

Ke et al. found that >70% of m6A residues are present in the 3′-most (last) exons, with a sharp rise within 150–400 nucleotides of the start of the last exon. This report also suggests a role of m6A modification in regulating proximal alternative polyA choice.
Keywords: m6A-CLIP/IP, last exon, alternative polyadenylation, microRNA
Abstract
We adapted UV CLIP (cross-linking immunoprecipitation) to accurately locate tens of thousands of m6A residues in mammalian mRNA with single-nucleotide resolution. More than 70% of these residues are present in the 3′-most (last) exons, with a very sharp rise (sixfold) within 150–400 nucleotides of the start of the last exon. Two-thirds of last exon m6A and >40% of all m6A in mRNA are present in 3′ untranslated regions (UTRs); contrary to earlier suggestions, there is no preference for location of m6A sites around stop codons. Moreover, m6A is significantly higher in noncoding last exons than in next-to-last exons harboring stop codons. We found that m6A density peaks early in the 3′ UTR and that, among transcripts with alternative polyA (APA) usage in both the brain and the liver, brain transcripts preferentially use distal polyA sites, as reported, and also show higher proximal m6A density in the last exons. Furthermore, when we reduced m6A methylation by knocking down components of the methylase complex and then examined 661 transcripts with proximal m6A peaks in last exons, we identified a set of 111 transcripts with altered (approximately two-thirds increased proximal) APA use. Taken together, these observations suggest a role of m6A modification in regulating proximal alternative polyA choice.
Methylation of bacterial rRNA and tRNA was studied beginning in the 1950s (Srinivasan and Borek 1964; for review, see Starr and Sells 1969; Klagsbrun 1973). Even more frequent methylation of rRNAs and tRNAs in animal cells was also established by 1970 (Greenberg and Penman 1966; Burdon et al. 1967; Bernhardt and Darnell 1969; for review, see Munns and Sims 1975). The possible role of eukaryotic RNA methylation in RNA processing (size reduction) of pre-rRNA and pre-tRNA was first suggested because methylation occurred to the larger precursor molecules, pre-rRNA and pre-tRNA, and methionine starvation (diminishing available S-adenosyl methionine, the methyl donor) greatly impeded processing in both cases (Vaughan et al. 1967; Bernhardt and Darnell 1969).
In 1971, it was discovered that the polysomal mRNA was polyadenylated (Darnell et al. 1971; Edmonds et al. 1971; Lee et al. 1971) and could be purified from rRNA and tRNA by attachment to polyU sepharose or oligo dT cellulose (for review, see Nakazato and Edmonds 1974). In light of the results on the possible connection of methylation to pre-rRNA and pre-tRNA processing, it became important and obvious to investigate the possible methylation of mRNA.
Perry and Kelley (1974) were the first to find that isolated polyA+ mRNA (from mouse L cells) could be methylated in cells from labeled methyl groups of methionine. Both a labeled alkali-resistant oligonucleotide and mononucleotides were present in alkali-degraded methyl-labeled mRNA. The oligonucleotide was identified later as the m7G-methylated 5′ cap structure—first in virus mRNA (Shatkin 1974; Wei and Moss 1974, 1975; Furuichi et al. 1975) and soon after in cell mRNA (Furuichi et al. 1975; Wei et al. 1975).
Both the Perry and the Rottman laboratories (Desrosiers et al. 1975; Perry et al. 1975) characterized by chromatography the methyl-labeled mRNA mononucleotides as 95% N6 methyladenylic acid and ∼5% 5′ methylcytidylic acid. There was a frequency of ∼1–2 m6A per 1000 nucleotides (nt) in mRNA (Desrosiers et al. 1975; Perry et al. 1975). From these original findings until the present, no function for any single m6A residue in mRNA has been clearly defined.
Additional notable findings concerning m6A in mRNA were made quite early. In bovine prolactin mRNA, Rottman and colleagues (Horowitz et al. 1984) found a single m6A that was located in the 3′ untranslated region (UTR). In late 16S and 19S SV40 virus mRNA, the ribonuclease T1-generated oligonucleotides that contained a single m6A residue were in the coding regions of the mRNAs (Canaani et al. 1979). In a number of early reports, the favored surrounding nucleotides were RACs (R: G or A) (Schibler et al. 1977; Wei and Moss 1977) and Kane and Beemon (1987) established RRACU as a larger consensus site for eight specific residues in Rous virus RNA.
With respect to the position within the mRNA (e.g., total HeLa cell mRNA), ∼15% of m6A was in the polyA-selected, 3′-terminal mRNA fragments of ∼500 nt, 200–250 of which were polyA (Salditt-Georgieff et al. 1976). This region of mRNAs is now recognized to be mainly the 3′ UTR. From the beginning of examination of m6A residues in mRNA, it was clear that this modification was not confined to a single location.
Bokar, first in the Rottman laboratory and later in his own group (Bokar et al. 1997), partially purified an ∼800-kDa complex with m6A methylase activity, from which a single 70-kDa enzyme was obtained and its gene was cloned (now known as METTL3 [methyltransferase-like 3]). In in vitro experiments with oligonucleotides of known sequences, Harper et al. (1990), using nuclear extracts, found a strong sequence preference of RRACU for adenylic acid methylation sites, strongly confirming the earlier studies identifying residues in isolated mRNAs. Recently, METTL3 has been found to be associated in cell extracts (for example, by coprecipitation) along with METTL14, a methylase detected by sequence similarity (Liu et al. 2014), and also WTAP (Wilms’ tumor-associated protein), a protein originally described as having some association with pre-mRNA splicing (Little et al. 2000). These three proteins presumably comprise the 800-kDa complex originally described by Bokar et al. (1997).
The first reported m6A-specific antibodies (Munns and Sims 1975) proved that they could be used to identify m6A residues in RNAs and specific RNA segments. In 1987, Lührmann and Bringmann (Bringmann and Luhrmann 1987) prepared highly specific m6A antibodies that they used to show the presence of m6A in U1, U2, U4, and U6 but not U5 spliceosomal RNAs. These early experiments finally stimulated commercial production of highly specific m6A antibodies. The availability of these reagents led to a plethora of studies, beginning in 2012, coupling m6A antibody precipitation of oligonucleotides derived from total mammalian and yeast cell polyA+ RNA to high-throughput sequencing (RNA-seq) (e.g., see Dominissini et al. 2012; Meyer et al. 2012; Fustin et al. 2013; Schwartz et al. 2013, 2014).
This recent work confirmed and extended the early conclusions that m6A is widely distributed in half or more of individual mRNA molecules at an average frequency of two to four m6A residues in most of the mRNAs possessing it, although a distribution of up to 10 or even more m6A sites in specific mRNAs was also demonstrated (Meyer et al. 2012). The earlier identified, preferred motif RAC/RRACU was reconfirmed within the precipitated m6A-containing oligonucleotides and served as the basis for assigning m6A residues to a particular mRNA segment but not identifying which RAC/RRACU motif was actually methylated. The majority of m6A was reported to be distributed in the body of mRNAs, but a substantial fraction was reported to be near the distal end of mRNAs (Dominissini et al. 2012; Meyer et al. 2012; Schwartz et al. 2014). These modifications in the 3′ region of mRNAs were described as enriched around the translation stop codon and in the 3′ UTR (Meyer et al. 2012; Schwartz et al. 2014).
While no single site-specific mutagenesis of m6A sites providing evidence of function has been reported in any mRNA, a global study suggests that m6A in general may play a role in mRNA stabilization (Wang et al. 2014). Complete knockout of METTL3 in mice is lethal, and recent reports show that early embryonic differentiation events are prevented by CRISPR removal of METTL3 in mouse embryonic stem cells (see the Discussion; Batista et al. 2014; Geula et al. 2015).
In the present work, modifying the HITS (high-throughput sequencing)-CLIP (cross-linking immunoprecipitation) protocol (Licatalosi et al. 2008; Darnell 2010) and selection of m6A antibody-bound oligonucleotides, we describe a precise and generally applicable method to locate specific m6A residues. Analysis of m6A distribution in RNA from a variety of tissues and cell types confirmed that 3′ UTRs of mRNAs are highly enriched in m6As, but a major new result is that the concentration of m6As rises sharply from the beginning of the last exon but is not concentrated around the stop codon. We found that the 3′-terminal exons, including the 3′ UTRs, have 70% of all m6As; more are in the 3′ UTR than in the last exon-coding region.
Bioinformatic analysis of the preferential locations of m6A within 3′ UTRs containing multiple alternative polyA (APA) sites suggests a possible connection of m6As to polyA site choices in mRNAs with APA sites. Furthermore, the uncommon mRNAs whose terminal exons contain no coding sequence but provide a polyA site also contain frequent m6A residues. It is known that brain mRNAs in general are longer for mRNAs expressed in both the liver and the brain (Wehrspaun et al. 2014), and there is a higher m6A content in the last exons of mRNAs from the brain than from the liver. Moreover, when we reduced m6A methylation by knocking down three components of the methylase complex and examined transcripts with proximal m6A peaks in terminal exons, a set of transcripts was identified that showed changes in APA usage after knockdown. Finally, there is an overlap in the 3′ UTRs of m6A residues with binding sites for the Ago complex that contains negative acting regulatory microRNAs. Taken together, these results strongly suggest that the signal for a rise in m6A is not in proximity to a stop codon but is triggered somehow by entry into the last exon of an mRNA, where it may play a regulatory role in determining APA and/or some role in access by the Ago/microRNA complex.
Results
With the commercial availability of m6A-specific antibodies and RNA-seq, thousands of m6A residues in human and yeast polyA+ RNA have been reported (Dominissini et al. 2012; Meyer et al. 2012; Fustin et al. 2013; Schwartz et al. 2013, 2014; Wang et al. 2014). All of these reports located the m6A residues in a region with a resolution of ∼200 nt, within which the sequence motifs RAC/RRCAU, known to be targets of the RNA methylase (Harper et al. 1990), were found, but specificity as to which site actually contained the m6A was not determined. However, to prove the function of any particular m6A addition to a specific mRNA, site-specific mutagenesis will ultimately be needed. Therefore, a method for finding as precise a location of m6A residues as possible in sequenced m6A-containing oligonucleotides was our first goal (Fig. 1).
Figure 1.

Single-nucleotide resolution achieved by m6A-CLIP/immunoprecipitation (IP). (A) m6A-CLIP/IP illustration. (B) The m6A PS (defined in the text) maps m6A sites at its 0 position. “Enrichment of RRACU or RAC” is the fold enrichment of RRACU (red) or RAC (blue) motif density at that position compared with the background motif density (horizontal gray dot line). The background motif density was calculated as the motif density of RRACU or RAC at regions 400 nt upstream of or downstream from PSs where the density value was flattened. The vertical black dotted line represents the PS 0 position. The motif logo with its P-value represents the de novo motif. (C) Substitution of cross-linking-induced mutation sites (CIMSs) maps m6A sites at its −1 position. (D) m6A-induced truncation sites (MITSs) map m6A sites at its +1 position. (E) Summary of m6A sites identified by different approaches. “All RACs” is the total number of RACs within m6A peak regions. (F) Precisely mapped m6A sites are conserved in vertebrates regardless of the mapping approaches. Box plot bars are the first quartile to third quartile; the black line in the middle is the median value. (**) P < 10−6; (*) P < 0.01, Wilcoxon rank sum test. (G) m6A-CLIP/IP mapped a precise m6A site in TUG1 RNA by two independent means: at −2 of a deletion CIMS and also at +1 of a MITS. “m6A-IP enrich” is m6A-IP reads normalized to its input. “m6A-CLIP/IP” is by m6A-CLIP/IP (the A marked in red). SCARLET (site-specific cleavage and radioactive labeling followed by ligation-assisted extraction and thin-layer chromatography) is by Liu et al. (2013) at the underlined motif. (H) m6A-CLIP/IP mapped a precise m6A site in BSG mRNA that is located at the +1 position of MITSs. (I) m6A-CLIP/IP mapped precise m6A sites in TPT1 mRNA. From left to right, these sites were located at −1 of substitution CIMSs, +1 of MITSs, and 0 of PSs and were co-mapped by −1 of substitution CIMSs and +1 of MITSs. See also Supplemental Figures 1 and 2.
The CLIP method (Fig. 1A; Licatalosi et al. 2008; Darnell 2010) of locating specific RNA–protein interaction sites has been adapted to assist in m6A site location in mRNA. We performed m6A-CLIP pilot experiments using a single 25-nt-long RNA oligonucleotide either with or without a single m6A at position 13 (Supplemental Fig. 1A,B, two bands matching the molecular sizes of antibody light and heavy chain, respectively, in complex with the RNA oligo, and there are similarly two bands in Supplemental Fig. 1D). A UV-induced covalent RNA/m6A antibody complex was formed with only the m6A-containing oligonucleotide, confirming the high specificity and stringency of m6A-CLIP. After protease removal of the attached antibody, we sequenced the RNA-cross-linked oligonucleotides and observed that UV-induced insertions were found in some of the cross-linked molecules at the +1 position; i.e., 3′ to the m6A site (Supplemental Fig. 1B). There are only two known m6A sites in human rRNAs: the A1832 site of 18S and the A4190 site of 28S. We carried out the m6A-CLIP procedure on total RNA where rRNA is predominant and, in both cases, found substitution mutations predominantly at the +1 position of the two known m6A sites in CLIP but not in a noncross-linked sample (Supplemental Fig. 1C). The observations on mutations in cross-linked synthetic RNA oligonucleotides and the rRNAs around m6A sites have been very useful in later mRNA studies.
Upon application of m6A-CLIP to fragments of polyA-selected RNAs from human cells (CD8+ T cells), the UV-induced m6A antibody covalent complex was apparent after UV but not in two negative controls (m6A antibody precipitation without irradiation and exposure to nonspecific rabbit IgG with irradiation) (Supplemental Fig. 1D), confirming the stringency and high signal to noise ratio of m6A-CLIP.
We then turned to locating m6A sites using the CLIP method on mRNA from human CD8+ T cells and mouse brains. PolyA+ RNA was broken, and oligonucleotides 20- to 80-nt-long were selected. A portion was saved as input. The remainder was precipitated with anti-m6A antibody. Half of this RNA was eluted from the antibody with m6Ap (m6A immunoprecipitation [IP]), and the other half was exposed to UV irradiation followed by digestion with proteinase (m6A-CLIP). Both RNA oligonucleotide preparations (m6A-IP and m6A-CLIP) were recovered for sequencing after conversion to cDNA libraries (Fig. 1A, m6A-CLIP/IP illustration; details in the Materials and Methods).
As found by others comparing sequences of the input RNA and m6A-IP RNA (e.g., Dominissini et al. 2012; Meyer et al. 2012), we identified thousands (38,164 in mouse brains and 27,495 in human CD8+ T cells) of m6A peak regions (with enriched m6A-IP reads relative to the input RNA sample) (Fig. 1A; see details in the Materials and Methods) that averaged ∼100–200 nt, and most of these regions had multiple RAC or RRACU sequences. At this stage, no positive identification of a precise m6A site was possible.
As done by Schwartz et al. (2013, 2014) with antibody-precipitated fragments, we identified a peak site (PS) within each m6A peak region observed in the nonirradiated m6A-IP data by locating the nucleotide position that had the highest enrichment of m6A-IP reads after normalization to its input reads (see details in the Materials and Methods). De novo motif analysis revealed that the RRACU and RAC motifs were significantly enriched around these PSs (Fig. 1B, P < 10−400). We searched for RRACU/RAC motifs that had the highest enrichment at the 0 position (the center of the PS) (Fig. 1B). These RAC/RRACU motifs located at the 0 positions of PSs had the best signal to noise ratio. This approach identified 7117 such specific m6A sites for mouse brains as an actual m6A site.
We next turned to the preparation and sequencing of the UV cross-linked m6A antibody-bound RNA sample (m6A-CLIP). When a protease was used to remove a cross-linked antibody prior to cDNA preparations and sequencing, a small peptide or amino acid remained attached to the m6A residue in the cross-linked RNA sample. The sequencing of the m6A-containing cross-linked fragment after protease digestion involved tailing with primer sites and reverse transcription into DNA for sequencing (details are given in the Materials and Methods). During this procedure, cross-linking-induced mutation sites (CIMSs; including single-base substitutions, deletions, and insertions) (Zhang and Darnell 2011) and cross-linking-induced truncation sites (CITSs) occur (Konig et al. 2010), which subsequently assist in more precise mapping of m6A. In the present study, we took advantage of a recent improvement involving circularization of the DNA copy of RNA fragments that enables capture of truncations (Weyn-Vanhentenryck et al. 2014).
By comparing mutations identified in RNA of the m6A-CLIP samples and the sequences from m6A-IP samples, we identified thousands of UV-induced CIMSs and CITSs in mouse polyA+ RNA (details are in the Materials and Methods). De novo motif analysis revealed that the same RAC/RRACU motif was significantly enriched around the CIMSs and CITSs (Fig. 1C, P < 10−400). The RAC/RRACU motifs around CIMSs were sharply enriched at the −1 position for the substitution mutations (1 nt in the 5′ direction) (Fig. 1C) and at −2 for the deletion mutations (Supplemental Fig. 1E). Insertion CIMS mutations, although rare, showed enrichment of the m6A motifs at the −1 position (Supplemental Fig. 1F). The CITS data showed enrichment of the RRACU/RAC at the −3 position (Supplemental Fig. 1G). Taking CIMSs and CITSs together, we precisely located 4305 putative m6A sites in a RAC/RRACU motif in mouse brain mRNA.
We discovered another aid to precise m6A site location. Reverse transcription across m6A-IP sites without cross-linking can also lead to truncation. By comparing cDNA truncation sites in the sequences of the m6A-IP samples with those of its input RNA samples, we identified m6A-induced truncation sites (MITSs) in m6A-IP samples, while the input RNA had a cDNA truncation rate at a background level (see the details in the Materials and Methods). De novo motif analysis around MITSs revealed the same RAC/RRACU motif (P < 10−400) (Fig. 1D). Enrichment of RAC/RRACU motifs occurred at the +1 position of the truncation (mouse) (Fig. 1D). MITSs precisely charted 21,779 m6A sites with the RAC/RRACU motif for mouse brains.
Combining PS, CIMS, CITS, and MITS information, we located 30,078 m6A sites for mouse brains (Fig. 1E), with a false discovery rate (FDR) of 17% (estimated by random site permutation, described in the Materials and Methods). For human CD8 cells, the numbers were 19,682 m6A sites, with an FDR of 16% (Supplemental Fig. 2A–D). m6A sites identified by each type of location method significantly overlapped with sites identified by other types of location methods (P < 10−100, hypergenometric test) (Supplemental Fig. 2E).
Furthermore, the potential functional importance of the tens of thousands of m6A sites that we precisely mapped with the various location methods in mice and humans (CIMSs, CITSs, MITSs, and PSs) was supported by the fact that the RAC/RRACU motifs that we called m6A sites were conserved in vertebrate evolution compared with other RAC/RRACU motifs within the same m6A peak regions (Fig. 1F; Supplemental Fig. 2F). Because our method achieves m6A single-nucleotide precision implementing m6A-CLIP and m6A-IP simultaneously, we term it m6A-CLIP/IP.
Comparing the present m6A site localization with the SCARLET (site-specific cleavage and radioactive labeling followed by ligation-assisted extraction and thin-layer chromatography) method
Previous work by Liu et al. (2013) located precisely eight specific m6A sites in specific polyA+ RNAs by a technique that they called SCARLET. This procedure selected m6A-containing oligonucleotides from mRNA by using specific antisense oligonucleotides to select a sequence region with an identified RAC/RRACU sequence that was therefore suspected of containing an m6A site. Specific localization of an m6A residue involved RNA cleavage, biochemical tailing to select mRNA fragments, and a final chromatographic base separation to prove the presence of individual specific m6A residues. SCARLET, although precise, is not practical for large-scale location of m6A sites. The precise m6A sites mapped here by m6A-CLIP/IP (the collective set of PS, CIMS, CITS, and MITS m6A sites) overlapped very well with those identified by the direct SCARLET technique. An m6A residue in TUG1, a polyA+ human noncoding RNA (NR_002323), was located at nucleotide 1552 by SCARLET, and our m6A localization mapped exactly the same site in human RNAs (Fig. 1G). Similarly, both methods identified an m6A at position 1335 in BSG, a polyA+ human mRNA (NM_198591) (Fig. 1H). There are three nearby m6A sites that were confirmed by SCARLET in TPT1 human polyA+ mRNA, and we precisely located all of these sites, which are crowded into a single m6A peak region (Fig. 1I). Four m6A sites within an ∼200-nt range were verified in MALAT1, a polyA+ long noncoding RNA; we again precisely mapped each of the four sites (Supplemental Fig. 2G). In total, Liu et al. (2013) identified within these RNAs eight m6A sites with high confidence, and our genome-wide strategy precisely located eight of the eight. As a negative control, two RRACU sites (positions 2953 and 5910 in TUG1) were found with no m6A methylation according to SCARLET, and neither of them was included by m6A-CLIP/IP. Although the number of sites examined by SCARLET is small, the comparison above suggests that m6A-CLIP/IP achieved high sensitivity and specificity for precise m6A mapping.
A large fraction of m6A modifications of mRNAs occurs in the last exon of mRNAs
In producing mRNAs, there are several processing variations of individual pre-mRNAs in any mammalian cell or tissue that are relevant to the following discussions: (1) The most common (>90%) are molecules with multiple exons necessitating one or more splices, some of which can be differential. (2) Recent attention has focused on the exons that, in any particular mRNA, all encode the same C-terminal amino acids but contain within their 3′ UTRs APA sites that lead to longer or shorter 3′ UTR sequences (Tian et al. 2005; Ji et al. 2009; Darnell 2013). It was found that metabolic conditions (e.g., fast or slow growth rates or a change in differentiation state) are correlated with choice between these alternative 3′ UTR polyA sites (Sandberg et al. 2008; Ji et al. 2009). (3) Exon junction sequence protein complexes (EJCs) are required in the vast majority of mRNAs (∼95%) at every splice junction to avoid nonsense-mediated decay (NMD) when the mRNA is first translated in the cytoplasm (Maquat 2015). Thus, in most mRNAs, the last exon contains a translation stop codon and untranslated 3′ sequences downstream before polyA addition terminates the mRNA. However ∼5% of mRNAs have a downstream noncoding exon spliced to the last coding exon and somehow escape NMD. These noncoding exons have various sequence lengths before a concluding 3′ polyA site.
Using the results, we obtained an m6A distribution in mRNA (as well as data previously published by other groups) and, anchoring the analysis of the m6A distribution at either the start of the last exons or the stop codons, obtained a very clear answer about the general distribution of m6A residues in mRNA (Fig. 2A; Supplemental Fig. 3A). As reported by other groups, m6A peak region density (m6A peak regions are defined as regions with enriched m6A-IP reads relative to the input RNA sample, referred to as “peak density” from now on) rises modestly (approximately threefold) approaching stop codons (Fig. 2A, top panel). In contrast, when the distribution of m6A is anchored at the beginning of the last exon, the m6A peak density gradually declines approaching the last exon. Upon entering the last exon, the peak density of m6A then rises sharply (at least a sixfold increase compared with the m6A level at the −200 position), reaching a maximum within 150–400 nt (Fig. 2A, bottom panel). This region is where the majority of 3′ UTRs begin and is therefore also the region that contains the majority of stop codons. The average distance from the 3′ splice site that begins the last exon to the stop codon that begins the 3′ UTR is reported to be only ∼208 nt, and 98% of all UTRs start by 400 nt (Fig. 2B, bottom panel; Furuno et al. 2003). Thus, the first 400 nt past the beginning of the last exon contain a mixture of the last coding region and the first 3′ UTR. The initial region of the last exon shows a very small rise in m6A deposition that accelerates to a peak between 150 and 400 nt from the last exon start (Fig. 2A). As the last exon sequence continues away from the stop codons, the m6A density remains high in what is now the sequence of the 3′ UTR. In total, about two-thirds of m6As in last exon are located in the 3′ UTR.
Figure 2.

m6A is enriched when entering last exons but not at stop codons. (A) Entering the last exon, m6A density increased sharply (bottom panel) in contrast to its lagging increase when approaching the stop codon (top panel). Data are from mouse brains. “m6A peak density” was calculated as the number of m6A peak regions in a 10-nt interval divided by the total number of mRNAs that contained this position. (B) m6A is enriched in the last exons but not around the stop codons. The top panel is the m6A peak distribution in the region around the stop codons; the two bottom panels are the distribution of the m6A peak regions and stop codons around the last exon start. mRNAs were grouped according to their stop codon locations to the last exon start. (C) m6A is enriched in the last exons but not around the stop codon when the stop codon is not in the last exon. (Top and bottom panels) mRNA with noncoding last exons anchored at the last exon start or the stop codon. (D) Most exonic m6As locate in last exon (mouse brains). The three pie graphs show the relative proportions of m6A peaks and the RAC and RRACU motifs in the last exon and other exons, with 100% representing all m6A peaks and RAC and RRACU sequences in mRNA. See also Supplemental Figure 3 for human and previously published data.
This situation is brought into sharper focus by examining mRNAs with stop codons at various distances from the last exon starts (Fig. 2B; Supplemental Fig. 3B). (1) There is little enrichment of m6A around the stop codon that is within 50 nt from the last exon start. Furthermore, the m6A density peaks ∼200 nt downstream from the stop codons (Fig. 2B, top panel, blue curve). (2) When the stop codon is located in the range of 51–150 nt from the last exon start, m6A rises before the stop codon and peaks after the stop codon (Fig. 2B, top panel, black curve); the same distribution is evident even when the stop codon is located in the range of 151∼250 nt from the last exon start (Fig. 2B, top panel, green curve). (3) Last, when the stop codon is located >250 nt away from the last exon start, the m6A density shows enrichment before the stop codon that continues at a high level into the 3′ UTR (Fig. 2B, top panel, purple curve). The m6A density always peaks in the 150- to 400-nt distance from the beginning of the last exon start regardless of the stop codon locations (Fig. 2B, middle panel). Thus, the apparent enrichment of m6A around the stop codons simply reflects the fact that they lie in the early region of the last exon (Dominissini et al. 2012; Meyer et al. 2012).
We next analyzed the ∼5% of mRNAs that have a stop codon in a next-to-last exon followed by a terminal noncoding 3′ exon. The terminal 3′ exon (untranslated) of these mRNAs is highly methylated, while the proximal exon harboring the stop codon is not. In these instances, the terminal exon is approximately as highly methylated as the last exon in the majority of mRNAs (Fig. 2C, bottom panel, black line; Supplemental Fig. 3C).
Finally, some noncoding RNAs have been described that are apparently spliced and are reported to have 3′ exons with polyA sites (Guttman et al. 2009). These molecules also have an elevated density of m6A residues in the last exons (Fig. 2B middle panel, gray line).
The last exons contain >70% of the total m6As in mRNAs even though the RAC/RRACU motifs in the last exons represented no more than 50% of the total RAC/RRACU motifs in mRNAs (Fig. 2D; Supplemental Fig. 3D). Normalizing m6A peaks to exon length or RAC/RRACU motifs, the m6A density in the last exons is higher than in the other exons (twofold to threefold, P < 10−100; i.e., m6A peak/exon length, m6A peak/RAC counts, or m6A peak/RRACU counts).
All of these results strongly emphasize that methylating adenosine residues in mRNA does not occur at or because of proximity to the stop codons but is higher at the beginning of the last exons and continues into the 3′ UTR, where most of the distal m6A residues lie.
Possible relation of m6A density to choice of a polyA site
Among the last exons with different lengths, longer last exons have a higher m6A density than shorter ones (long last exons: >850 nt, 5916 mRNAs; short last exons: ≤850 nt, 3624 mRNAs) (Fig. 3; Supplemental Fig. S4A). However, RAC/RRACU motifs were equally prevalent in all last exons (Supplemental Fig. S4B). As noted earlier, many pre-mRNAs offer the possibility/necessity of choosing between APA sites in the last exons (Tian et al. 2005). The fact that the majority of m6As locate in the last exons and particularly in the long last exons led us to suspect that m6A might contribute to choosing polyA sites. If m6A is inhibitory for polyadenylation, then short last exons might contain fewer m6As so as to allow early polyadenylation. Longer last exons might acquire more m6As so as to inhibit proximal polyadenylation. The distribution of m6A density in Figure 3 agrees with this reasoning.
Figure 3.
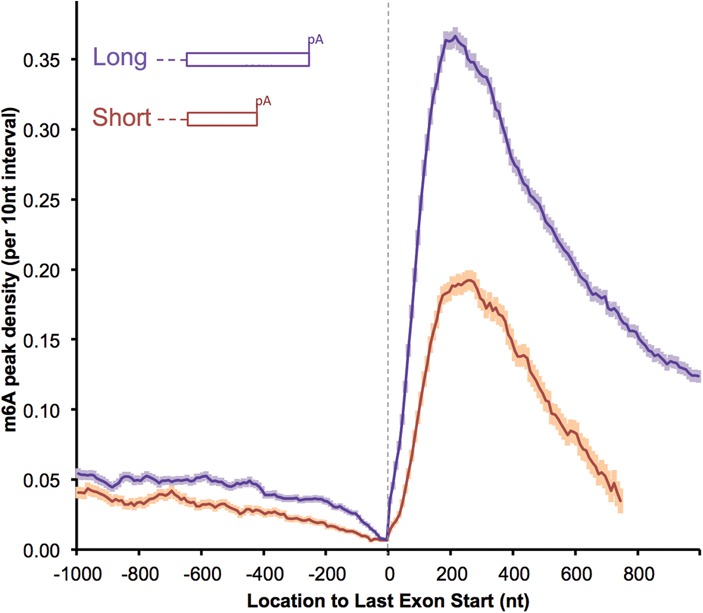
Short last exons have a lower m6A density (mouse brains). The m6A density in the last exons were compared between longer and shorter last exons (long: >850 nt, 5916 genes; short: ≤850 nt, 3624 genes). All of the genes considered are multiexon-coding genes with RPKM (reads per kilobase per million mapped reads) ≥ 1 in the mouse brain. The results showing more proximal m6A in transcripts using distal polyA sites are normalized to transcript abundance: “m6A peak density” was calculated as the number of m6A peak regions in a 10-nt interval divided by the total number of mRNAs that contained this position (the same definition is used throughout the text). The error bar indicates the standard error of the mean at each position. See also Supplemental Figure 4 for human data and RAC motif controls.
Furthermore, we examined the m6A density in the formation of ∼2000 such mRNAs with at least two APA sites in the last exon and compared cases in which a proximal or distal polyA site was used. Only those cases in which APA sites were used at least 10% of the time were examined. When a proximal polyA site was chosen more frequently than a distal site, the m6A density both before and after the proximal polyA site was lower, whereas if a proximal polyA site was used less frequently, the m6A density around this site was greater (“used more“ means ≥60% usage of all polyA sites in last exon, and “used less” means ≤40% usage; other cutoffs produce similar results) (Fig. 4A, left panel). The reverse was true: If a distal polyA site was used more frequently, there was more m6A prior to the site (Fig. 4A, right panel). These relationships held for both mouse brains (Fig. 4A) and human CD8 T cells (Supplemental Fig. 5A). These results are in accord with the possibility that the m6A modification may be associated with the decision not to use a polyA site.
Figure 4.

Higher m6A levels in the last exons correlates with more distal polyA site usage. (A) m6A is more abundant near proximal sites that are used less frequently (black line; left panel); when distal sites are used more, the m6A level is higher until the polyA site is reached (black line; right panel). The error bar indicates the standard error of the mean at each position. The shaded area highlights the major area of difference between two groups. (**) P < 10−35, Fisher's exact test. “Used more“ means ≥60% usage of all polyA sites in the last exon, and “used less” means ≤40% usage. Other cutoffs, including 50% or 70%, produce the same findings. The expression of mRNAs in regions between proximal and distal polyA sites is adequate for m6A detection (RPKM ≥ 1). Mouse brain data are presented here; see also Supplemental Figure 5A for human data. (B) Distal APA sites in the last exons are used more often in brains for coexpressed mRNAs. APA site pairs with statistically significant differences in usage are highlighted in orange (higher usage of distal sites in the brain) or blue (higher usage of distal sites in the liver). FDR = 20%, Fisher's exact test. The same conclusion is true for FDR = 5% (Supplemental Fig. 5B). (C) m6A peak regions in the last exons have a higher enrichment value in the brain. “m6A peak enrich. value” is the m6A-IP reads normalized to the input reads for each m6A peak region. m6A peak regions with statistically significant differences in “m6A peak enrich. value” are highlighted in orange (higher in the brain) or blue (higher in the liver). FDR = 5%, Fisher's exact test. (D) Positional plot of m6A peaks around APA sites in the last exons. “m6A peak density (brain–liver)” for each position was calculated as the number of m6A peak regions that are significantly higher in the brain (orange points in C) in a 10-nt interval divided by the total number of mRNAs that contained this position. (Orange lines) More distal usage; (gray lines) no change in the brain. The expression of mRNAs in regions between the proximal and distal polyA sites is adequate for m6A detection in both tissues (RPKM ≥ 1). The error bar indicates the standard error of the mean. The shaded area highlights the major area of difference between two groups. (**) P < 10−20, Fisher's exact test.
Brain vs. liver: differential usage of the available 3′ polyA sites
The earlier findings (Sandberg et al. 2008; Ji et al. 2009) on the choice of polyA sites and the consequent exchange in the length of 3′ UTRs that occurs with differentiation and the more proximal site usage with rapid growth led us to examine the density of m6A residues in the last exons in the same mRNA in different tissues: the liver and the brain. Furthermore, it has been reported frequently that mRNAs in the brain have unusually long UTRs both in general and in specific genes (Wang and Yi 2014; Wehrspaun et al. 2014).
We compared the 9000 mRNAs that are expressed in both the liver and the brain (total polyA+ RNA from total organs in both cases). Among these mRNAs, there were 2879 with multiple polyA sites detected in the last exons in both tissues. Among these mRNAs, we found more than three times as many dually expressed mRNAs that use more distal polyA sites in the brain compared with the liver (Fig. 4B). Such a choice was not universal, as 527 brain mRNAs used a more distal site, 152 liver mRNAs used a more distal site, and 2200 mRNAs that used multiple sites used the same sites in both tissues (Fig. 4B). Correlated with the more frequent distal polyA choice and longer mRNAs in the brain, there was a much higher m6A peak density in the last exons of mRNAs when expressed in the brain than when expressed in the liver (Fig. 4C).
We compared the m6A density in UTRs as a function of whether a proximal or a distal site was used to complete the mRNA. It was clear that when a proximal polyA site was used less in the brain, there was a higher m6A density close to the polyA site (Fig. 4D, left panel, gray shading; Supplemental Fig. 5C, left panel, gray shading). The converse was also true: When the distal site was used more in the brain, more m6A was found upstream of that site (within 1 kb) (Fig. 4D, right panel; Supplemental Fig. 5C, right panel). All of these results support the possibility that at least some of the m6A in the 3′ UTR is inhibitory for proximal polyadenylation.
The effects of global reduction of m6A on alternative polyadenylation
Several recent experiments have used siRNA mRNA knockdown experiments with somewhat irregular success at reducing proteins of the m6A methylase complex (METTL3, METTL14, and WTAP, defined above) (Schwartz et al. 2014). We used this approach in cultured human cell line A549, which had been used before (Schwartz et al. 2014). Simultaneous RNA oligonucleotide suppression of all three m6A methylase complex proteins achieved virtually complete reduction in the three proteins’ levels (judged by antibody staining) after 96 h of siRNA treatment (Fig. 5A). The total m6A content in the polyA+ RNA fell by ∼75%.
Figure 5.
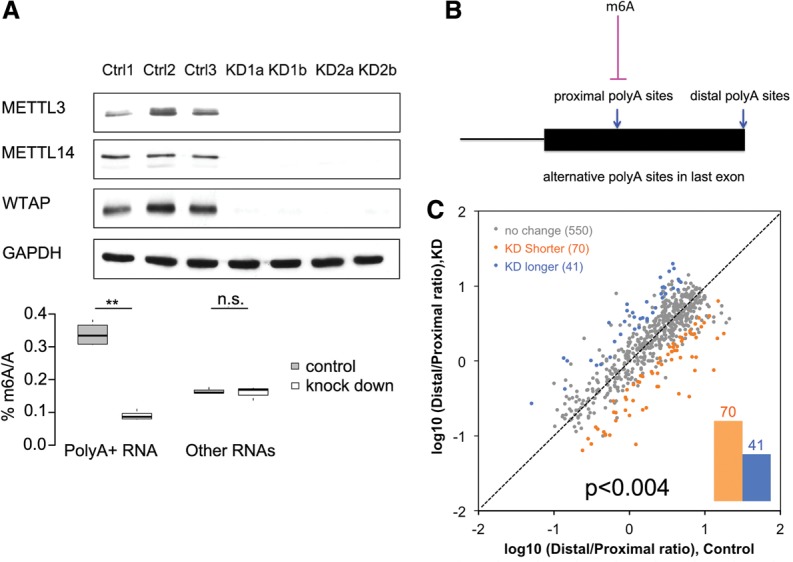
The effects of global limitation of m6A on alternative polyadenylation. (A) Triple knockdown of METTL3, METTL14, and WTAP reduced m6A of polyA+ RNAs to 26% in human A549 cells. The three different controls are biological replicates of control siRNA. The four different knockdown samples are as follows: KD1a and KD1b are biological replicates of one siRNA set, KD2a and KD2b are biological replicates of the other siRNA set, and the two siRNA sets generate essentially the same result. (**) P < 10−7, Student's t-test. (B) m6A may contribute to the inhibition of proximal polyA sites in the last exons. (C) The global limitation of m6A changed the choice of APA sites in the last exons. APA site pairs with statistically significant differences in usage are highlighted in orange (higher usage of proximal sites in knockdown) or blue (higher usage of proximal sites in the control). FDR = 20%, Fisher's exact test (the same conclusion is true for FDR = 5%, as shown in Supplemental Fig. S6). The mRNAs using two APA sites in the last exon and with m6A present around the proximal polyA sites are summarized. See also Supplemental Figures S6 and S7.
A group of polyA+ mRNA from these treated (knockdown) and untreated (control siRNA) A549 cells was analyzed to test the hypothesis that m6A may be inhibitory for proximal polyadenylation (Fig. 5B). We found ∼2500 mRNAs that had only two polyA sites, both of which were used. Of these, we chose 661 mRNAs that had an m6A within ∼150 nt upstream of or downstream from the proximal site. After knockdown, about one-sixth of these 661 mRNAs (111 total) changed the proximal versus the distal choice site (Fig. 5C; Supplemental Fig. S6), with 70 switching to the more proximal site, and 41 switching to the more distal site. This result at least allows the possibility that some m6A residues are inhibitory for proximal polyadenylation (70 vs. 41; P < 0.004, binomial test) (see Supplemental Fig. 7 for individual examples).
Thus, in the examination of liver and brain polyA choices and in the global knockdown results, it seems that one function of the m6A residue in the last exon may be a role in choosing a polyA site (Fig. 5B).
Overlap of m6A and Ago-binding sites in 3′ UTRs
MicroRNAs are perhaps the best-studied instance of regulatory binding sites in the 3′ UTRs of mRNAs. MicroRNAs are delivered into the cytoplasm as part of Ago protein–RNA complexes. UV irradiation cross-linked the Ago protein–microRNA complex to sites where RNA complementarity occurs, allowing precipitation with Ago antibodies. CLIP analysis then identified the presumed regulatory sites in the 3′ UTRs (Chi et al. 2009). We therefore examined microRNA/Ago-binding sites for a possible overlap with m6A residues by m6A-CLIP/IP. In these two now recognizable sites, there is indeed overlap in the 3′ UTR. For example, using a window of ±200 nt (200 nt on either side of m6A sites), there was an Ago site within ∼60% of an m6A site, significantly higher than that of randomly permuted controls (Fig. 6; note in Fig. 3 that a large amount of m6A lies in mRNAs with long 3′ UTRs). The binding sites in the 3′ UTRs for several other brain proteins known to bind pre-mRNAs and the 3′ UTRs of mRNAs were not enriched for m6A sites. In particular, m6A residues were less abundant around Hu sites (located by Hu-CLIP peaks) compared with controls (Fig. 6).
Figure 6.
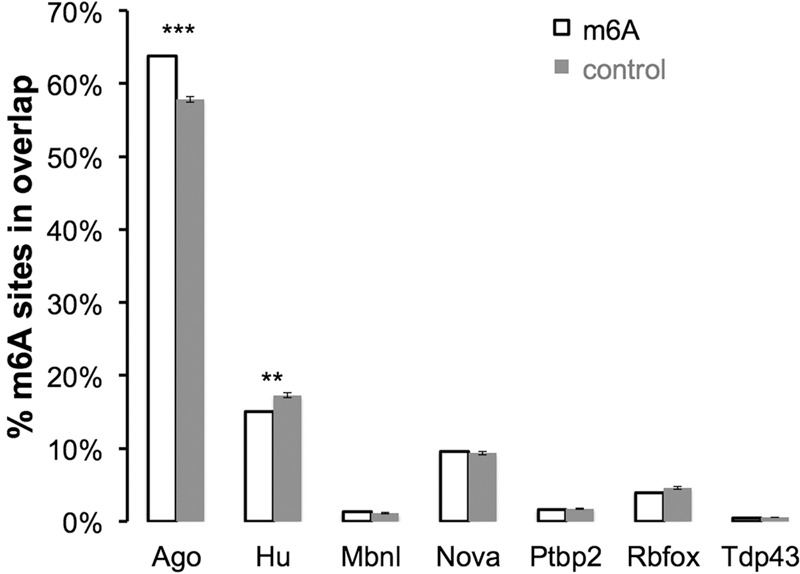
Overlap of m6A and Ago-binding sites in 3′ UTRs of mouse brains. (% m6A sites in overlap) The percentage of m6A sites (±200-nt flank) overlapped with the RNA-binding protein-binding site in mouse brains (mapped by published HITS-CLIP data) (see the Materials and Methods for reference details). The control was the randomly picked RAC motif (±200-nt flank) in the same 3′ UTR as m6A. Ten independent controls were performed to estimate the statistics. The error bar indicates the standard error of the mean. (***) P < 10−50; (**) P < 10−10, one sample t-test.
Discussion
We described here the use of biochemical and bioinformatic strategies that can locate very accurately individual m6A residues in the total polyA+ RNA molecules featuring, of course, mRNA molecules. First, we described a general conclusion that m6A density rises abruptly in the terminal exons regardless of whether the final exon is conventional in containing coding sequences and a stop codon. The density of m6A residues is distinctly less in the last 200 nt before the last exon and rises to a sixfold higher level in the first 150–400 nt of the last exon (Fig. 2A), a region that actually includes the last of the coding sequences and the beginning of 3′ UTR sequences. Thus, there is no preference for m6A residues at or around a stop codon. The last exon, including the 3′ UTR, has >70% of the total m6A residues, two-thirds of which are in the 3′ UTRs. Importantly, even if the last exon is entirely noncoding (∼5% of all mRNAs), the same level of m6A methylation occurs in these last exons.
Upon entering the last exon, an impending event for an RNA polymerase complex within <1 min (RNA moves at ∼3 kb/min) (for review, seeDarnell 2013) is that the polyA-related protein complexes (some portion of which has been apparently associated with the polymerase throughout its journey) (Hsin and Manley 2012) are about to cause primary transcript cleavage, polyA addition, and polymerase release. It is therefore tempting to speculate that a substantial fraction of m6A methylation events would occur close to (but probably before) the time of the actual polyadenylation event.
The new findings described here are relevant to a possible role of m6A in choosing APA sites, since at least 70% of m6A sites in mRNA are in the last exons, with about two-thirds in the 3′ UTRs where APA sites exist. Likewise, at least for some long brain mRNAs that use more distal available alternative sites, there is more extensive methylation of proximal adenosine residues, again offering support for a role of m6A sites in polyA choice. However, this possible role does not seem to be true for all mRNAs or all m6A sites. Only 527 mRNAs expressed in both the brain and the liver used a more distal site in the brain than in the liver, 152 mRNAs used a more distal site in the liver, and ∼2200 mRNAs used multiple sites approximately equally in both tissues.
Use of mutagenesis by siRNA knockdown shed some light on the situation. We did achieve a fourfold reduction in m6A in mRNA. However, the result of a knockdown of METTL3/METTL14/WTAP also gave no support that all of the m6A additions in the 3′ UTR exists to promote a distal polyA choice. In the knockdown cells, ∼110 out of ∼660 mRNAs using dual APA sites and m6A around the proximal sites did change the use of the polyA sites, with approximately two-thirds shifting from distal sites to proximal sites, consistent with an inhibitory effect of m6A on APA in these transcripts.
While no general rule is immediately evident from these observations, abundant evidence exists that polyA site choice involves a variety of mechanisms, since many proteins are involved in polyadenylation (Hsin and Manley 2012). For example, CstF64 (a cleavage stimulation factor), the concentration of which controls polyA site selection in B cells (Takagaki and Manley 1998, 2000), can be largely removed by siRNA knockdown with only a slight effect on global APA choice (Yao et al. 2012).
Much of what has been written in recent studies dealing with m6A has sought to connect m6A residues to other events in mRNA formation or function. For example, recent results described a generally more rapid mRNA turnover. One study used actinomycin to stop transcription (Wang et al. 2014), which alters cells drastically. Moreover, these claims contradict a previous experiment using radiolabeled methionine and a methylase inhibitor, S-tubercidinylhomocysteine (STH), an analog of S-adenosylhomocysteine. In HeLa cells, this agent stopped methylation ∼80% but had no effect on the half-life of new RNA that was formed in its presence (Camper et al. 1984). To study m6A function, site-specific mutagenesis to replace m6A will be required. Such experiments must begin with site-specific identification of m6A residues.
To our knowledge, only one report exists in which individual A residues known to be methylated in mRNA were changed to study the subsequent function of a specific RNA. Kane and Beemon (1985) found that the purified, sequenced genomic ∼7-kb RNA released from Rous sarcoma virus (RSV) contained m6A in at least seven specific protein-coding sites, all in the 3′ half of the full-length viral RNA. However, not all m6A sites in this genomic RNA were equally methylated, and the content in individual molecules therefore was not identical. This puzzling finding, which was recognized also using the SCARLET technique for individual animal cell RNAs (Liu et al. 2013), could obviously alter the frequency of any event dependent on an m6A residue. In a second study, Kane and Beemon (1987) mutated two adenylic acid residues known to be methylated in RSV in a region of an exon that is retained in the spliced product that encodes the src mRNA. This mutant virus was as infectious as the wild type and produced an equal amount of src mRNA and an equal number of colonies in soft agar (the assay for transformation).
There are nevertheless several facts currently available that attest to the physiological importance of m6A methylation. The first to be clearly delineated is the necessity for m6A addition to a single mRNA for yeast to undergo meiosis (Clancy et al. 2002). Second, mRNA demethylases exist in mammals and mammalian cells. Furthermore, mutations in one of the human demethylase genes is associated with a phenotype (overweight) (Jia et al. 2011; Zheng et al. 2013).
Finally, a METTL3 knockout was recently described (Geula et al. 2015) in naïve pluripotent stem cells that are at the preimplantation epiblast stage. These “naïve” embryonic stem cells are alive and grow without this enzyme. However, when triggered to terminate the naïve stage and enter the post-implantation stage, they fail. These experiments illustrate a necessary role for m6A mRNA addition but also show the viability of some cells without METTL3.
Thus, a fair statement at the moment is that the precise role of any particular m6A residue is unknown, but many such sites must be important.
Materials and methods
RNA extraction, DNase I treatment, and polyA selection
Total RNA was isolated from cells and tissues according to the manufacturer's instructions using TRIzol reagent (Life Technologies, catalog no. 15596), resuspended in RNA-free water, digested with RQ1 RNase-free DNase (Promega, catalog no. M6101) for 20 min at 37°C, and then subjected to phenol/chloroform extraction and ethanol precipitation. The resulting total RNA pallet was resuspended in RNA-free water. PolyA+ RNAs were selected using Dynabeads mRNA purification kit (Invitrogen, catalog no. 61006) according to the manufacturer's protocol.
m6A-IP
The PolyA+ RNA was fragmented to 20- to 80-nt fragments by alkaline hydrolysis (1 mM EDTA, 6 mM Na2CO3, 44 mM NaHCO3 at pH∼9.3) for 18 min at 95°C, and then the RNA fragments were precipitated following standard ethanol precipitation. Dynabeads Protein A beads (250 µL; Life Technologies, catalog no. 10008D) was pretreated with BSA (New England Biolabs, catalog no. B9000S) for 45 min at room temperature to reduce background. Anti-m6A rabbit polyclonal antibody (25 µg; Synaptic Systems, catalog no. 202003) was then conjugated to Dynabeads protein for 1 h at room temperature. The fragmented polyA+ RNAs (3 µg) were subject to PAGE selection for 20- to 80-nt size and then incubated with the antibody in 1× PXL buffer (1× PBS, 0.1% SDS, 0.5% sodium deoxycholate, 0.5% NP-40) supplemented with 2% RNasin Plus RNase inhibitor (Promega, catalog no. N2615) for 2 h at 4°C. After incubation, the beads and antibody were washed five times by four different wash buffers: twice by ice-cold Nelson low-salt buffer (15 mM Tris at pH 7.5, 5 mM EDTA), once by ice-cold Nelson high-salt buffer (15 mM Tris at pH 7.5, 5 mM EDTA, 2.5 mM EGTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 1 M NaCl), once by ice-cold Nelson stringent wash buffer (15 mM Tris at pH 7.5, 5 mM EDTA, 2.5 mM EGTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 120 mM NaCl, 25 mM KCl), and last by ice-cold NT-2 buffer (50 mM Tris at pH 7.4, 150 mM NaCl, 1 mM MgCl2, 0.05% NP-40). After the stringent wash series, the beads and antibody were split into two halves: One half was used to perform the m6A-CLIP leg, and the other half had its bounded RNA eluted with 0.5 mg/mL N6-methyladenosine sodium salt (Sigma-Aldrich, catalog no. M2780) for 1 h at 4°C (the m6A-IP leg). The eluted RNA was ethanol-precipitated and resuspended in RNase-free water for downstream cDNA library preparation.
m6A-CLIP
The remaining half of the m6A antibody was incubated with 1× immunoprecipitation buffer (0.3× SSPE, 1 mM EDTA, 0.05% Tween-20), subjected to UV cross-link (UV Stratalinker 2400, Stratagene), and then washed again five times by four different wash buffers—twice by Nelson low-salt buffer, once by Nelson high-salt buffer, once by Nelson stringent wash buffer, and once by NT-2 buffer—to further reduce background. After this wash series, the m6A antibody received on-bead PNK treatment (T4 PNK; New England Biolabs, catalog no. M0201L) for 20 min at 37°C to remove the 3′ phospho group of its UV cross-linked RNAs. After three washes with 1× PNK buffer (50 mM Tris-HCl at pH 7.4, 10 mM MgCl2, 0.5% NP-40) to remove the remaining PNK enzyme, the m6A antibody was subjected to on-bead linker ligation with T4 RNA ligase 2, truncated KQ (New England Biolabs, catalog no. M0373L), and 68 pmol preadenylated DNA linker (L32N, ordered from IDT) overnight at 16°C. The ligated product were subjected to 32P-γATP PNK hot labeling, and then the m6A antibody and its cross-linked RNA targets were eluted off beads by incubating with 6 µL of 1 M DTT (Sigma-Aldrich, catalog no. D0632), 27 µL of 1× PNK buffer, and 30 µL of Invitrogen 4× LDS sample buffer (Invitrogen, catalog no. NP0007) for 10 min at 70°C. The eluted product received SDS-PAGE and was then transferred to a nitrocellulose membrane (Bio-Rad, catalog no. 162-0112) to harvest RNAs UV cross-linked to the m6A antibody. The membrane bound with RNAs was cut out and digested of the protein by proteinase K (Roche, catalog no. 03115828001). The freed RNA (already ligated to L32N linker previously) was purified by phenol/chloroform extraction and ethanol precipitation and then resuspended in RNA-free water ready for downstream BrdU-CLIP library preparation.
BrdU-CLIP cDNA library protocol
The eluted RNA from the m6A-IP step and the input RNA fragments were first treated with T4 PNK to remove its 3′ phospho group and then ligated to 68 pmol preadenylated DNA linker (L32N from IDT) with T4 RNA ligase 2 and truncated KQ (New England Biolabs, catalog no. M0373L) overnight at 16°C. This ligation mixture was subject to 8% PAGE purification to harvest the ligated product. The purified RNAs from m6A-IP and m6A-CLIP and their input RNA fragments were all subjected to the same BrdU-CLIP cDNA library preparation. The detailed BrdU-CLIP cDNA library protocol was described previously (Weyn-Vanhentenryck et al. 2014), and we practiced a similar procedure with an improved RT primer set that starts with three “D” (no C) nucleotides on the 5′ end and improves ligation efficiency.
m6A-IP and m6A-CLIP pilot experiments
We applied exactly the same procedure to the polyA+ RNAs as we did to the RNA oligo test and total RNA pilot to investigate for m6A in mRNA. All of the libraries were sent for 100-nt single-end deep sequencing on an Illumina HiSeq 2000 machine. Because of the nature of our library preparation, all libraries were strand-specific.
siRNA knockdown
Human A549 cells were plated in 100-mm dishes with regular DMEM (with 10% FBS and 1× Penstrep) at 20% confluency. Using Lipofectamine RNAiMax transfection reagent (Invitrogen Life Technologies, catalog no. 13778-150), siRNAs were transfected. One additional siRNA boost was delivered at 48 h following transfection. Cells were harvested at 96 h after transfection.
Mass spectrometric quantification of m6A
A protocol similar to that described in Zheng et al. (2013) was practiced. RNA samples were digested by incubation with 0.2 U of nuclease P1 (Sigma, N8630) in ammonium acetate buffer (pH 5.3) containing 5 nM magnesium chloride for 40 min at 45°C and following incubation with 0.5 U of alkaline phosphatase (Sigma, P5931) for 40 min at 37°C. Contaminants were removed by precipitation with ice-cold methanol and centrifugation at 16,000g for 30 min. Supernatants were collected in new tubes and subsequently subjected to desalting by vacuum centrifugation at room temperature until dry, redissolved in 100 µL of water, and vacuum dried one more time. The samples were then finally redissolved in 50 µL of water. Nucleosides were separated on an Ascentis Express C18 column, and online mass spectrometry detection was performed by an API 5000 triple quadrupole (ABSciex) operating in positive electrospray ionization mode. Quantification was performed by comparison with the standard curve obtained from pure nucleoside standards.
m6A-IP data analysis
Libraries generated with BrdU-CLIP adaptors were first filtered according to their quality scores. The BrdU-CLIP reads had a 4-nt sample multiplexing index plus a 10-nt random barcode at the 5′ end. We required a minimum quality score of 20 at these index and barcode positions in addition to an average score of 20 for the remaining positions. Barcode and index sequences were removed and recorded separately. The remaining read sequences (actual RNA tags) were mapped to the reference genomes (mm9 and hg18) by the Novoalign program (http://www.novocraft.com) with no more than two mismatches (substitutions, insertions, or deletions) allowed per read. A minimum of 20 nucleotide matches was required, and only those reads mapped uniquely to the genome (single hits) were kept. To remove PCR duplicates, we collapsed reads mapped to the same genomic locations (the same start and end) into a single read if they had the same barcode sequence; on the other side, reads mapped to the same location but with distinct barcodes were kept. This step removed PCR duplicates while retaining genuinely unique reads representing independent RNA fragments (the same analysis pipeline was used for m6A IP and CLIP and the input RNA fragments). To ensure reads unambiguously mapped in the genome, we further mapped the reads by two additional alignment programs (Bowtie 2 and TopHat 2). Unlike Bowtie 2 and Novoalign, TopHat 2 allowed mapping across exon–exon junctions. The mapping parameter for Bowtie 2 was -k 4, and those for TopHat 2 were -library-type fr-firststrand -p 10, with other parameters in the default setting. We kept reads uniquely mapped in the genome by all three programs and the reads that were not mappable by Novoalign and Bowtie 2 but uniquely mapped to exon–exon junctions by TopHat 2 (requiring an at least 4-nt overlap on each exon).
To make read coverage tracks for figure display, we calculated the m6A-IP enrichment value at each nucleotide position: We first normalized the read coverage at each nucleotide position to library size for m6A-IP and its input accordingly before averaging these normalized read densities for biological replicates. We then divided the normalized read density for m6A-IP to that of the input to calculate the m6A-IP enrichment value.
m6A peak region calling
Similar to previous m6A studies (Meyer et al. 2012; Batista et al. 2014), we searched for the enriched m6A peak regions by scanning the genome using 20-nt sliding windows and evaluating the statistical significance of enrichment by Fisher's exact test (m6A-IP vs. input). Benjamini-Hochberg was implemented to adjust the P-value to the FDR for multiple testing. We identified a window as being significantly m6A-enriched when its FDR was <0.05 and the fold of enrichment (m6A-IP vs. input) was at least fourfold in at least two out of three biological replicates. Significantly, m6A-enriched 20-nt windows were concatenated if they were adjacent to each other. Because we performed size selection for 20- to 80-nt RNA fragments, the resulting m6A peak regions would be mostly 40–200 nt. We filtered out m6A peak regions that were <40 nt. In some cases, m6A peak regions were >200 nt, suggesting multiple m6A sites within those peak regions. Thus, we split them into multiple m6A peak regions with the minimum number of m6A peaks that could form such a concatenated window.
Precise m6A site mapping by CIMSs, CITSs, MITSs, and PSs
CIMS analysis
Mutations (deletions, insertions, and substitutions) identified in uniquely mapped reads (supported by Novoalign, Bowtie 2, and TopHat) were used for CIMS analysis. CIMS analysis was performed as described previously (Moore et al. 2014) for reads of m6A-CLIP, m6A-IP, and its input. CIMSs identified only in m6A-CLIP reads (not in m6A-IP or its input) were defined as UV-induced CIMSs. Furthermore, we required that reads containing these UV-induced CIMS mutations could be identified from at least two of the three biological replicates. In m6A peak regions, we found that the RRACU and RAC motifs were significantly enriched at the −1 position of UV-induced substitution CIMSs and at the −2 position of UV-induced deletion CIMSs (we did not use UV-induced insertion CIMSs, as they were rare compared with substitution and deletion, although the RAC motif did show significant enrichment at its −1 position). In m6A peak regions, we defined precisely mapped m6A sites (in the RAC motif) as those located exactly at the −1 position of UV-induced substitution CIMSs and exactly at the −2 position of UV-induced deletion CIMSs and defined precisely mapped m6A sites (in the RRACU motif) as those located exactly within 1 nt of the −1 position for UV-induced substitution CIMSs (i.e., the −2, −1, and 0 positions) and those exactly at the −2 position of UV-induced deletion CIMSs. For figure plots, “position to CIMSs” was the nucleotide position of the A base (in the RAC or RRACU motif) to the CIMSs. “Enrichment of RAC or RRACU” was the fold of enrichment of the RAC/RRACU motif density at that position compared with the background motif density. The background motif density was calculated as the motif density of RAC/RRACU at the regions 400 nt upstream of and downstream from substitution CIMSs, where its value was flattened.
CITS analysis
CITS analysis was performed similarly to that of Weyn-Vanhentenryck et al. (2014) with some improvements. Similar to Weyn-Vanhentenryck et al. (2014), we removed all m6A-CLIP reads with UV-induced CIMSs, as most of them were reads in which the cross-link sites were read through during reverse transcription. We identified the potential sites of truncation on the remaining m6A-CLIP reads as the nucleotides right 5′ of each read. Because we sequenced the same samples immunoprecipitated by the same m6A antibody with no UV (the m6A-IP leg), we also identified the potential truncation sites on m6A-IP reads under this “no UV” condition. We then performed a stringent test to examine directly whether a potential truncation site in the m6A-CLIP reads was truly a CITS by requiring their observed truncation frequency to be significantly higher than that of truncations in the m6A-IP reads under the “no UV” condition. The P-values (calculated by Fisher's exact test) were adjusted to the FDR for multiple testing by Benjamini-Hochberg. We required FDR ≤ 0.05 for all CITSs identified. Furthermore, we required that reads truncated at these CITSs could be identified in at least two of the three biological replicates. In m6A peak regions, we found that the RRACU and RAC motifs were enriched at the −3 position of CITSs. We defined precisely mapped m6A sites (in the RRACU motif) as those located exactly within 1 nt of the −3 nt position of CITSs (i.e., the −4, −3, and −2 positions) in m6A peak regions. We did not use precisely mapped m6A sites (in the RAC motif), as the enrichment of RAC at the −3 nt position for CITSs was considerably weaker compared with that of RRACU at the −3 nt position for CITSs or that of RRACU/RAC at precise positions for CIMSs (i.e., the −1 nt of substitution CIMSs or the −2 nt of deletion CIMSs).
MITS analysis
Because we sequenced both the RNA fragments immunoprecipitated by the m6A antibody with no UV (m6A-IP leg) and the input RNA fragments, we were able to identify the potential truncation sites in the m6A-IP reads and the input reads. By directly comparing the truncation frequencies between the two, we were able to identify stringently potential truncation sites with significantly higher truncation frequency in m6A-IP reads than those in the input (e.g., 16-fold higher, FDR ≤ 0.01, Fisher's exact test). To be more stringent, we clustered these sites if they were within 1 nt of each other and, for each cluster, kept the site with the highest truncation frequency in m6A-IP reads compared with the input reads. Because these truncation sites existed in m6A-enriched RNA fragments but not in its input RNA fragments and because both RNAs were subjected to the same reverse transcription, these truncations were likely induced by m6A. Thus, we named these sites as MITSs. Furthermore, we required that reads containing these MITS truncations could be identified in at least two of the three biological replicates. In m6A peak regions, we found that the RRACU and RAC motifs were significantly enriched at the +1 position of MITSs. Thus, we defined precisely mapped m6A sites (in the RAC motif or RRACU motif) as those located exactly at the +1 position of MITSs in m6A peak regions.
PS analysis
For each m6A peak region, m6A PS was defined as the nucleotide position that had the highest enrichment of m6A-IP reads compared with its input reads (Schwartz et al. 2013). As described previously by Schwartz et al. (2013), we found that the RRACU and RAC motifs were significantly enriched at the 0 position of the PSs. Thus, we defined precisely mapped m6A sites (in the RAC motif) as those located exactly at the 0 position of the PSs and defined precisely mapped m6A sites (in the RRACU motif) as those located exactly within 1 nt of to 0 position for the PSs (i.e., the −1, 0, and 1 positions).
FDR estimation for precisely mapped m6A sites
To estimate the background statistics that the RRACU or RAC motif happened by chance to be at a fixed position to these CIMSs (positions −1 for substitution and −2 for deletion), CITSs (position −3), MITSs (position +1), and PSs (position 0), we randomly permuted these sites (the same number of sites) in transcripts that were adequately expressed in mouse brains and human CD8 T cells accordingly (RPKM [reads per kilobase per million mapped reads] ≥ 1). We enumerated these events as the total number of false positive events. The FDR was estimated by dividing the total number of false positive events by the total number of precisely mapped m6A sites identified in each case.
Determination of differentially methylated m6A peak regions between mouse brains and livers
To identify m6A peak regions that were differentially methylated in mouse brains and livers, we considered the combined set of m6A peak regions identified in both tissues. For each m6A peak region of the combined set, we enumerated reads for m6A-IP and the input for brain and liver tissues (scaled to each library's size accordingly) to evaluate the statistical significance for the fold change of enrichment between the two tissues (Fisher's exact test). Benjamini-Hochberg was implemented to adjust the P-value to the FDR for multiple testing. The requirements for a differentially methylated m6A peak region included (1) that the genes containing these m6A peak regions should be adequately expressed in both brains and livers (RPKM ≥ 1), (2) that the expression of mRNAs in m6A peak regions is adequate for m6A peak region detection in both brains and livers (RPKM ≥ 1), and (3) an at least twofold change of peak region enrichment between brain and liver tissues in addition to the requirement of FDR ≤ 0.05.
Determination of m6A peak regions lost in simultaneous knockdown of METTL3, METTL14, and WTAP
To determine m6A peak regions that were lost due to simultaneous knockdown of METTL3, METTL14, and WTAP, we performed an analysis similar to the brain versus liver m6A peak region comparison described above. The requirement that an m6A peak region be lost due to knockdown of METTL3, METTL14, and WTAP included that (1) the genes containing these m6A peak regions should be adequately expressed in both knockdowns and the controls (RPKM ≥ 1), (2) the expression of mRNAs in m6A peak regions is adequate for m6A peak region detection in both knockdowns and the controls (RPKM ≥ 1), and (3) the m6A peak regions being lost due to simultaneous knockdown of METTL3, METTL14, and WTAP were determined by requiring FDR ≤ 0.05 and an at least 1.5-fold decrease of peak region enrichment in knockdown compared with control.
Analysis of published m6A peak-calling data
Data from the previous m6A publications were downloaded directly from the NCBI Gene Expression Omnibus (GEO) and aligned to the same reference genomes by TopHat/Bowtie according to Meyer et al. (2012) and Batista et al. (2014) for mm9 and Schwartz et al. (2014) for hg19. We determined m6A peak regions using either the methods described in these studies or a strategy similar to that described above except implementing a 100-nt sliding window (as their RNA fragments were all longer); the same findings were obtained for the figure citing these data (Supplemental Fig. 3).
Analysis of published RNA-binding protein CLIP data
Data of the published RNA-binding protein CLIP (Fig. 6) were downloaded directly from the NCBI GEO (Ago [Chi et al. 2009; MJ Moore, TKH Scheel, JM Luna, CY Park, JJ Fak, E Nishiuchi, CM Rice, and RB Darnell, in prep.], Hu [Ince-Dunn et al. 2012], Mbnl [Charizanis et al. 2012], Nova [Zhang et al. 2010], Ptbp2 [Licatalosi et al. 2012], Rbfox [Weyn-Vanhentenryck et al. 2014], and Tdp43 [Lagier-Tourenne et al. 2012]), and the analysis pipeline was described previously (Moore et al. 2014).
Analysis of polyA sequencing (polyA-seq) data
PolyA-seq experiments for mouse brains and livers and human CD8 T cells and A549 cells (control vs. knockdown) were performed according to Derti et al. (2012). We followed a polyA-seq data analysis pipeline similar to that of previous studies (Shepard et al. 2011; Almada et al. 2013). The sequenced library reads were first filtered by trimming Ts off the beginning of reads and removing reads that had a string of ≥12 Ts. The reads were then aligned to the reference genomes (mm9 for mouse brains and livers, and hg18 for human CD8 T cells and A549 cells) by TopHat 2 with default settings. Only uniquely mapped reads were kept for downstream analysis. Reads that had more than five consecutive As or more than six As in the 10 nt immediately downstream from the polyA junction were removed, as they were likely due to internal priming. We then clustered reads that were within 50 nt from each other by Bedtools merge and kept the polyA sites (i.e., read clusters) that had at least four reads total, with reads drawn from at least two biological replicates.
Identified APA sites in the last exons significantly changed among samples
To avoid ambiguity in assigning polyA sites to mRNAs according to gene annotations, we used a subset of the University of California at Santa Cruz (UCSC) RefSeq gene annotations by taking only one transcript isoform of each mRNA: We used the isoform with the most distal 3′ end (i.e., the polyA site) of the mRNA. Next, we removed overlapping transcript variants from the set to avoid any ambiguity in determining which transcript the polyA site was from. To be consistent, this set of transcript annotations was exactly the same set that we used for m6A peak annotation. We examined APA sites in the last exons, and polyA sites in two samples (e.g., brains vs. livers) that were within 50 nt of each other were considered the same polyA site. We focused on polyA sites that were adequately used, as they contained at least 5% of the total polyA-seq reads in the mRNA; choosing a different arbitrary cutoff (e.g., 10%) generated essentially the same results for downstream analysis. The polyA-seq read counts for one polyA site between two samples were compared with those of another polyA site found in the same last exon. Fisher's exact test was used to evaluate the statistical significance of the difference of a proximal–distal polyA site pair between two samples. Benjamini-Hochberg procedure was implemented to adjust P-values to the FDR for multiple testing. To determine a significantly changed proximal and distal polyA site pair between two samples, we required (1) FDR ≤ 20% (shown in Figs. 4B,D, 5C; the same findings were obtained for FDR ≤ 5%, as shown in Supplemental Figs. 5B,C, 6), (2) an at least twofold change in the ratios of polyA-seq read counts, and (3) that the transcript should have adequate expression in both samples (RPKM ≥ 1).
Evolutionary conservation and de novo motif statistics
Analysis of phylogenetic conservation was done by comparing phyloP scores of precisely mapped m6A sites with the A nucleotides in the RAC motifs in the same m6A peak regions. PhyloP scores were downloaded from the UCSC genome browser (phyloP score of 30 vertebrate genomes for mouse mm9; phyloP score of 44 vertebrates for human hg18). The statistical significance for phyloP score differences between groups was determined by Wilcoxon rank sum test (similar results were obtained with other tests, including Student's t-test). De novo motif analysis was done using the HOMER package (Heinz et al. 2010) with default parameters for RNA motifs. The inputs for CIMSs, CITSs, MITSs, and PSs were their adjacent 20-nt flanking sequences, and the control in each case was the region with the same size located 400 nt upstream of or downstream from the site (randomly picked between the two).
Distribution of m6A peak regions around the stop codons, the start of the last exons, and the proximal/distal polyA sites
To avoid ambiguity in assigning m6A peak regions to mRNAs according to gene annotations, we used a subset of UCSC RefSeq gene annotations by taking only one transcript isoform of each mRNA and no overlapping regions. To be consistent, this set of transcript annotations for m6A peak regions was exactly the same set that we used for polyA site annotation. We classified genes into coding and noncoding and further classified coding genes into those with (1) the stop codon in the last exon and (2) the stop codon not in the last exon.
We considered m6A peak regions that were within 1 kb mRNA distance to the stop codons and took into account that mRNAs of interest may have different lengths. We generated 100 intervals, each with a 10-nt size for 1 kb upstream of and downstream from the stop codons. For any particular interval, we computed the “m6A peak region density” (i.e., “m6A peak density”) as follows: We scanned through all mRNAs of interest that contained this interval and checked whether an m6A peak region landed in this interval. We then counted those cases that contained m6A peak regions and divided this value by the total number of mRNAs that contained this interval.
For the plot of m6A peak region density around the start of the last exons and the proximal/distal polyA sites, we performed the same calculation except anchoring at the start of the last exons and the proximal/distal polyA sites. For the m6A peak region density plot around the proximal/distal polyA sites, we focused on the last exons with dual polyA sites that were determined by polyA site mapping in our samples using polyA-seq (Derti et al. 2012), and each site had adequate polyA site usage (e.g., 10% of total polyA site usage in the last exon shown in Fig. 4A). The two sites were named the proximal site and the distal site based on their locations to the last exon start site. If the distal site was used >60%, it was defined as “distal polyA site used more,” and its proximal site (used <40%) was defined as “proximal polyA site used less,” and vice versa. We also tried other cutoffs (50% and 70%) and obtained the same findings. “Position to proximal/distal polyA site (nt)” was defined as the nucleotide position to the proximal/distal polyA site on mRNAs, respectively. “m6A peak region density” for each 10-nt interval was calculated as the count of a position when in an m6A peak region divided by the total count of mRNAs containing this position.
Data deposition
The data reported in this study have been deposited in the GEO database, accession no. GSE71154.
Supplementary Material
Acknowledgments
We thank the Rockefeller University Genomics Resource Center and New York Genome Center for their support in HITS and data analysis input. We thank Dennis Weiss for comments and suggestions. We are grateful to members of the R.B. Darnell and J.E. Darnell laboratories for thoughtful intergenerational discussions, and members of the A. Klungland laboratory for comments and suggestions. S.K. is a Cancer Research Institute Irvington Post-doctoral Fellow. This work was also supported in part by grants from the National Institutes of Health (NS034389 and NS081706) and the Simons Foundation (SFARI 240432) to R.B.D. R.B.D. is an Investigator of the Howard Hughes Medical Institute. S.K., R.B.D., and J.E.D. designed the experiments. S.K., J.E.D., and R.B.D. wrote the manuscript. S.K. performed all of the experimental work and data analysis reported here except the following: E.A.A. and A. Klungland contributed the mouse brain and liver samples, C.M. and B.H. carried out the methyltransferase complex component triple knockdown, E.C.G. contributed the human CD8 T-cell samples, J.J.F. performed the RNA-seq experiment, A.M. contributed the improved BrdU-CLIP procedure, I.Z.-S. contributed the mouse colony maintenance, M.J.M. and C.Y.P. shared the RNA-binding protein CLIP data, and C.B.V. and A. Kus´nierczyk performed m6A mass spectrometric quantification.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.269415.115.
References
- Almada AE, Wu X, Kriz AJ, Burge CB, Sharp PA. 2013. Promoter directionality is controlled by U1 snRNP and polyadenylation signals. Nature 499: 360–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, et al. 2014. m6A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15: 707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt D, Darnell JEJ. 1969. tRNA synthesis in HeLa cells: a precursor to tRNA and the effects of methionine starvation on tRNA synthesis. J Mol Biol 42: 43–56. [DOI] [PubMed] [Google Scholar]
- Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. 1997. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 3: 1233–1247. [PMC free article] [PubMed] [Google Scholar]
- Bringmann P, Luhrmann R. 1987. Antibodies specific for N6-methyladenosine react with intact snRNPs U2 and U4/U6. FEBS Lett 213: 309–315. [DOI] [PubMed] [Google Scholar]
- Burdon RH, Martin BT, Lal BM. 1967. Synthesis of low molecular weight ribonucleic acid in tumour cells. J Mol Biol 28: 357–371. [DOI] [PubMed] [Google Scholar]
- Camper SA, Albers RJ, Coward JK, Rottman FM. 1984. Effect of undermethylation on mRNA cytoplasmic appearance and half-life. Mol Cell Biol 4: 538–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canaani D, Kahana C, Lavi S, Groner Y. 1979. Identification and mapping of N6-methyladenosine containing sequences in simian virus 40 RNA. Nucleic Acids Res 6: 2879–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charizanis K, Lee KY, Batra R, Goodwin M, Zhang C, Yuan Y, Shiue L, Cline M, Scotti MM, Xia G, et al. 2012. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron 75: 437–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Zang JB, Mele A, Darnell RB. 2009. Argonaute HITS-CLIP decodes microRNA–mRNA interaction maps. Nature 460: 479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy MJ, Shambaugh ME, Timpte CS, Bokar JA. 2002. Induction of sporulation in Saccharomyces cerevisiae leads to the formation of N6-methyladenosine in mRNA: a potential mechanism for the activity of the IME4 gene. Nucleic Acids Res 30: 4509–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell RB. 2010. HITS-CLIP: panoramic views of protein-RNA regulation in living cells. Wiley Interdiscip Rev RNA 1: 266–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JEJ. 2013. Reflections on the history of pre-mRNA processing and highlights of current knowledge: a unified picture. RNA 19: 443–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JE, Wall R, Tushinski RJ. 1971. An adenylic acid-rich sequence in messenger RNA of HeLa cells and its possible relationship to reiterated sites in DNA. Proc Natl Acad Sci 68: 1321–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derti A, Garrett-Engele P, Macisaac KD, Stevens RC, Sriram S, Chen R, Rohl CA, Johnson JM, Babak T. 2012. A quantitative atlas of polyadenylation in five mammals. Genome Res 22: 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desrosiers RC, Friderici KH, Rottman FM. 1975. Characterization of Novikoff hepatoma mRNA methylation and heterogeneity in the methylated 5′ terminus. Biochemistry 14: 4367–4374. [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. 2012. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485: 201–206. [DOI] [PubMed] [Google Scholar]
- Edmonds M, Vaughan MHJ, Nakazato H. 1971. Polyadenylic acid sequences in the heterogeneous nuclear RNA and rapidly-labeled polyribosomal RNA of HeLa cells: possible evidence for a precursor relationship. Proc Natl Acad Sci 68: 1336–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuichi Y, Morgan M, Muthukrishnan S, Shatkin AJ. 1975. Reovirus messenger RNA contains a methylated, blocked 5′-terminal structure: m-7G(5′)ppp(5′)G-MpCp-. Proc Natl Acad Sci 72: 362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuno M, Kasukawa T, Saito R, Adachi J, Suzuki H, Baldarelli R, Hayashizaki Y, Okazaki Y. 2003. CDS annotation in full-length cDNA sequence. Genome Res 13: 1478–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fustin JM, Doi M, Yamaguchi Y, Hida H, Nishimura S, Yoshida M, Isagawa T, Morioka MS, Kakeya H, Manabe I, et al. 2013. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell 155: 793–806. [DOI] [PubMed] [Google Scholar]
- Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. 2015. Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 347: 1002–1006. [DOI] [PubMed] [Google Scholar]
- Greenberg H, Penman S. 1966. Methylation and processing of ribosomal RNA in HeLa cells. J Mol Biol 21: 527–535. [DOI] [PubMed] [Google Scholar]
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, et al. 2009. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458: 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JE, Miceli SM, Roberts RJ, Manley JL. 1990. Sequence specificity of the human mRNA N6-adenosine methylase in vitro. Nucleic Acids Res 18: 5735–5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz S, Horowitz A, Nilsen TW, Munns TW, Rottman FM. 1984. Mapping of N6-methyladenosine residues in bovine prolactin mRNA. Proc Natl Acad Sci 81: 5667–5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsin JP, Manley JL. 2012. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev 26: 2119–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince-Dunn G, Okano HJ, Jensen KB, Park WY, Zhong R, Ule J, Mele A, Fak JJ, Yang C, Zhang C, et al. 2012. Neuronal Elav-like (Hu) proteins regulate RNA splicing and abundance to control glutamate levels and neuronal excitability. Neuron 75: 1067–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, Lee JY, Pan Z, Jiang B, Tian B. 2009. Progressive lengthening of 3′ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc Natl Acad Sci 106: 7028–7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, et al. 2011. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 7: 885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane SE, Beemon K. 1985. Precise localization of m6A in Rous sarcoma virus RNA reveals clustering of methylation sites: implications for RNA processing. Mol Cell Biol 5: 2298–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane SE, Beemon K. 1987. Inhibition of methylation at two internal N6-methyladenosine sites caused by GAC to GAU mutations. J Biol Chem 262: 3422–3427. [PubMed] [Google Scholar]
- Klagsbrun M. 1973. An evolutionary study of the methylation of transfer and ribosomal ribonucleic acid in prokaryote and eukaryote organisms. J Biol Chem 248: 2612–2620. [PubMed] [Google Scholar]
- Konig J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J. 2010. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol 17: 909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM, Ling SC, Liang TY, Mazur C, et al. 2012. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci 15: 1488–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Mendecki J, Brawerman G. 1971. A polynucleotide segment rich in adenylic acid in the rapidly-labeled polyribosomal RNA component of mouse sarcoma 180 ascites cells. Proc Natl Acad Sci 68: 1331–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Schweitzer AC, Blume JE, Wang X, et al. 2008. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456: 464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, Yano M, Fak JJ, Mele A, Grabinski SE, Zhang C, Darnell RB. 2012. Ptbp2 represses adult-specific splicing to regulate the generation of neuronal precursors in the embryonic brain. Genes Dev 26: 1626–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little NA, Hastie ND, Davies RC. 2000. Identification of WTAP, a novel Wilms’ tumour 1-associating protein. Hum Mol Genet 9: 2231–2239. [DOI] [PubMed] [Google Scholar]
- Liu N, Parisien M, Dai Q, Zheng G, He C, Pan T. 2013. Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA 19: 1848–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. 2014. A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol 10: 93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquat LE. 2015. The amazing web of post-transcriptional gene control: the sum of small changes can make for significant consequences. RNA 21: 488–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. 2012. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149: 1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Zhang C, Gantman EC, Mele A, Darnell JC, Darnell RB. 2014. Mapping Argonaute and conventional RNA-binding protein interactions with RNA at single-nucleotide resolution using HITS-CLIP and CIMS analysis. Nat Protoc 9: 263–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munns TW, Sims HF. 1975. Methylation and processing of transfer ribonucleic acid in mammalian and bacterial cells. J Biol Chem 250: 2143–2149. [PubMed] [Google Scholar]
- Nakazato H, Edmonds M. 1974. Purification of messenger RNA and heterogeneous nuclear RNA containing poly(A) sequences. Methods Enzymol 29: 431–443. [DOI] [PubMed] [Google Scholar]
- Perry RP, Kelley DE. 1974. Existence of methylated messenger RNA in mouse L cells. Cell 1: 37–42. [Google Scholar]
- Perry RP, Kelley DE, Friderici K, Rottman F. 1975. The methylated constituents of L cell messenger RNA: evidence for an unusual cluster at the 5′ terminus. Cell 4: 387–394. [DOI] [PubMed] [Google Scholar]
- Salditt-Georgieff M, Jelinek W, Darnell JE, Furuichi Y, Morgan M, Shatkin A. 1976. Methyl labeling of HeLa cell hnRNA: a comparison with mRNA. Cell 7: 227–237. [DOI] [PubMed] [Google Scholar]
- Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB. 2008. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science 320: 1643–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schibler U, Kelley DE, Perry RP. 1977. Comparison of methylated sequences in messenger RNA and heterogeneous nuclear RNA from mouse L cells. J Mol Biol 115: 695–714. [DOI] [PubMed] [Google Scholar]
- Schwartz S, Agarwala SD, Mumbach MR, Jovanovic M, Mertins P, Shishkin A, Tabach Y, Mikkelsen TS, Satija R, Ruvkun G, et al. 2013. High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell 155: 1409–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, Mertins P, Ter-Ovanesyan D, Habib N, Cacchiarelli D, et al. 2014. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep 8: 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatkin AJ. 1974. Methylated messenger RNA synthesis in vitro by purified reovirus. Proc Natl Acad Sci 71: 3204–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard PJ, Choi EA, Lu J, Flanagan LA, Hertel KJ, Shi Y. 2011. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA 17: 761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan PR, Borek E. 1964. Enzymatic alteration of nucleic acid structure. Science 145: 548–553. [DOI] [PubMed] [Google Scholar]
- Starr JL, Sells BH. 1969. Methylated ribonucleic acids. Physiol Rev 49: 623–669. [DOI] [PubMed] [Google Scholar]
- Takagaki Y, Manley JL. 1998. Levels of polyadenylation factor CstF-64 control IgM heavy chain mRNA accumulation and other events associated with B cell differentiation. Mol Cell 2: 761–771. [DOI] [PubMed] [Google Scholar]
- Takagaki Y, Manley JL. 2000. Complex protein interactions within the human polyadenylation machinery identify a novel component. Mol Cell Biol 20: 1515–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, Hu J, Zhang H, Lutz CS. 2005. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res 33: 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan MHJ, Soeiro R, Warner JR, Darnell JEJ. 1967. The effects of methionine deprivation on ribosome synthesis in HeLa cells. Proc Natl Acad Sci 58: 1527–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Yi R. 2014. 3′UTRs take a long shot in the brain. Bioessays 36: 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. 2014. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505: 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrspaun CC, Ponting CP, Marques AC. 2014. Brain-expressed 3′UTR extensions strengthen miRNA cross-talk between ion channel/transporter encoding mRNAs. Front Genet 5: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CM, Moss B. 1974. Methylation of newly synthesized viral messenger RNA by an enzyme in vaccinia virus. Proc Natl Acad Sci 71: 3014–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CM, Moss B. 1975. Methylated nucleotides block 5′-terminus of vaccinia virus messenger RNA. Proc Natl Acad Sci 72: 318–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CM, Moss B. 1977. Nucleotide sequences at the N6-methyladenosine sites of HeLa cell messenger ribonucleic acid. Biochemistry 16: 1672–1676. [DOI] [PubMed] [Google Scholar]
- Wei CM, Gershowitz A, Moss B. 1975. Methylated nucleotides block 5′ terminus of HeLa cell messenger RNA. Cell 4: 379–386. [DOI] [PubMed] [Google Scholar]
- Weyn-Vanhentenryck SM, Mele A, Yan Q, Sun S, Farny N, Zhang Z, Xue C, Herre M, Silver PA, Zhang MQ, et al. 2014. HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep 6: 1139–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao C, Biesinger J, Wan J, Weng L, Xing Y, Xie X, Shi Y. 2012. Transcriptome-wide analyses of CstF64–RNA interactions in global regulation of mRNA alternative polyadenylation. Proc Natl Acad Sci 109: 18773–18778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Darnell RB. 2011. Mapping in vivo protein–RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat Biotechnol 29: 607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Frias MA, Mele A, Ruggiu M, Eom T, Marney CB, Wang H, Licatalosi DD, Fak JJ, Darnell RB. 2010. Integrative modeling defines the Nova splicing-regulatory network and its combinatorial controls. Science 329: 439–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang C-M, Li CJ, Vågbø CB, Shi Y, Wang W-L, Song S-H. 2013. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 49: 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.