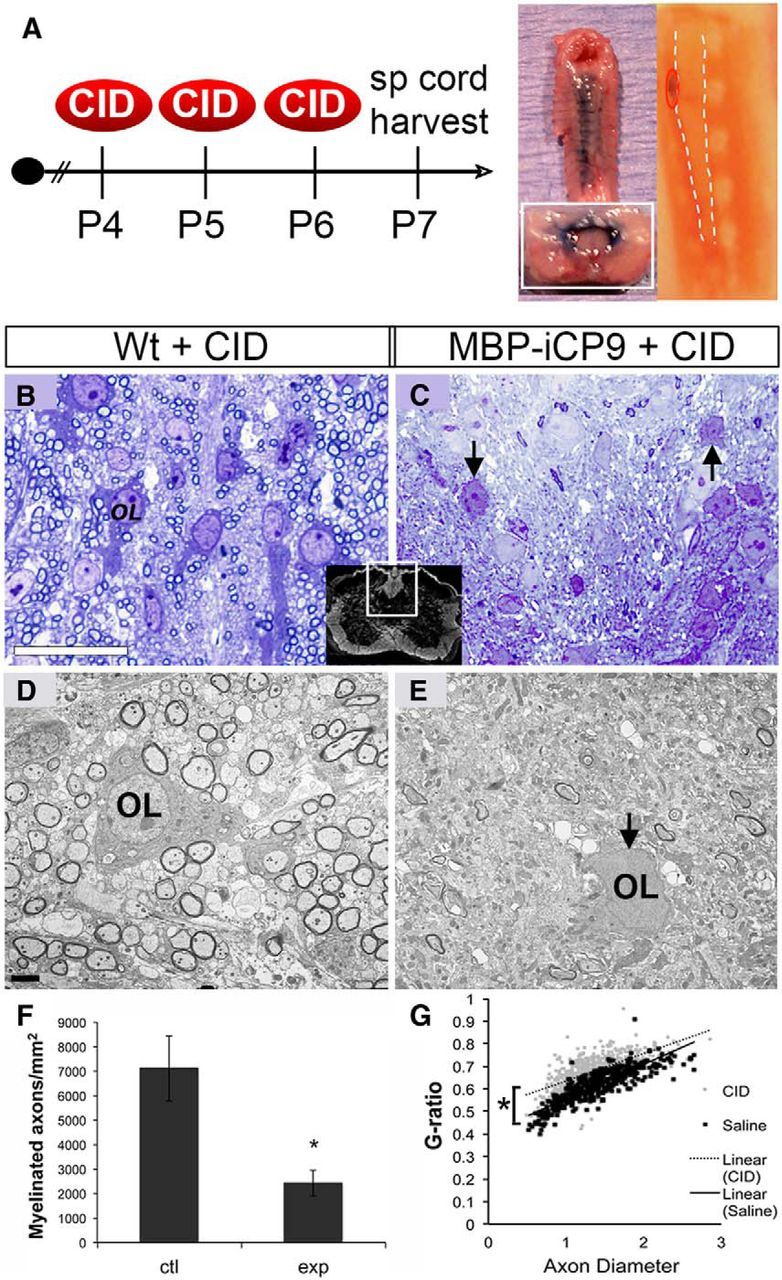
Oligodendrocyte ablation during early spinal cord development delays myelination. A, The 2 μl CID (0.25 mg/kg) was administered intrathecally daily from P4 to P6. Surrogate Evans blue injections (middle-right panel) demonstrated the feasibility and extent of coverage following thoracic intrathecal injections. Charcoal applied to the tip of the needle verified proper targeting of CID (far-right panel). P7 tissue sections stained with toluidine blue (B, C) or analyzed by electron microscopy (D, E) revealed a sharp decrease in the number of myelinated axons (F) and a reduction in the G-ratio of residual myelin (G) in the dorsal columns of MBP-iCP9 animals treated with CID compared with identically treated WT littermate controls. F, Myelinated axons were counted from EM images within a similar defined area of the spinal cord white matter in both control and experimental groups. *p < 0.05. Error bars indicate SEM. G, Each dot in the G-ratio plot represents a measurement of myelin thickness versus axon diameter in the white matter of either control (black dot) or experimental (gray dot) mice. A total of 50–100 different axons were counted per animal from a defined area in 3 or 4 mice per condition. Best-fit lines were drawn for WT (solid black) and MBP-iCP9 (dotted) G-ratios. n = 4 for control; n = 5 for experimental. Scale bars: B, 25 μm; D, 5 μm.
Figure 4.
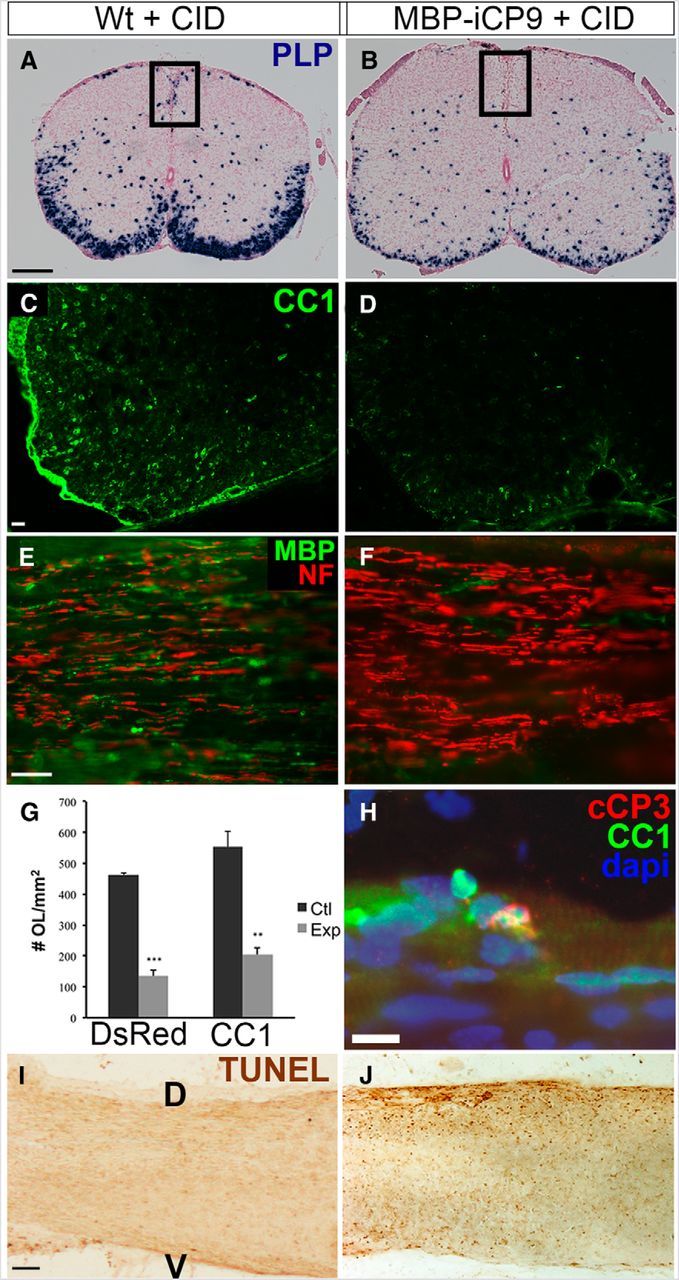
Intrathecal CID induces widespread, acute oligodendrocyte apoptosis. A, B, In situ hybridization for proteolipid protein (PLP) indicates reduced PLP mRNA in the dorsal column. C, D, CID treatment induces loss of CC1+ oligodendrocytes from MBP-iCP9 spinal cord cross sections. E, F, Although CID significantly reduced MBP expression in longitudinal sections of MBP-iCP9 spinal cords, NF+ axons were preserved. G, Oligodendrocyte death was verified by reduction of two different markers: DsRed and CC1. **p < 0.01, ***p < 0.001. Error bars indicate SEM. H–J, Cleaved Caspase-3 (cCP3) and TUNEL staining of longitudinal sections indicate extensive oligodendrocyte apoptosis, particularly toward the superficial (pial) surface of the spinal cord, following intrathecal CID treatment. Scale bars: A, I, 100 μm; C, 30 μm; E, H, 25 μm.
Robust recovery of myelination deficits following postnatal oligodendrocyte apoptosis
To determine whether the disruption of developmental myelination resulting from the induction of oligodendrocyte apoptosis during the first postnatal week was permanent or transient, CID was again delivered intrathecally from P4-P6 into either MBP-iCP9 or control animals and the levels of myelination compared 2 weeks later at P21. As expected, in both WT and MBP-iCP9 animals, the levels of myelination had increased significantly from that seen at P7 (compare Fig. 3A and Fig. 5A). More importantly, the level of myelination in treated MBP-iCP9 animals dramatically recovered to levels that were not substantially different from those in untreated or CID-treated WT controls (Fig. 5B). In MBP-iCP9 animals, both dorsal (Fig. 5B) and lateral (Fig. 5D) regions of the spinal cord contained relatively normal levels of myelination at P21, although some residual elevated cellularity remained. Quantitative analyses demonstrated that the relative thickness of the myelin sheaths (Fig. 5E) and the number of myelinated axons (Fig. 5F) in MBP-iCP9 transgenic animals were comparable with controls, suggesting that delayed developmental myelination through oligodendrocyte apoptosis was transient and that myelination largely recovered by P21. However, not all aspects of MBP-iCP9 tissues were normal at P21. Small-caliber axons particularly in lateral spinal cord regions remained unmyelinated and others appeared unhealthy with intracellular debris suggestive of early degeneration (Fig. 5D, arrows). Nevertheless, g-ratios were virtually identical between conditions (Fig. 5E) as were the number of myelinated axons (Fig. 5F). Unexpectedly, the number of oligodendrocytes detected in semithin sections stained with toluidine blue was significantly increased in MBP-iCP9 animals at P21 (785.0 ± 35 for control vs 1130 ± 52 for experimental; Fig. 5G), suggesting that delaying the timing of myelination alters the dynamics and final number of oligodendrocytes.
Figure 5.

Robust recovery of myelination following early postnatal oligodendrocyte apoptosis. CID (0.25 mg/kg) was administered intrathecally daily from P4 to P6, after which animals were survived to P21 to assess the extent of recovery from early oligodendrocyte apoptosis. A–D, Toluidine blue-stained thin sections from P21 MBP-iCP9 revealed significant remyelination in various spinal cord regions compared with littermate controls. E, Myelin g-ratios verified that remyelination was of appropriate thickness for given axon diameters. Solid line indicates a best-fit line for control conditions. Dotted line indicates a best-fit line for experimental conditions. F, The number of myelinated axons also recovered to levels observed in WT littermate controls. Myelinated axons were counted as described in Figure 2. G, Unlike myelinated axons, the number of oligodendrocytes, counted from frozen sections labeled with antibodies to CC1 or epon sections stained with toluidine blue, significantly increased following the stark reduction observed at P7. n = 3 control; n = 3 experimental. Scale bar, 10 μm. *p < 0.05. Error bars indicate SEM.
Early oligodendrocyte ablation impairs adult myelin repair
In young rodents, remyelination is robust following local injection of the gliotoxin lysophosphatidylcholine (LPC) into spinal cord white matter with repair beginning at days ∼7–10 and largely complete by 21–23 dpl (Jeffery and Blakemore, 1995; Woodruff and Franklin, 1999; Woodruff et al., 2004). To determine whether oligodendrocyte apoptosis during early development altered the capacity for subsequent remyelination, MBP-iCP9 or WT littermates that were both CID treated daily from P4-P6 were matured to P42 at which time a local demyelinating lesion was induced by injection of LPC into the dorsal columns white matter (Fig. 6A). Animals were recovered for 5, 14, or 28 dpl and the levels of remyelination assayed by electron microscopy. In adult, WT littermate controls at 14 d after LPC, significant levels of remyelination identified by characteristically thin myelin sheaths were detectable throughout the lesion at both the ultrastructural and light level (Fig. 6B, insets). By contrast, in LPC-lesioned adult MBP-iCP9 animals in which oligodendrocytes were ablated during early development, very limited remyelination was detectable at both the ultrastructural and light level (Fig. 6C). The few remyelinated axons that were present were found at the edge of the lesion. Although the density of axons in the lesions did not appear to be significantly different between control and MBP-iCP9 animals, there was a significant reduction in the density of remyelinated axons within the lesions in MBP-iCP9 animals (20. ± 2.0 axons/μm2) compared with controls (130 ± 20 axons/μm2; Fig. 6D). No significant differences in lesion size were apparent between treatment groups. Either a delay or an inhibition of remyelination may explain the reduction in myelinated axons in CID-treated MBP-iCP9 transgenic animals at 14 dpl. To address this question, post-LPC survival times were doubled to 28 dpl, a time at which remyelination is largely complete in control animals (Jeffery and Blakemore, 1995). Remyelination in CID-treated MBP-iCP9 animals remained incomplete in the lesion even at 28 dpl, with sparsely distributed myelinated fibers present throughout the lesion (Fig. 6E), suggesting an inhibition or profound delay in the rate of remyelination in animals subjected to early postnatal oligodendrocyte apoptosis. One factor that might compromise the ability of repair in CID-treated MBP-iCP9 animals is that oligodendrocyte-lineage cells or the pattern of myelination was unstable and became disrupted after P21 but before the LPC lesion at P42. Several lines of evidence suggest that this is not the case. Comparison of the prelesion density of Olig2+ cells between identically treated WT and MBP-iCP9 animals showed no significant difference (Fig. 6F). Likewise, comparing the relative density of CC1+ cells revealed a small but not significant increase in the number of oligodendrocytes in MBP-iCP9 versus WT but no change in the density of NG2+ OPCs (Fig. 6F). Finally, comparing the prelesion pattern of myelination in WT versus MBP-iCP9 animals revealed no obvious differences (Fig. 6G,H) similar to the results at P21. The compromise in myelin repair in the MBP-iCP9 animals is therefore unlikely to reflect inherent instability in white matter structure but rather a differential response to the LPC lesion.
Figure 6.
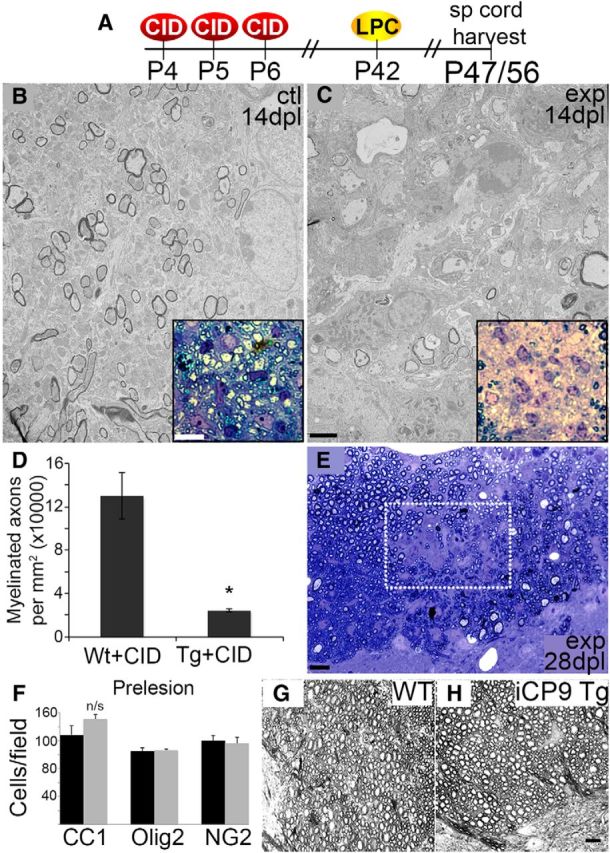
Oligodendrocyte ablation during early CNS development impairs remyelination of adult lesions. Remyelination was assessed in the LPC model in which the time course of myelin repair is well defined. A, LPC lesions were induced at day P42 in the dorsal columns of the thoracic spinal cords of MBP-iCP9 or WT littermates, both of which were CID treated from P4 to P6. Animals were killed at 5, 14, and 28 dpl. B, C, Electron micrographs demonstrate that induced oligodendrocyte apoptosis during development reduced the number of remyelinated axons following adult demyelination only in adult MBP-iCP9 animals, an effect quantified in D. Remyelinated axons were identified by stereotypically thin myelin sheaths relative to surrounding axons that were never demyelinated. A total of 75–100 axons were counted per animal in 3 WT and 6 MBP-iCP9. *p < 0.05. Error bars indicate SEM. E, Remyelination remains sparse at 28 dpl when remyelination is largely complete in WT mice. F, Before LPC lesion induction, no significant difference in the distribution or density of CC1+, Olig2+, or NG2+ cells was detected in WT versus MBP-iCP9 animals. n/s, Not significant. G, H, The pattern of myelination in grayscale images of toluidine blue-stained sections also shows no prelesion difference in myelination between treatment conditions (n = 4). Scale bars: C, 2 μm; E, 10 μm.
Lesion-induced proliferation of oligodendrocyte progenitor cells is reduced in animals subjected to developmental oligodendrocyte apoptosis
Demyelinating lesions induced by LPC stimulate a migration of OPCs from surrounding regions into the lesion around days 3–5, followed by proliferation, differentiation, and remyelination of the lesion (Crang and Blakemore, 1989; Keirstead et al., 1998). To determine the extent to which developmental oligodendrocyte apoptosis compromised the response of OPCs to subsequent demyelinating lesions, at various post-lesion time points, the density and proliferation of OPCs within the lesions were compared between MBP-iCP9 and WT littermates, all of which were CID treated from P4-P6. At 5 dpl, there was a significant reduction in the density of Olig2+ oligodendrocyte-lineage cells in MBP-iCP9 compared with WT littermates, an unexpected finding given that there was no difference in Ki67-labeled cell proliferation between treatment groups (Fig. 7A,C,E). The difference in post-LPC responses between WT and MBP-iCP9 animals were more pronounced at 14 dpl (Fig. 7B,D). In control animals, 70 ± 10% of the Olig2+ cells were Ki67+ at 14 dpl; whereas in MBP-iCP9 animals, only 30 ± 4% of the Olig2+ cells were Ki67+ (Fig. 7F), suggesting that the selective loss of mature oligodendrocytes during early development truncates an otherwise-sustained proliferation of oligodendrocyte lineage cells to an LPC-induced demyelinating lesion. Together, these data suggest that the induction of oligodendrocyte apoptosis in the thoracic spinal cord during early development results in a delay in myelination that largely recovers but leaves the tissue compromised in its ability to rapidly remyelinate following a subsequent demyelinating LPC lesion.
Figure 7.
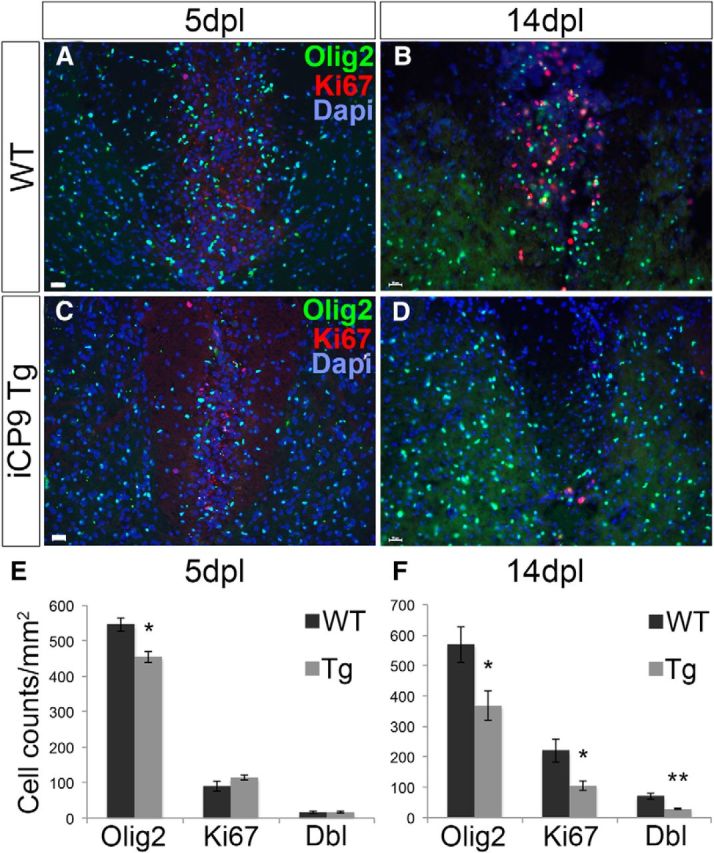
The neonatal loss of oligodendrocytes correlates with a truncated OPC proliferative response to adult demyelination. A, B, LPC injected into WT animals resulted in a surge of OPC proliferation (Olig2+Ki67+) from 5 to 14 dpl. C, D, By contrast, a proliferative surge in MBP-iCP9 animals was conspicuously absent during this postlesion time frame despite an equivalent initial proliferative response from 0 to 5 dpl. E, However, at 5 dpl, there was a significant reduction in dorsal column Olig2+ cells in MBP-iCP9 versus WT that could not be explained by a decrease in the number of Ki67+ proliferating cells. F, By 14 dpl, however, there was a decrease in Olig2+ cells, Ki67+ cells, and double-labeled cells, suggesting an unsustained recruitment of proliferating OPCs from 5 to 14 dpl in MBP-iCP9 animals compared with WT littermate controls. n = 3 control. n = 5 experimental. Scale bar, 25 μm. *p < 0.05, **p < 0.01. Error bars indicate SEM.
Discussion
The regulation of myelination during development and repair has been extensively studied; however, it remains unknown how perturbations of developmental myelination affect the capacity for subsequent repair. Here we showed, using a novel transgenic mouse model (MBP-iCP9), that although the developing CNS was robust in its ability to cope with extensive oligodendrocyte apoptosis and resultant myelin loss, it significantly impaired the ability of the adult CNS to remyelinate subsequent lesions. Activation of iCP9 with the small-molecule CID from P4-P6 resulted in a delay in myelination that largely recovered by P21. Although myelination mostly returned to normal, there was a residual increase in the number of oligodendrocytes in affected regions. The long-term consequences of early oligodendrocyte apoptosis were revealed in second lesion experiments. In WT littermate controls, missing the MBP-iCP9 transgene and therefore nonresponsive to CID, robust remyelination was observed following LPC-induced demyelination of the thoracic spinal cord. Remyelination was easily detectable by EM at 14 dpl and was largely complete by 21 dpl. By contrast, in MBP-iCP9 animals exposed to CID between P4 and P6, remyelination was largely absent at 14 dpl and incomplete even at 28 dpl. Impaired remyelination correlated with an unsustained proliferative response in OPCs to LPC-mediated demyelination. Based on these results, we propose that apoptosis of myelinating oligodendrocytes during early postnatal development generates a local tissue memory that inhibits full recovery from a second unrelated demyelinating insult by modulating the proliferative response of local OPCs responding to the demyelinating lesion.
The mechanisms responsible for the timing of myelination during normal CNS development are incompletely understood. One concept suggests that the OPCs possess an inherent timing mechanism modulated by proliferative behavior that regulates differentiation and myelination (Temple and Raff, 1986). The ability of the developing spinal cord to recover from early depletion of mature oligodendrocytes suggests that there is significant plasticity in the oligodendrocyte lineage, consistent with earlier studies in forebrain (Kessaris et al., 2006). The increase in the oligodendrocyte density in regions of the spinal cord at P21 following developmental apoptosis may reflect the delay in onset of myelination. For example, if the timing of myelination depends in part on the maturation level of target axons, possibly through the reduced expression of inhibitors, such as LINGO-1 (Mi et al., 2005) or PSA-NCAM (Charles et al., 2002), then delaying the appearance of mature oligodendrocytes may present a larger number of mature, “myelin ready” axons to an individual maturing oligodendrocyte, resulting in more, shorter internodes and thus a higher density of oligodendrocytes. Alternatively, delaying the appearance of mature oligodendrocytes compromises their capacity for myelination. Unlike remyelination in the adult CNS, however, the delay of developmental myelination does not appear to significantly impact the relative thickness of the myelin sheath, and g-ratios are relatively normal.
A significant consequence of perturbing developmental myelination is that recovery from a subsequent demyelinating insult is compromised. A number of factors may contribute to this repair failure, including changes in astrocytes (Zhang and Barres, 2010) and microglia cells (Williamson et al., 2011). In a different model, for example, neonatal bacterial infection in rats leads to significant hippocampal-dependent memory deficits only if the same rats were exposed to a subsequent LPS challenge, which triggered exaggerated Il-1β release from CD11-b+ microglia. Early oligodendrocyte apoptosis and resultant astrocyte and/or microglia activation in the MBP-iCP9 mice (data not shown) may prime the system in a similar way, rendering susceptibility to a prolonged or exaggerated immune response and excessive myelin damage, even if potentially reparative OPCs are abundant in or around lesions (Chang et al., 2002). Alternatively, early OL depletion may exhaust the supply of lineage-specific cell divisions (Temple and Raff, 1986), leading to premature cell senescence and lack of repair. Consistent with this possibility, a recent study suggested that stress early in life, including insults known to affect white matter development (Makinodan et al., 2012), resulted in a reduction in telomere length (Mitchell et al., 2014). An unexpected finding in our study was the increase in GFAP+ cells after oligodendrocyte ablation. In a model of fetal ischemia, the loss of oligodendrocytes led to an increase in expression of bone morphogenetic protein 4 (BMP4) and a failure to effectively respond to later insults (Reid et al., 2012). Because BMP4 induces a GFAP+ phenotype in OPCs (Miller et al., 2004), it is possible that induction of apoptosis in oligodendrocytes releases BMP4 that induces GFAP expression in OPC, which accounts for the increase in astrocytes and contributes to subsequent failure of repair. Additional studies are required to define the extent of changes in neural cells as a result of early oligodendrocyte apoptosis.
The timing or nature of the developmental insult appears to specifically influence adult myelin repair. Despite an apparent return to equilibrium in terms of oligodendrocyte cell replacement and myelination, a second demyelinating challenge revealed susceptibility to nonrepairing myelin loss in the mature CNS. Indeed, even when post-lesion survival was extended to a time point at which myelin regeneration is essentially complete after a single LPC lesion, remyelination in animals subjected to developmental oligodendrocyte ablation remained incomplete. These data are inconsistent with a previous report in which repeated episodes of focal demyelination in the adult CNS neither depleted the OPC pool nor impaired remyelination (Penderis et al., 2003). One important difference between these studies is the animal age at the onset of the first demyelinating stimulus. In the report by Penderis et al. (2003), the first demyelinating insult was not administered until 10 weeks of age when the animal is fully mature. By contrast, based on the current data, we argue that induction of oligodendrocyte apoptosis specifically during early postnatal development impairs adult oligodendrogenesis and myelin repair. A second difference is the mechanism of the initial insult. Apoptotic cells are known to release a number of unique signals, such as ATP, which trigger responses in surrounding cells, particularly microglia. The signals that result from induced apoptosis are likely distinct from those following toxin-induced demyelination and may lead to permanent tissue changes. Whatever the signals, these results identify a potentially novel mechanism underlying remyelination failure in multiple sclerosis, an inflammatory demyelinating disease of the CNS. According to the classification proposed by Lucchinetti et al. (2000), the dominant pathomechanism in a subset of active MS lesions, so-called Type III, is one of primary oligodystrophy (Barnett and Prineas, 2004). Follow-up studies involving various mouse models of primary oligodendrocyte death, however, have failed to demonstrate that such an injury is sufficient to initiate an autoimmune response, arguing against an oligodystrophy pathomechanism of immune-mediated demyelination. Our data identify developmental oligodendrocyte death as a potential contributing factor to remyelination failure in the adult CNS. Combined with epidemiological and proteomic studies of MS purporting childhood environmental factors as predisposing factors for MS (Kurtzke, 2000; Ascherio et al., 2007), our data open a potentially new avenue of exploration in MS (Dhaunchak et al., 2012).
These data add novel insights to our current understanding of factors that influence adult oligodendrocyte-lineage cell dynamics and remyelination. Using newly generated MBP-iCP9 transgenic mice, we showed that induced apoptosis of early oligodendrocytes predisposes impaired adult remyelination, which correlated with a truncated proliferation of responding OPCs. Experiments are ongoing to identify the molecular underpinnings of delayed adult remyelination secondary to developmental oligodendrocyte apoptosis.
Footnotes
This work was supported by the Myelin Repair Foundation and National Institutes of Health Grant NS30800 to R.H.M. The CWRU Transgenic Core generated the MBP-iCP9 transgenic mouse line. We thank Sara VanDommelen for help with manuscript preparation and Anita Zaremba for help with neurosphere preparations.
The authors declare no competing financial interests.
References
- Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis: I. The role of infection. Ann Neurol. 2007;61:288–299. doi: 10.1002/ana.21117. [DOI] [PubMed] [Google Scholar]
- Asipu A, Mellor AL, Blair GE. The specificity of the myelin basic protein gene promoter studied in transgenic mice. Biochem Biophys Res Commun. 2001;288:809–818. doi: 10.1006/bbrc.2001.5837. [DOI] [PubMed] [Google Scholar]
- Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci. 2001;21:1302–1312. doi: 10.1523/JNEUROSCI.21-04-01302.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol. 2004;55:458–468. doi: 10.1002/ana.20016. [DOI] [PubMed] [Google Scholar]
- Barres BA, Hart IK, Coles HS, Burne JF, Voyvodic JT, Richardson WD, Raff MC. Cell death in the oligodendrocyte lineage. J Neurobiol. 1992;23:1221–1230. doi: 10.1002/neu.480230912. [DOI] [PubMed] [Google Scholar]
- Blakemore WF. Observations on remyelination in the rabbit spinal cord following demyelination induced by lysolecithin. Neuropathol Appl Neurobiol. 1978;4:47–59. doi: 10.1111/j.1365-2990.1978.tb00528.x. [DOI] [PubMed] [Google Scholar]
- Cai J, Qi Y, Hu X, Tan M, Liu Z, Zhang J, Li Q, Sander M, Qiu M. Generation of oligodendrocyte precursor cells from mouse dorsal spinal cord independent of Nkx6 regulation and Shh signaling. Neuron. 2005;45:41–53. doi: 10.1016/j.neuron.2004.12.028. [DOI] [PubMed] [Google Scholar]
- Calver AR, Hall AC, Yu WP, Walsh FS, Heath JK, Betsholtz C, Richardson WD. Oligodendrocyte population dynamics and the role of PDGF in vivo. Neuron. 1998;20:869–882. doi: 10.1016/S0896-6273(00)80469-9. [DOI] [PubMed] [Google Scholar]
- Caprariello AV, Mangla S, Miller RH, Selkirk SM. Apoptosis of oligodendrocytes in the central nervous system results in rapid focal demyelination. Ann Neurol. 2012;72:395–405. doi: 10.1002/ana.23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A, Tourtellotte WW, Rudick R, Trapp BD. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med. 2002;346:165–173. doi: 10.1056/NEJMoa010994. [DOI] [PubMed] [Google Scholar]
- Charles P, Reynolds R, Seilhean D, Rougon G, Aigrot MS, Niezgoda A, Zalc B, Lubetzki C. Re-expression of PSA-NCAM by demyelinated axons: an inhibitor of remyelination in multiple sclerosis? Brain. 2002;125:1972–1979. doi: 10.1093/brain/awf216. [DOI] [PubMed] [Google Scholar]
- Crang AJ, Blakemore WF. The effect of the number of oligodendrocytes transplanted into X-irradiated, glial-free lesions on the extent of oligodendrocyte remyelination. Neurosci Lett. 1989;103:269–274. doi: 10.1016/0304-3940(89)90111-0. [DOI] [PubMed] [Google Scholar]
- Dhaunchak AS, Becker C, Schulman H, De Faria O, Jr, Rajasekharan S, Banwell B, Colman DR, Bar-Or A Canadian Pediatric Demyelinating Disease Group. Implication of perturbed axoglial apparatus in early pediatric multiple sclerosis. Ann Neurol. 2012;71:601–613. doi: 10.1002/ana.22693. [DOI] [PubMed] [Google Scholar]
- Foran DR, Peterson AC. Myelin acquisition in the central nervous system of the mouse revealed by an MBP-Lac. J Neurosci. 1992;12:4890–4897. doi: 10.1523/JNEUROSCI.12-12-04890.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goujet-Zalc C, Babinet C, Monge M, Timsit S, Cabon F, Gansmüller A, Miura M, Sanchez M, Pournin S, Mikoshiba K. The proximal region of the MBP gene promoter is sufficient to induce oligodendroglial-specific expression in transgenic mice. Eur J Neurosci. 1993;5:624–632. doi: 10.1111/j.1460-9568.1993.tb00528.x. [DOI] [PubMed] [Google Scholar]
- Gow A, Friedrich VL, Jr, Lazzarini RA. Myelin basic protein gene contains separate enhancers for oligodendrocyte and Schwann cell expression. J Cell Biol. 1992;119:605–616. doi: 10.1083/jcb.119.3.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery ND, Blakemore WF. Remyelination of mouse spinal cord axons demyelinated by local injection of lysolecithin. J Neurocytol. 1995;24:775–781. doi: 10.1007/BF01191213. [DOI] [PubMed] [Google Scholar]
- Keirstead HS, Levine JM, Blakemore WF. Response of the oligodendrocyte progenitor cell population (defined by NG2 labelling) to demyelination of the adult spinal cord. Glia. 1998;22:161–170. doi: 10.1002/(SICI)1098-1136(199802)22:2<161::AID-GLIA7>3.0.CO%3B2-A. [DOI] [PubMed] [Google Scholar]
- Kessaris N, Fogarty M, Iannarelli P, Grist M, Wegner M, Richardson WD. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci. 2005;9:173–179. doi: 10.1038/nn1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtzke JF. Epidemiology of multiple sclerosis: does this really point toward an etiology? Lectio Doctoralis. Neurol Sci. 2000;21:383–403. doi: 10.1007/s100720070055. [DOI] [PubMed] [Google Scholar]
- Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47:707–717. doi: 10.1002/1531-8249(200006)47:6<707::AID-ANA3>3.0.CO%3B2-Q. [DOI] [PubMed] [Google Scholar]
- MacCorkle RA, Freeman KW, Spencer DM. Synthetic activation of caspases: artificial death switches. Proc Natl Acad Sci U S A. 1998;95:3655–3660. doi: 10.1073/pnas.95.7.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinodan M, Rosen KM, Ito S, Corfas G. A critical period for social experience-dependent oligodendrocyte maturation and myelination. Science. 2012;337:1357–1360. doi: 10.1126/science.1220845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlow N, Wolke D, Bracewell MA, Samara M EPICure Study Group. Neurologic and developmental disability at six years of age after extremely preterm birth. N Engl J Med. 2005;352:9–19. doi: 10.1056/NEJMoa041367. [DOI] [PubMed] [Google Scholar]
- Matsushima GK, Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001;11:107–116. doi: 10.1111/j.1750-3639.2001.tb00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi S, Miller RH, Lee X, Scott ML, Shulag-Morskaya S, Shao Z, Chang J, Thill G, Levesque M, Zhang M, Hession C, Sah D, Trapp B, He Z, Jung V, McCoy JM, Pepinsky RB. LINGO-1 negatively regulates myelination by oligodendrocytes. Nat Neurosci. 2005;8:745–751. doi: 10.1038/nn1460. [DOI] [PubMed] [Google Scholar]
- Miller RH. Regulation of oligodendrocyte development in the vertebrate CNS. Prog Neurobiol. 2002;67:451–467. doi: 10.1016/S0301-0082(02)00058-8. [DOI] [PubMed] [Google Scholar]
- Miller RH, Dinsio K, Wang R, Geertman R, Maier CE, Hall AK. Patterning of spinal cord oligodendrocyte development by dorsally derived BMP4. J Neurosci Res. 2004;76:9–19. doi: 10.1002/jnr.20047. [DOI] [PubMed] [Google Scholar]
- Mitchell C, Hobcraft J, McLanahan SS, Siegel SR, Berg A, Brooks-Gunn J, Garfinkel I, Notterman D. Social disadvantage, genetic sensitivity, and children's telomere length. Proc Natl Acad Sci U S A. 2014;111:5944–5949. doi: 10.1073/pnas.1404293111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CS, Milner R, Nishiyama A, Frausto RF, Serwanski DR, Pagarigan RR, Whitton JL, Miller RH, Crocker SJ. Astrocytic tissue inhibitor of metalloproteinase-1 (TIMP-1) promotes oligodendrocyte differentiation and enhances CNS myelination. J Neurosci. 2011;31:6247–6254. doi: 10.1523/JNEUROSCI.5474-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble M, Murray K, Stroobant P, Waterfield MD, Riddle P. Platelet-derived growth factor promotes division and motility and inhibits premature differentiation of the oligodendrocyte/type-2 astrocyte progenitor cell. Nature. 1988;333:560–562. doi: 10.1038/333560a0. [DOI] [PubMed] [Google Scholar]
- Penderis J, Shields SA, Franklin RJ. Impaired remyelination and depletion of oligodendrocyte progenitors does not occur following repeated episodes of focal demyelination in the rat central nervous system. Brain. 2003;126:1382–1391. doi: 10.1093/brain/awg126. [DOI] [PubMed] [Google Scholar]
- Reid MV, Murray KA, Marsh ED, Golden JA, Simmons RA, Grinspan JB. Delayed myelination in an intrauterine growth retardation model is mediated by oxidative stress upregulating bone morphogenetic protein 4. J Neuropathol Exp Neurol. 2012;71:640–653. doi: 10.1097/NEN.0b013e31825cfa81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruckh JM, Zhao JW, Shadrach JL, van Wijngaarden P, Rao TN, Wagers AJ, Franklin RJ. Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell. 2012;10:96–103. doi: 10.1016/j.stem.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Q, Ma CY, Lv N, Lv ZL, Yan YB, Wu ZR, Li JJ, Duan JL, Zhu CL. Clinical study of cerebral palsy in 408 children with periventricular leukomalacia. Exp Ther Med. 2015;9:1336–1344. doi: 10.3892/etm.2015.2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temple S, Raff MC. Clonal analysis of oligodendrocyte development in culture: evidence for a developmental clock that counts cell divisions. Cell. 1986;44:773–779. doi: 10.1016/0092-8674(86)90843-3. [DOI] [PubMed] [Google Scholar]
- Traka M, Arasi K, Avila RL, Podojil JR, Christakos A, Miller SD, Soliven B, Popko B. A genetic mouse model of adult-onset, pervasive central nervous system demyelination with robust remyelination. Brain. 2010;133:3017–3029. doi: 10.1093/brain/awq247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Nishiyama A, Cheng D, Macklin W. Differentiation and death of premyelinating oligodendrocytes in developing rodent brain. J Cell Biol. 1997;137:459–468. doi: 10.1083/jcb.137.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HH, Frost E, To V, Robinson S, ffrench-Constant C, Geertman R, Ransohoff RM, Miller RH. The chemokine receptor CXCR2 controls positioning of oligodendrocyte precursors in developing spinal cord by arresting their migration. Cell. 2002;110:373–383. doi: 10.1016/S0092-8674(02)00838-3. [DOI] [PubMed] [Google Scholar]
- Vallstedt A, Klos JM, Ericson J. Multiple dorsoventral origins of oligodendrocyte generation in the spinal cord and hindbrain. Neuron. 2005;45:55–67. doi: 10.1016/j.neuron.2004.12.026. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res. 2001;50:553–562. doi: 10.1203/00006450-200111000-00003. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009;8:110–124. doi: 10.1016/S1474-4422(08)70294-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson LL, Sholar PW, Mistry RS, Smith SH, Bilbo SD. Microglia and memory: modulation by early-life infection. J Neurosci. 2011;31:15511–15521. doi: 10.1523/JNEUROSCI.3688-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff RH, Franklin RJ. Demyelination and remyelination of the caudal cerebellar peduncle of adult rats following stereotaxic injections of lysolecithin, ethidium bromide, and complement/anti-galactocerebroside: a comparative study. Glia. 1999;25:216–228. doi: 10.1002/(SICI)1098-1136(19990201)25:3<216::AID-GLIA2>3.0.CO%3B2-L. [DOI] [PubMed] [Google Scholar]
- Woodruff RH, Fruttiger M, Richardson WD, Franklin RJ. Platelet-derived growth factor regulates oligodendrocyte progenitor numbers in adult CNS and their response following CNS demyelination. Mol Cell Neurosci. 2004;25:252–262. doi: 10.1016/j.mcn.2003.10.014. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Barres BA. Astrocyte heterogeneity: an underappreciated topic in neurobiology. Curr Opin Neurobiol. 2010;20:588–594. doi: 10.1016/j.conb.2010.06.005. [DOI] [PubMed] [Google Scholar]