

In this study, Martinez et al. find a positive feedback loop in the JNK signaling pathway through the alternative splicing of MKK7, identify the RNA-binding protein CELF2 as a major regulator of MKK7 splicing, and show that ∼25% of T-cell receptor-mediated alternative splicing events are dependent on JNK signaling. This study provides insight into a novel paradigm for the reciprocal interplay of signaling and splicing.
Keywords: alternative splicing, JNK, MKK7, positive feedback, CELF2
Abstract
Alternative splicing is prevalent among genes encoding signaling molecules; however, the functional consequence of differential isoform expression remains largely unknown. Here we demonstrate that, in response to T-cell activation, the Jun kinase (JNK) kinase MAP kinase kinase 7 (MKK7) is alternatively spliced to favor an isoform that lacks exon 2. This isoform restores a JNK-docking site within MKK7 that is disrupted in the larger isoform. Consistently, we show that skipping of MKK7 exon 2 enhances JNK pathway activity, as indicated by c-Jun phosphorylation and up-regulation of TNF-α. Moreover, this splicing event is itself dependent on JNK signaling. Thus, MKK7 alternative splicing represents a positive feedback loop through which JNK promotes its own signaling. We further show that repression of MKK7 exon 2 is dependent on the presence of flanking sequences and the JNK-induced expression of the RNA-binding protein CELF2, which binds to these regulatory elements. Finally, we found that ∼25% of T-cell receptor-mediated alternative splicing events are dependent on JNK signaling. Strikingly, these JNK-dependent events are also significantly enriched for responsiveness to CELF2. Together, our data demonstrate a widespread role for the JNK–CELF2 axis in controlling splicing during T-cell activation, including a specific role in propagating JNK signaling.
Signal-induced alternative splicing is a common mechanism of gene regulation that expands protein inventory and shapes cellular function in response to a changing environment (Nilsen and Graveley 2010). For example, antigen-induced signaling through the T-cell receptor (TCR) controls the splicing of hundreds of genes involved in a large array of T-cell effector functions, including the secretion of cytokines and cytotoxins (Ip et al. 2007; Martinez et al. 2012; Martinez and Lynch 2013). Interestingly, genes encoding signaling molecules are particularly enriched among targets of T-cell activation-induced alternative splicing, suggesting a complex interplay of signaling and splicing (Martinez et al. 2012). However, we still know little about the functional consequences of regulated splicing on specific signaling pathways (Lynch 2004; Martinez and Lynch 2013).
A particularly intriguing but unexplored example of alternative splicing of a signaling molecule is that of the gene encoding MAP kinase kinase 7 (MKK7, also called MAP2K7) (Martinez et al. 2012). MKK7 activates the c-Jun kinases JNK1 and JNK2 in response to T-cell activation signals, leading to phosphorylation and activation of the cJun transcription factor that mediates T-cell gene expression programs and cytokine expression (Fig. 1A; Tournier et al. 1997; Matsuda et al. 1998; Sasaki et al. 2001). Consistently, CD4+ T cells from MKK7- and JNK-deficient mice fail to differentiate into appropriate T-helper subsets or carry out appropriate effector functions (Dong et al. 1998, 2000). Beyond T cells, JNK and MKK7 are broadly important kinases involved in cellular responses to stresses such as UV damage and oxidative stress as well as cell growth and apoptosis in a variety of cell types (Wang et al. 2007; Asaoka and Nishina 2010; Haeusgen et al. 2011).
Figure 1.
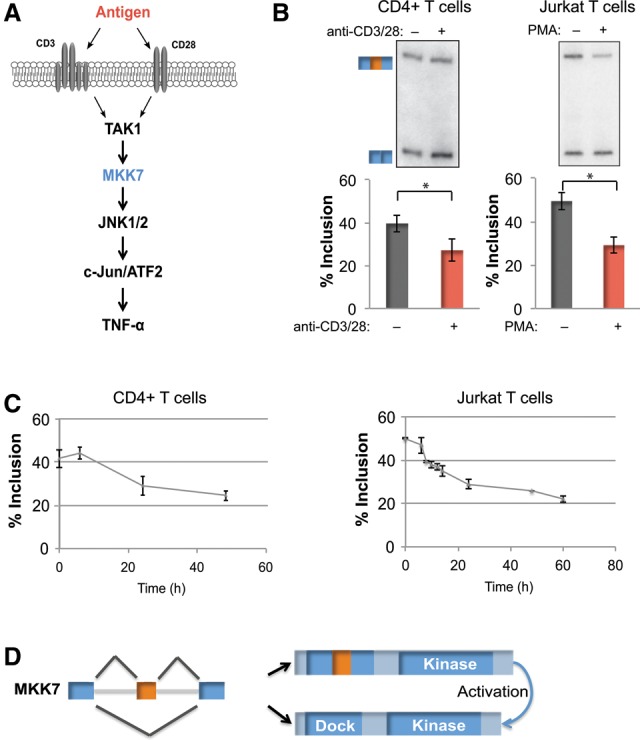
MKK7 is alternatively spliced in response to activation of primary human CD4+ T cells and a model Jurkat T-cell line. (A) Schematic of JNK pathway activation in the context of TCR signaling. (B, left panel) Representative RT–PCR gel and quantification of MKK7 exon 2 (MKK7-E2) inclusion in unstimulated and activated (anti-CD3/CD28, 48 h) primary human CD4+ T cells. n = 9. (Right panel) Representative RT–PCR gel and quantification of MKK7-E2 inclusion in unstimulated and activated (PMA, 48 h) Jurkat T cells. n = 14. In all of the figures, error bars represent standard deviation. (C) Time course of average MKK7-E2 inclusion in response to activation in primary human CD4+ T cells (anti-CD3/CD28) (left) or Jurkat T cells (PMA) (right) as quantified by RT–PCR. n = 3. (D) MKK7 domain schematic displaying the predicted outcome of MKK7-E2 inclusion on its MAPK-docking site. (*) P < 0.005, Student's t-test. See also Supplemental Figure S1.
In a recent global transcriptome analysis, we detected that exon 2 of MKK7 (MKK7-E2) is among the many exons that are differentially included in response to activation of a cultured T-cell line (Martinez et al. 2012). Interestingly, inclusion of exon 2 disrupts the second and highest affinity of three canonical MAPK-docking sites within MKK7 through which it interacts with JNK1/2 (Supplemental Fig. S1A,B; Ho et al. 2006). Based on our understanding of docking sites, the long isoform including exon 2 (MKK7-L) is predicted to be less effective in activating JNK than the short isoform (MKK7-S), although the functional consequence of MKK7-E2 inclusion has not been directly studied, Moreover, the mechanism by which the MKK7-S isoform is generated is also unknown. Alternative splicing is typically controlled by cis-regulatory elements within the alternative exon and/or flanking introns (Fu and Ares 2014). These regulatory elements commonly serve as binding sites for trans-acting proteins that can promote or repress alternative exon inclusion, including members of the SR and hnRNP families of proteins as well as additional RNA-binding proteins such as CELF, MBNL, and RbFox proteins (Fu and Ares 2014). However, there have been no studies linking any of these proteins to MKK7 splicing and no information regarding how T-cell signaling regulates this process.
Here we demonstrate that expression of MKK7-S increases upon activation of both cultured and primary human T cells and that increased expression of MKK7-S enhances JNK pathway activity and cytokine production. Remarkably, we found that, in turn, JNK activity is both necessary and sufficient to promote skipping of MKK7-E2 via TCR-initiated signaling. Thus, we identified a positive feedback loop via regulated alternative splicing of MKK7 to enhance the JNK pathway. We further characterized the mechanistic basis for JNK-dependent repression of MKK7-E2, demonstrating the critical role of the intronic sequences flanking exon 2 and recognition of these regulatory elements by JNK-induced CELF2. Finally, we show that JNK signaling mediates activation-induced alternative splicing of not just MKK7 but approximately one-quarter of TCR-induced splicing surveyed in primary human CD4+ T cells, thus serving as a widespread mechanism for controlling gene expression. Strikingly, JNK-induced splicing events are significantly enriched for responsiveness to CELF2, supporting a pervasive role for the JNK–CELF2 signaling arm in the regulation of gene expression in response to T-cell activation.
Results
MKK7 is alternatively spliced in response to activation of primary human CD4+ T cells and a model Jurkat T-cell line
We previously detected signal-induced skipping of MKK7-E2 during a transcriptome-wide analysis of splicing in a Jurkat T-cell line stimulated with PMA, a commonly used mimic for antigen signaling in T cells (Fig. 1B; Martinez et al. 2012). To confirm that MKK7 is also regulated in primary T cells, we assessed inclusion of MKK7-E2 in primary human CD4+ T cells following activation with antibodies that engage the physiologic T-cell surface receptors CD3 and CD28 (Fig. 1B). Importantly, both the extent and time course of the change in MKK7-E2 inclusion between stimulated and unstimulated cells are highly similar in both Jurkat and primary T cells (Fig. 1B,C), suggesting a common mechanism of regulation in these related cell types.
Alternative splicing of MKK7 positively regulates JNK signaling
Given the conserved regulation of MKK7 splicing upon activation of both primary and cultured T cells and the role of JNK signaling in T-cell biology, we wanted to determine the functional relevance of this isoform switch. We predicted that skipping of exon 2 increases MKK7's ability to activate JNK by generating a smaller isoform that contains a more optimal JNK-docking site (MKK7-S) (Fig. 1D; Supplemental S1A,B). Consistently, we found that MKK7-S coprecipitates with JNK1 more efficiently than MKK7-L in cotransfection studies (Supplemental Fig. S1C). To directly test the functional consequence of MKK7-E2 skipping, we used a splice site-blocking antisense morpholino oligo (AMO) complementary to the 5′ splice site of MKK7-E2 (Fig. 2A). Although the minimal size difference between the MKK7-L and MKK7-S proteins precludes their discrimination by Westerns, we confirmed by RT–PCR that transfection of the AMO results in forced skipping of MKK7-E2 and preferential production of the MKK7-S isoform (Fig. 2B). To activate JNK signaling, MKK7 needs to be phosphorylated and activated by an upstream kinase such as TAK1 before activating JNK (Fig. 1A; Wan et al. 2006; Blonska et al. 2007). Consequently, we found no phosphorylation of the JNK target c-Jun in unstimulated cells even in the presence of the AMO (Fig. 2C). However, AMO treatment results in a marked increase in c-Jun phosphorylation when MKK7 has been activated by PMA (Fig. 2C). Furthermore, we observed a threefold increase in TNF-α mRNA, a target of c-Jun-mediated transcription (Tsai et al. 1996a,b; Lawrence et al. 2011), in activated cells that have been treated with the MKK7 AMO (Fig. 2D). Importantly, the increased c-Jun phosphorylation and TNF-α expression cannot be attributed to increased MKK7 protein levels, as the AMO does not alter total MKK7 protein levels (Fig. 2C). Together, these data demonstrate that skipping of MKK7-E2 generates an isoform that is more effective in activating JNK and enhancing JNK pathway activity, including increased c-Jun activation and up-regulation of the immune cytokine TNF-α.
Figure 2.
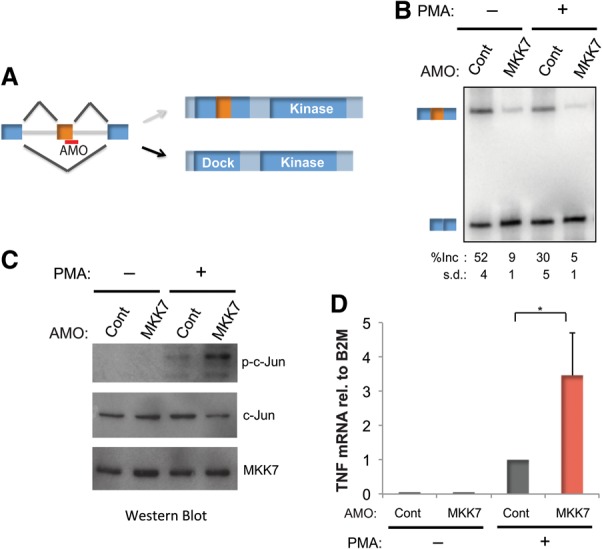
Alternative splicing of MKK7 positively regulates JNK signaling. (A) Schematic of the MKK7 5′ splice site-blocking AMO on the pre-mRNA and its effect on the MKK7 protein. (B) Representative RT–PCR gel of MKK7-E2 and average percent inclusion (n > 3) for Jurkat T cells transfected with a control AMO against a splice site in an unrelated gene (Cont) or the AMO blocking the 5′ splice site of MKK7-E2 (MKK7) in the presence and absence of PMA (48 h). (C) Corresponding Western blot of Jurkat T cells treated as in B, blotted for phospho-c-Jun (Ser73), total c-Jun ,and total MKK7 (PMA, 2 h). (D) Quantification of TNF-α mRNA normalized to B2M by RT–PCR (n = 4) in cells treated as in B (PMA, 48 h). Error bars represent standard deviation. (*) P < 0.005, Student's t-test.
JNK activity is necessary and sufficient for activation-induced MKK7-E2 skipping
Having determined the functional significance of MKK7 alternative splicing, we next sought to identify the signaling pathway that triggers repression of MKK7-E2. Multiple signaling pathways are activated both downstream from antigen engagement in T cells and upon PMA stimulation of Jurkat cells, including those involving Erk, Akt, mTOR, and JNK (Su et al. 1994; Harris and Lawrence 2003; Zheng et al. 2007; Smith-Garvin et al. 2009). Strikingly, among these signaling pathways, only inhibition of JNK signaling blocks PMA-induced MKK7-E2 skipping (Fig. 3A). In contrast, inhibition of the Erk (DeSilva et al. 1998), Akt (Yang et al. 2004), and mTOR (Brown et al. 1994) pathways has no significant effect on PMA-induced repression of MKK7-E2 (Fig. 3A). Consistent with previous reports (Jin et al. 2009), we did observe that the JNK inhibitor used (SP600125) partially inhibits mTOR, as indicated by a decrease in phospho-S6K. However, since direct inhibition of mTOR with rapamycin has no effect on MKK7-E2 repression (Fig. 3A), we conclude that the effect of SP600125 on MKK7-E2 splicing is not mediated via mTOR. We also found that inhibition of GSK3 signaling, previously shown to be responsible for the PMA-induced regulation of CD45 alternative splicing in Jurkat and primary T cells (Heyd and Lynch 2010), has no effect on MKK7-E2 inclusion (Supplemental Fig. S2A).
Figure 3.

JNK activity is necessary and sufficient for activation-induced MKK7-E2 skipping. (A, top) Average percent MKK7-E2 inclusion (n = 3) in Jurkat T cells pretreated with the following inhibitors prior to PMA treatment: 50 μM JNKi (SP600125), 20 μM MEKi (U0126), 1 μM Akti (triciribine), and 100 nM mTORi (rapamycin). (Bottom) Western blot analysis of downstream substrates phospo-c-Jun (JNK), phospho-Erk (MEK), and phospho-S6K (mTOR); the Akt-activating autophosphorylation site Ser473 (Akt); and hnRNPL as a loading control. Protein samples were harvested 2 h after PMA, while RNA was harvested after 48 h. Note that the samples from mTOR inhibition were run on the same gels as the other samples but with a spacer lane that has been removed. (B) Average MKK7-E2 inclusion (n = 3) with 12.5, 25, 50, and 100 μM JNK inhibitor SP600125 prior to PMA treatment (48 h). (C) Average MKK7-E2 percent inclusion (n = 2) of primary human CD4+ T cells treated with JNK inhibitor SP600125 and/or anti-CD3/CD28 (48 h). See also Supplemental Figure S2B. (D) Average percent MKK7-E2 inclusion (top) and Western analysis (bottom) in two independent Jurkat T lines depleted of JNK by an shRNA grown in the absence or presence of PMA (48 h). (E) Representative (n = 3) RT–PCR gel of MKK7-E2 inclusion (top) and the corresponding Western analysis of Flag-CAJNK expression (bottom) from HEK293 cells transfected with vector control or constitutively active JNK1 and JNK2 (CAJNK1 or CAJNK2). (F) Same as in E but with 0.01, 0.1, and 1 μg of CAJNK1. Phospho-c-Jun, as a marker for CA-JNK activity, is also shown. Error bars represent standard deviation. (*) P < 0.005, Student's t-test. See also Supplemental Figure S2.
Importantly, SP600125 blocks activation-induced MKK7-E2 skipping in a dose-dependent manner, plateauing near 50 μM, consistent with the cellular IC50 for this compound (Fig. 3B; Bennett et al. 2001). Moreover, inhibition of JNK in primary human CD4+ T cells is also sufficient to significantly block the anti-CD3/CD28 enhanced repression of MKK7-E2 (Fig. 3C; Supplemental Fig. S2B). Finally, as an additional test of the requirement for JNK in the regulation of MKK7 splicing, we generated stable Jurkat T-cell lines expressing an shRNA targeting JNK. As shown by Western blot, JNK2 and at least one isoform of JNK1 are dramatically depleted from the cells expressing the JNK shRNA (Fig. 3D, bottom). While we cannot differentiate whether JNK1 or JNK2 is the primary driver, we found that JNK depletion in Jurkat cells largely abrogates MKK7-E2 repression in response to PMA activation (Fig. 3D, top). We thus conclude, based on both genetic and pharmacologic studies, that JNK signaling is necessary for antigen-promoted skipping of MKK7-E2.
To determine whether JNK activity is also sufficient to promote MKK7-E2 skipping, we expressed constitutively active JNK1 or JNK2 (CAJNK1/2) (Lei et al. 2002) in HEK293 cells, as these cells are more amenable to transient transfection and protein expression than Jurkat cells. Inclusion of MKK7-E2 in untransfected HEK293 cells is less than that observed in unstimulated Jurkat cells but is still readily detectable (Fig. 3E). Strikingly, the presence of either Flag-tagged CAJNK1 or CAJNK2 is sufficient to completely inhibit inclusion of MKK7-E2 (Fig. 3E). Expression of CAJNK1 and CAJNK2 similarly induces the expected activity, as assessed by c-jun phosphorylation (Supplemental Fig. S2C). The induced skipping of MKK7-E2 is observed even at the lowest amounts and activity of CAJNK1 detectable (Fig. 3F). Thus, we conclude that JNK signaling is both necessary and sufficient for MKK7 alternative splicing. Moreover, the fact that JNK signaling is sufficient to induce MKK7-E2 skipping in HEK293 cells highlights that the regulation of MKK7 by JNK is not cell type-specific but rather is a general feature of this signaling pathway.
MKK7 intronic sequences are required for activation-induced skipping of exon 2
We next sought to determine the molecular mechanism by which T-cell activation leads to skipping of MKK7-E2. As a first step to identifying the sequences and trans-acting factors by which MKK7 is regulated in response to T-cell activation, we made a chimeric MKK7 minigene containing MKK7-E2 and ∼100 nucleotides (nt) of the flanking introns, inserted between exons from the heterologous β-globin gene (Fig. 4A; Supplemental Fig. S3A). The minigene was transfected into Jurkat T cells, and inclusion of MKK7-E2 was quantified by RT–PCR with minigene-specific primers. This minigene (MKK7) recapitulates the activation-induced MKK7-E2 skipping observed in the endogenous gene (eMKK7) (Fig. 4A,B) and exhibits a similar level of basal inclusion in unstimulated cells (Supplemental Fig. S3B). Swapping out the exonic or intronic sequences from MKK7 with those from the β-globin gene demonstrates that the introns alone are sufficient for activation-induced skipping of a constitutive heterologous exon, while the exonic sequence does not seem to contain any signal-responsive element (Fig. 4A,B, introns). A control minigene containing the heterologous exon placed between β-globin introns shows that this exon does not contribute to the signal responsiveness (Fig. 4A,B, control). Strikingly, we found that replacing the upstream or downstream intron with a corresponding β-globin intron abrogates MKK7-E2 skipping in activated cells, suggesting that the MKK7 introns act cooperatively (Fig. 4A,B, up or down). Together, these results demonstrate that ∼100 nt of the upstream and downstream introns is necessary and sufficient to enhance MKK7-E2 skipping in activated cells.
Figure 4.

MKK7 intronic sequences are required for activation-induced skipping of exon 2 and recruit RNA-binding proteins. (A) Schematic of MK7 minigenes. MKK7 sequences are orange, and β-globin sequences are gray. (B) Graph of average fold repression (ratio of included to skipped isoforms in stimulated over unstimulated conditions) as determined by RT–PCR of MKK7 minigenes with minigene-specific primers. (C) UV cross-linking with nuclear extracts from Jurkat T cells (±PMA, 60 h) to radiolabeled in vitro transcribed MKK7 RNAs followed by RNase digestion and resolution on an SDS-PAGE. Note that the +PMA sample for the MKK7 construct was run on the same gel as the −PMA sample but with a spacer lane that has been removed. (D) Immunoprecipitation of UV cross-linking reactions of the intron constructs in C after RNase digestion with the antibodies shown.
Trans-acting factors recognize the regulatory intronic sequences
To identify proteins that bind in trans to the MKK7 introns, we performed UV cross-linking with radiolabeled in vitro transcribed RNA, including MKK7-E2 and the flanking introns, and nuclear extract made from unstimulated (−PMA) and activated (+PMA) Jurkat T cells. Strikingly, we observed a dramatic difference between these two extracts in the binding pattern of four protein bands on an SDS-PAGE gel (Fig. 4C). Three of these proteins exhibit stronger binding in the activated versus unstimulated extracts, while the fourth protein is enriched in the unstimulated condition. At least one additional protein binds with similar intensity in activated relative to unstimulated nuclear extracts (Fig. 4C). Importantly, UV cross-linking with the intronic sequences that are necessary and sufficient for activation-induced skipping reveal the same binding pattern as with the same construct that includes MKK7-E2, while a construct lacking these introns lacks the binding pattern (Fig. 4C). These results are consistent with our minigene data, showing that both intronic sequences are important for signal-responsive regulation, while exon 2 is dispensable.
To determine the identity of the proteins associating with the MKK7 introns, we performed the UV cross-linking assay followed by immunoprecipitation of candidate proteins. Candidates were chosen based on the size of the cross-linked species and the sequences of the MKK7 introns (Supplemental Fig. S3A). By this method, we identified the four differential MKK7 intron-binding proteins as CELF2 (50 kDa), hnRNPC (40 kDa), HuR (30 kDa), and SRp20 (20 kDa) (Fig. 4D). As predicted from the total binding pattern, binding of CELF2 and SRp20 increases, binding of hnRNP C decreases, and binding of HuR is similar in activated compared with unstimulated nuclear extracts (Fig. 4D). Importantly, antibodies to several additional potential candidates, including PSIP1, hnRNPA1, hnRNPE, SRSF1, and 9G8, do not precipitate any bound species (Fig. 4D; data not shown).
As an additional approach to identify proteins bound to the MKK7 intronic sequences, we performed an RNA affinity purification experiment followed by mass spectrometry (Supplemental Fig. S4A). We filtered the list for proteins with at least 10 spectral counts across conditions and sorted them based on fold change in activated compared with unstimulated extract (Supplemental Table S1). Remarkably, the top induced binding protein from the mass spectrometry list was CELF2, consistent with the observed binding pattern by UV cross-linking. Furthermore, hnRNPC, HuR, and SRp20 were also identified in this list. A Western blot of the RNA affinity purification experiment confirmed the increase in CELF2 protein in activated nuclear extract compared with HuR, which is not induced in activated nuclear extract, supporting the mass spectrometry analysis (Supplemental Fig. S4B). Subsequent UV cross-linking immunoprecipitation and Western blot analysis of RNA affinity purification experiments failed to validate any proteins other than CELF2, hnRNPC, HuR, and SRp20 (Supplemental Table S1).
JNK induces CELF2 protein expression through mRNA stabilization, which drives activation-induced skipping of MKK7-E2
Having identified several proteins that bind differentially to the MKK7 introns in activated compared with unstimulated states, we sought to determine whether any of these proteins drives activation-induced skipping of MKK7-E2 in the endogenous gene. We therefore knocked down each of the proteins that binds to the MKK7 intronic regulatory sequences and examined endogenous MKK7-E2 inclusion levels by RT–PCR (Fig. 5A–D). We found that depletion of HuR, SRp20, or hnRNP C results in only modest changes in the inclusion of MKK7-E2 (Fig. 5A–C). In stark contrast, knockdown of CELF2 with either of two shRNAs targeted to distinct sequences dramatically increases basal inclusion levels of MKK7-E2 and abolishes activation-induced skipping (Fig. 5D; Supplemental Fig. S5A). As a complementary approach, we tested whether an increase in CELF2 protein is sufficient to promote skipping of MKK7-E2 through the identified intronic binding sites by coexpressing CELF2 cDNA and the MKK7 miningenes in HEK293 cells. Notably, overexpression of CELF2 is sufficient to promote exon skipping of the parental MKK7 minigene (Fig. 5E). Moreover, we found that the repressive activity of CELF2 is mediated through the introns with no effect on MKK7-E2 itself or the control minigene (Fig. 5E). We therefore conclude that CELF2 acts as a primary repressor of MKK7-E2 inclusion.
Figure 5.

JNK induces CELF2 protein expression, which drives activation-induced skipping of MKK7-E2. (A–D, top) Western blot showing mock-treated or knockdown of each MKK7 intron-binding protein left unstimulated or activated with PMA (48 h). n = 3. (Bottom) Graph of endogenous percent MKK7-E2 inclusion as determined by RT–PCR for the corresponding knockdown. (A) HuR knockdown. (B) SRp20 knockdown. (C) hnRNPC knockdown. (D) CELF2 knockdown. (E, top) RT–PCR analysis of MKK7 minigenes that were cotransfected with Flag-CELF2 in HEK293 cells. (Bottom) Corresponding Western blot for CELF2 overexpression. (F) Representative Western blot analysis of proteins that bind MKK7 introns in wild-type Jurkat cells or Jurkat cells depleted of JNK by shRNA grown under unstimulated conditions or activated with PMA (48 h). See also Supplemental Figure S5.
We showed previously that CELF2 protein expression increases following T-cell activation and that inhibition of the Erk pathway does not impact this response (Mallory et al. 2011, 2015). Strikingly, however, JNK knockdown or inhibition markedly diminishes the increase in CELF2 protein expression that is observed upon activation with PMA, with little or no effect on the protein levels of the other MKK7 intron-binding proteins (Fig. 5F; Supplemental Fig. S5B). Importantly, the decrease in CELF2 upon JNK knockdown or inhibition correlates with a decrease in CELF2 mRNA levels (Supplemental Fig. S6A). We recently published that the TCR signaling-induced expression of CELF2 is driven by two mechanisms: an initial increase in transcription followed by an increase in the stability of the CELF2 mRNA via a yet unknown pathway (Mallory et al. 2015). We also showed that the increase in transcription is not sensitive to JNK inhibition (Mallory et al. 2015). Therefore, we hypothesized that JNK may be regulating CELF2 protein expression through controlling stabilization of the CELF2 mRNA. Consistently, upon inhibition of transcription with actinomycin, we observed that JNK depletion or inhibition blocks the increase in message stability that is induced upon PMA activation of cells, bringing CELF2 mRNA to unstimulated levels (Supplemental Fig. S6B). In sum, we conclude that, upon T-cell activation, a JNK-dependent increase in CELF2 mRNA stability results in an increase in CELF2 protein. Moreover, this increase in CELF2 protein is sufficient to account for increased binding of CELF2 to the MKK7 intronic regulatory sequences to repress MKK7-E2 inclusion.
JNK signaling mediates a network of TCR-induced alternative splicing in primary T cells
The JNK-dependent control of MKK7-E2 skipping raises the question of whether JNK signaling is a dedicated pathway for regulated splicing of MKK7 in the context of the positive feedback loop or whether JNK has a broader role in regulating TCR-induced splicing. We thus wanted to test JNK-dependent splicing changes in the physiological context of signal transduction initiating at the TCR in primary human T cells. As these cells are not readily amenable to genetic manipulation, we took advantage of the widely used JNK inhibitor SP600125 (Bennett et al. 2001) to identify antigen-induced alternative splicing events that require JNK signaling in primary human T cells. To query widespread JNK-dependent splicing changes, we used RNA-mediated oligonucleotide annealing, selection, and ligation (RASL) coupled with sequencing (RASL-seq) (Li et al. 2001; Zhou et al. 2012) on RNA from primary CD4+ T cells that were unstimulated, stimulated with anti-CD3 and anti-CD28, or pretreated with the JNK inhibitor prior to stimulation with anti-CD3 and anti-CD28. This method uses primers complementary to sequences at ∼5600 alternative spliced junctions to quantify changes in isoform ratio between two conditions. Focusing on the events for which we obtained an average of >10 reads across samples (∼3000 events) (Supplemental Table S2), we identified those that have an absolute differential inclusion (change in percent spliced in of variable exon [ΔPSI]) of >10, comparing unstimulated with anti-CD3/CD28-stimulated conditions (|PSIunstim − PSIαCD3/CD28| > 10; P < 0.05, paired t-test). This analysis yielded 420 anti-CD3/CD28-responsive alternative exons (Fig. 6A,B; Supplemental Table S3). This number of antigen-induced splicing events in primary human CD4+ T cells is consistent with our previous reports that ∼10%–15% of known alternative exons exhibit altered splicing upon activation of Jurkat cells (Ip et al. 2007; Martinez et al. 2012) and greatly expands our knowledge of the overall scope of alternative splicing in normal human T cells.
Figure 6.

JNK signaling mediates a network of TCR-induced alternative splicing in primary T cells. (A,B) Venn diagram (A) and heat map (B) of total significant alternative splicing events induced by anti-CD3/CD28 (48 h) and those dependent on JNK. Anti-CD3/CD28 sensitivity and JNK dependence are as defined in the text. The color scale for the heat map is at the left. See also Supplemental Table S3. (C) Pie chart comparing JNK-dependent versus GSK3-sensitive anti-CD3/CD28-induced events. See also Supplemental Table S3. (D) Gene ontology (GO) terms and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways enriched (P-value < 0.05) in significant JNK-dependent alternative splicing events as determined by DAVID analysis. (E) Scatter plot comparing RASL-seq-calculated changes in isoform expression versus those determined by RT–PCR. (F) Representative RT–PCR gel of RASL-seq-identified JNK-dependent alternative splicing events upon inhibition of JNK with either SP600125 or IN-8. Percent inclusion (% inc) is the average of three independent experiments. See also Supplemental Figure S7.
To quantify the effect of JNK inhibition on anti-CD3/CD28-induced alternative splicing, we next identified from among the list of 420 anti-CD3/CD28-responsive events those that showed a significant difference (|ΔPSI| > 10; P < 0.05) when treated with anti-CD3/CD28 in the absence versus presence of the JNK inhibitor. This led to the identification of 99 alternative splicing events for which JNK inhibition significantly reduces stimulation-responsive changes in splicing (“JNK-dependent”) (Fig. 6A; Supplemental Table S3). For the majority of these 99 events, the magnitude of the JNK dependence is similar to the magnitude of the CD3/CD28-induced change (Pearson's, r = 0.94; P < 1.3 × 10−51) (Fig. 6B; Supplemental Table S3). Thus, we conclude that ∼25% of the splicing changes induced upon TCR signaling are primarily driven through the JNK signaling pathway. The specific dependence of these events on JNK signaling is underscored by a parallel analysis with the GSK3 pathways that we implicated previously in T-cell signal-induced splicing (Heyd and Lynch 2010) in which we found that only a fifth of the JNK-dependent exons are also responsive to GSK3 (Fig. 6C; Supplemental Table S3).
Gene ontology (GO) analysis reveals that JNK-dependent alternative splicing events are enriched for spliceosomal components and mRNA-binding/processing activity, as compared with the ∼3000 genes queried by RASL-seq. Additional categories involved in gene regulation, such as chromatin-related factors, metal ion-binding proteins, and transcription factors, are also enriched among JNK-dependent genes (Fig. 6D; Supplemental Tables S4, S5A). This “JNK-dependent” GO enrichment is distinct from the “GSK3-dependent” enrichment (Supplemental Table S5B), consistent with JNK and GSK3 controlling different target genes. Importantly, 11 of 14 RASL-identified JNK-dependent alternative splicing events were also observed in RT–PCR assays (Fig. 6E,F; Supplemental Fig. S7), while the other three events show a more modest JNK dependence by RT–PCR but still trending with the RASL data (Supplemental Fig. S7A,B). This high correlation between RASL-seq and RT–PCR (R2 = 0.80) (Fig. 6E) is consistent with other studies using RASL-seq (Zhou et al. 2012; Shao et al. 2014). We further confirmed the JNK dependence of these events with a second highly selective JNK inhibitor (JNK-IN-8) that covalently inhibits JNK through a mechanism distinct from SP600125 (Zhang et al. 2012). Importantly, we found that for 10 of the 10 JNK targets tested, the antigen-induced splicing change is equally inhibited by either SP600125 or JNK-IN-8 (Fig. 6F; Supplemental Fig. S7C). Together, our data demonstrate that JNK serves as a broad regulator of alternative splicing in T cells, which is likely to influence a wide range of cellular functions in addition to amplification of its own signaling pathways through the alternative splicing of MKK7.
JNK signaling and CELF2 coregulate alternative splicing in T cells
Interestingly, we noticed that among the JNK-dependent alternative splicing events were genes that we previously found to be regulated by the splicing factor CELF2 (Fig. 7A; Mallory et al. 2015). In particular, we found that about one-third of JNK-dependent splicing events overlap with our recently published set of CELF2-regulated exons (Fig. 7A; Mallory et al. 2015). Our published set of CELF2-regulated exons used a high threshold for CELF2 dependence to limit false positives (Mallory et al. 2015); therefore, we recognize that we may be underestimating the number of JNK-responsive exons that are sensitive to CELF2. We thus used the AVISPA (advanced visualization of splicing prediction and analysis) algorithm (Barash et al. 2013; Gazzara et al. 2014) to identify motifs enriched among all JNK-responsive events (Supplemental Table S3) relative to the alternative cassette exons queried by RASL-seq that are not responsive to JNK (|ΔPSI| < 3; P > 0.5) (Supplemental Table S2). Remarkably, we found CELF2-binding sites to be highly enriched in both the upstream and downstream introns (Fig. 7B), consistent with the pattern of CELF2 binding in MKK7. Indeed, CELF2-binding sites are the top-scoring motif in the downstream introns of JNK-responsive events (Fig. 7B). The functional significance of these CELF2 motifs was validated in two JNK-dependent splicing events in DDR1 and MTHFD2L (Fig. 7C), which exhibit altered splicing upon knockdown of CELF2 despite not having met the threshold for CELF2 responsiveness in our previous RASL-seq study (Mallory et al. 2015). Together, our data demonstrate that CELF2 not only regulates MKK7 but also mediates a substantial proportion of JNK-dependent splicing in T cells.
Figure 7.
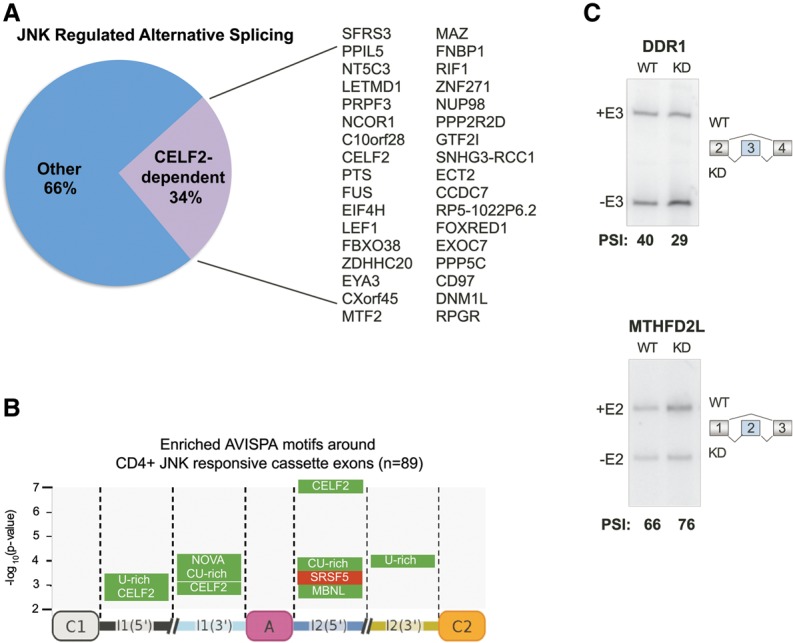
JNK signaling and CELF2 coregulate alternative splicing in T cells. (A) Pie chart of the overlap of significant JNK-dependent alternative splicing events from Figure 6 with published CELF2-dependent alternative splicing events (Mallory et al. 2015) and a list of genes containing alternative exons that are coregulated by JNK and CELF2. (B) Enriched motifs around JNK-responsive exons in primary CD4+ T cells as determined by the AVISPA algorithm. (C) Analysis of two cassette exons in two JNK-dependent alternative splicing events that are predicted to be coregulated by CELF2 on the basis of containing an AVISPA-determined CELF2 motif. Representative RT–PCR gel quantifying the percent inclusion in wild-type (WT) versus CELF2 knockdown (KD) samples from Jurkat T cells.
Discussion
Previous studies have established that alternative splicing is widely regulated in response to various cell stimuli, such as T-cell activation (Martinez and Lynch 2013; Fu and Ares 2014). Conversely, genes encoding signaling proteins are themselves subject to such regulation (Martinez and Lynch 2013), implying reciprocal regulation between signaling and splicing. However, specifics regarding the signaling pathways that connect T-cell activation to alternative splicing as well as the functional consequences of signal-induced splicing changes remain poorly characterized. In this study, we demonstrate that JNK signaling is a major pathway through which T-cell activation induces alternative splicing and provide the first link between JNK signaling and a splicing factor (CELF2) that regulates a sizeable fraction of JNK-responsive exons. Moreover, we found that one of the targets of JNK/CELF2-dependent splicing is the JNK kinase MKK7. While it remains possible that other signaling pathways fine-tune the regulation of MKK7, we show that JNK activation is necessary and sufficient for MKK7 alternative splicing and that MKK7 alternative splicing in turn augments JNK signaling.
Negative feedback loops involving alternative splicing have previously been reported. For example, activation of the IL-1 receptor promotes expression of a small isoform of Myd88 that in turn negatively regulates IL-1 signaling (Janssens et al. 2002, 2003). There are also examples of indirect feed-forward loops, such as Ras-mediated inclusion of CD44 exon 6 to generate an isoform that serves as a coreceptor for growth factors that in turn trigger Ras signaling (Cheng 2006). In contrast, we identified here a direct positive feedback loop in which signaling through JNK promotes alternative splicing of its upstream kinase, MKK7. The increase in c-Jun phosphorylation and TNF-α expression upon forced MKK7-E2 skipping, together with the fact that this splicing event peaks at ∼48 h following T-cell activation, strongly suggests a role for alternative splicing of MKK7 in propagating JNK signaling and augmenting T-cell effector functions following initial activation. Thus, the interplay of JNK and MKK7 provides both a new type of connectivity between signaling and splicing and one that is critical for full T-cell responses.
MKK7 alternative splicing also highlights the breadth of mechanisms through which splicing can modulate kinase activity. In the examples of A-Raf, IRAK1, and TrkB/C, the kinase domain itself is subject to alternative splicing (Middlemas et al. 1991; Allen et al. 1994; Tsoulfas et al. 1996; Palko et al. 1999; Jensen and Whitehead 2001; Rauch et al. 2011; Esteban 2006). In contrast, inclusion of alternative exons encoding the C terminus of ROCK2 during muscle differentiation causes inhibition of a distal kinase domain (Singh et al. 2014). Moreover, cancer cells express a spliced form of the MAP kinase MNK2 that lacks its docking domain (Maimon et al. 2014). This docking domain-deficient isoform of MNK2 was shown to be oncogenic, as it is unable to activate the downstream kinase p38 (Maimon et al. 2014). We now show that alternative splicing of MKK7 favors MKK7-S, the isoform with an intact MAPK-docking domain. The discovery of alternative splicing to modulate MAPK-docking sites in both MNK2 and MKK7 suggests that this may be a common mechanism to regulate MAPK activity.
In addition to providing insight into the functional impact of splicing on signaling, our studies here of MKK7 also revealed novel mechanistic insights into how JNK signaling impacts splicing. First, we defined intronic regulatory elements within the upstream and downstream introns that promote activation-induced skipping of MKK7-E2. These intronic sequences are highly conserved across vertebrates, implying that these are important for proper regulation of MKK7 in other cell types. Second, we identified several proteins that bind to the intronic regulatory sequences and contribute to overall inclusion levels of MKK7-E2, including HuR, SRp20, hnRNPC, and CELF2. We note that, while the effects of HuR, SRp20, and hnRNPC knockdown are subtle, these changes are at least consistent with their binding patterns in activated compared with unstimulated nuclear extracts. However, while these and additional factors may influence MKK7-E2 inclusion, we found that CELF2 is a major repressor of MKK7-E2 inclusion upon T-cell activation. It also remains to be determined how CELF2 promotes MKK7-E2 skipping; however, we note that CELF2 regulates at least one other target in T cells, LEF1 exon 6, through binding to both of the flanking introns (Mallory et al. 2011). Therefore, cooperativity or competition across an exon may be a common theme through which CELF2 functions.
Consistent with the role of CELF2 in regulating MKK7, a JNK-dependent increase in CELF2 protein following T-cell activation correlates with an increase in CELF2 protein binding to the MKK7 intronic regulatory elements. We recently published that TCR signaling-induced expression of CELF2 protein is driven by two mechanisms: an initial increase in transcription that is driven by NF-kB (JNK-independent) and an increase in the stability of the CELF2 mRNA that contributes to the increase in the stability of the CELF2 mRNA by an uncharacterized mechanism. We now show that, in response to T-cell activation, JNK signaling stabilizes CELF2 mRNA to increase CELF2 protein expression and directly impact alternative splicing. Thus, we provide a novel mechanism by which extracellular signals can be transmitted from the TCR through the JNK signaling cascade to the splicing factor CELF2, resulting in changes in gene expression.
Coincident with regulation of MKK7, JNK signaling also controls ∼25% of activation-induced alternative splicing events in primary T cells, with at least one-third of these events also responsive to CELF2. Because previous large-scale studies of alternative splicing in primary human T cells generated only limited events, our identification of ∼400 TCR-responsive splicing events greatly expanded our knowledge of splicing regulation during the human immune response. Furthermore, the specific identification that ∼100 of these TCR-responsive events are dependent on JNK activation adds new insight into the role of JNK signaling in biology. Through regulating alternative splicing of factors involved in different aspects of gene expression, such as mRNA processing, chromatin remodeling, and transcription, JNK signaling potentially affects a greater breadth of biological processes than previously appreciated. For example, JNK-dependent alternative splicing of apoptosis regulators such as cell cycle and apoptosis regulator 1 (CCAR1) and of BCL2-associated X protein (BAX) may serve to turn off apoptosis and promote T-cell proliferation, a hallmark of T-cell activation (Czabotar et al. 2013).
The regulation of MKK7 and other target genes in response to JNK signaling is unlikely to be restricted to T cells. We show here that activation of JNK regulates MKK7 splicing in HEK293 cells as well as in T cells. Moreover, other studies have implicated JNK in the regulation of fibronectin splicing in epithelial cells and have shown JNK to interact with and phosphorylate at least a few known splicing factors, although the impact of this regulation has not been linked to changes in splicing (Habelhah 2001; Pelisch et al. 2005; Hutchins and Szaro 2013). As JNK is activated by many cellular stress conditions, including oxidative stress and DNA damage, these data suggest that JNK-mediated alternative splicing may play a much broader role in the cellular response to a wide range of stress conditions and cell types. Whether CELF2 contributes to JNK-dependent splicing in other cell types remains to be determined. Clearly, proteins other than CELF2 additionally contribute to JNK-dependent splicing in T cells and may also do so in other cell types. However, we note that CELF2 is one of the splicing factors most frequently down-regulated in multiple cancer types (Sveen et al. 2015). Given the role of CELF2 in MKK7 splicing shown here and the role of JNK in proapoptotic signaling (Dhanasekaran and Reddy 2008), these data suggest the possibility that loss of CELF2 leads to decreased JNK activity in cancer.
In sum, we demonstrated here a broad role for JNK signaling in controlling splicing in human T cells and specifically showed that an important functional consequence of JNK-induced splicing is amplification of its own activity through the alternative splicing of MKK7. These data provide an important precedent for the regulation of JNK signaling in other cell types and the reciprocal regulation of signaling and splicing in general.
Materials and methods
Cell culture
The JSL1 clonal population of Jurkat cells was cultured in RPMI supplemented with 5% heat-inactivated fetal bovine serum (FBS) (Lynch and Weiss 2000). HEK293 cells were cultured in DMEM with 5% heat-inactivated FBS. For stimulations, Jurkat T cells were cultured with 20 ng/mL PMA (Sigma-Aldrich) for 48 h (RT–PCR), 2 h (Western analysis of phosphoproteins), or the indicated time points. CD4+ peripheral blood T cells were obtained from the University of Pennsylvania Human Immunology Core (IRB protocol #811028) with an average purity of 90%–95%. CD4+ cells were maintained in RPMI supplemented with 10% heat-inactivated FBS for unstimulated conditions or were additionally stimulated with anti-CD3 (BD Biosciences, 555336) and anti-CD28 (BD Biosciences, 348040) for 48 h. For stimulations, plate wells were coated with anti-CD3 at 2.5 μg/mL, and additional soluble anti-CD3 and anti-CD28 were added to a final concentration of 2.5 μg/mL. For inhibition studies, cells were pretreated for 1 or 3 h (JNK-IN-8) with the following inhibitors before stimulating as described above: 50 μM SP600125 (Calbiochem, 420119), 3 μM JNK-IN-8 (Calbiochem, 420150), 1 μM triciribine (Calbiochem, 124012), 100 nM rapamycin (LC Laboratories, R-5000), and 20 μM U0126 (Invitrogen, PHZ1283). Transcription inhibition was achieved by incubation of cells with actinomycin D at a final concentration of 5 μg/mL.
Transfections
Jurkat T cells (10 × 106) were transfected with 15 nmol of a 5′ splice site-blocking AMO MKK7 (5′-CGATGATAAAAAGGCTGGTACCTGC-3′; GeneTools) or control oligo against the 3′ splice site of exon 11 of NGLY1 (5′-CAGTTGCCTCTGTAATTCATGTTTT-3′) by electroporation as previously described (Rothrock et al. 2003). Cells were incubated with AMO for 24 h after transfection and then stimulated with PMA as described above. Knockdown of HuR, SRp20, hnRNPC, and CELF2 was done by transfection with translation-blocking morpholino oligos (GeneTools) hnRNPC (5′-ATCGTGTTTGATGGTAAGGTTTCTC-3′), HuR (5′-TGTGGTCTTCATAACCATTAGACAT-3′), and SRp20 (5′-AAAATGCGGCGGCTCAAATCCACAC-3′) as described above. HEK293 cells were transfected by Lipofectamine 2000 (Invitrogen, no. 11668-019) transfection according to the manufacturer's instructions and harvested after 48 h. Constitutively active JNK1 and JNK2 (pCDNA3 Flag MKK7B2Jnk1a1 and pCDNA3 Flag MKK7B2Jnk2a2) were a gift from Roger Davis (Addgene, plasmid nos. 19726 and 19727). HEK293 cells were cotransfected with 2 μg of Flag-CELF2 and 2 μg of corresponding MKK7 minigenes as above. The CELF2 expression vector pEFnFlag-CELF2 was generated previously (Mallory et al. 2015). Jurkat stable cell lines that were depleted of JNK were generated using lentiviral transfection of a shRNA targeting JNK2a2 cDNA (GenBank accession no. NM_002752.4) corresponding to nucleotides 462–480. Similarly, depletion of CELF2 was done by lentivirus encoding hairpins targeting nucleotides 171–189 (A) or 966–990 (B).
RT–PCR
RNA was isolated using RNABee (Tel-Test, Inc.) according to the manufacturer's protocol. RT–PCR was performed and analyzed as previously described in detail (Lynch and Weiss 2000; Rothrock et al. 2003; Ip et al. 2007) using sequence-specific primers for individual genes and a low cycle number. Primer sequences for all RT–PCR events are provided in Supplemental Table S6.
Western blots
Western blots were carried out as previously described (Melton et al. 2007). Briefly, 10 µg of total protein lysates was loaded into 10% 37.5:1 bis-acrylamide SDS-PAGE gels. Antibodies used for Western blot were as follows: phospho-c-Jun (Ser73) (#9164), c-Jun (L70B11) (#2315), MKK7 (#4172, p44/42 MAPK (Erk1/2) (137F5) (#4695), phosph-p70 S6 kinase (Thr389) (108D2) (#9234), phospho-Akt (Ser473) (#9271), SAPK/JNK (#9252), and Flag (#2368) from Cell Signaling. Antibodies to hnRNPL (ab6106) and U2AF35 (ab86305) were from Abcam. Antibody to c-myc (9E10) was from Roche, and CELF2 antibody was from the University of Florida Hybridoma Laboratory (HL1889).
Minigenes
MKK7 minigenes were generated by using PCR with primers to isolate the indicated sequences of the endogenous MKK7 gene and flank them with suitable restriction sites for insertion between β-globin exons 1 and 2 within the parental vector pAT7-SCglo (Rothrock et al. 2003). Primer sequences are listed in Supplemental Table S6.
UV cross-linking
UV cross-linking assays were performed as previously described (Mallory et al. 2011). In brief, MKK7 constructs were in vitro transcribed from a T7 promoter and body-labeled with 32P α-UTP. Nuclear extracts from unstimulated or stimulated Jurkat cells were incubated with MKK7 RNAs, UV cross-linked, RNase-digested, and resolved on an SDS-PAGE. For UV cross-linking followed by immunoprecipitation, UV cross-linking reactions after RNase digestion were incubated with corresponding primary antibodies overnight at 4°C followed by incubation with protein G sepharose beads (GE) and elution. Antibodies used in immunoprecipitation were as follows: HuR (Santa Cruz Biotechnology, sc-5261), hnRNPC (Abcam, ab10294), CELF2 (University of Florida Hybridoma Laboratory, HL1889), SRp20 (Thermo, 33-4200), and PSIP1 (Bethyl Laboratories, A300-847A).
RASL-seq
RASL-seq was performed as previously described using a set of probes that interrogate ∼5600 specific splicing events (Li et al. 2001; Mallory et al. 2011; Zhou et al. 2012). In brief, total RNA was harvested from human CD4+ T cells from six independent donors, all of which were cultured in RPMI alone (unstimulated), stimulated with anti-CD3/CD28, and pretreated with SP600125 prior to stimulation with anti-CD3/CD28 or treated with the GSK3 inhibitor SB216763 to determine dependence on JNK or sensitivity to GSK3, respectively. RASL libraries were made and barcoded by PCR for subsequent multiplexed sequencing on a HiSeq 2000. On average, 2 million reads were obtained for each RNA sample. Splicing events were filtered for a minimum of 10 reads averaged across all samples, and the percent long isoform (PSI) for each event in each sample was calculated. The ΔPSI was then calculated for each pairwise comparison, which corrects for any possible bias in the absolute detection of one isoform to another, as it relies on differences in the isoform ratio between samples. CD3/CD28-dependent splicing events were defined as those for which the ΔPSI was significant (|ΔPSI| > 10; P < 0.05, paired Student's t-test) across at least three donors, with consistent data among all six donors. JNK-dependent splicing events were identified as CD3/CD28-dependent splicing events for which the addition of the JNK inhibitor significantly returned the CD3/CD28-induced PSI back toward the unstimulated pattern (i.e., |PSICD3/CD28 − SP600125 − PSICD3/CD28 + SP600125| > 10 and in the same direction as PSICD3/CD28 − SP600125 − PSIunstim; P < 0.05, paired Student's t-test across all three donors). GSK3-sensitive events were calculated in a similar manner.
GO
GO and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis were performed using DAVID version 6.7 (http://david.abcc.ncifcrf.gov). The list of JNK-dependent changes was submitted as the gene list (Supplemental Table S3), with the total queried RASL-seq events with >10 reads across conditions (Supplemental Table S2) submitted as the background list. The same background was used for the GSK3-sensitive events. We selected significant categories with a P-value of <0.05 and a DAVID enrichment score of >1.
Motif enrichment
Hg19 coordinates for JNK-dependent splicing events (described above) and JNK-nonresponsive events (|ΔPSI| < 3; P > 0.5) were extracted; non-cassette events (e.g., alternative 5′ splice sites) were filtered out, leaving 89 and ∼950 cassette exons, respectively; and these events were run through AVISPA. AVISPA has been described in detail previously (Barash et al. 2013) and has been used to predict known and novel regulators of alternative splicing (Gazzara et al. 2014). Briefly, given a cassette exon, AVISPA extracts a set of >1400 features and uses an ensemble of splicing code models to classify exons as alternative or constitutive, predict their tissue-specific splicing patterns, and suggest motifs important to splicing outcome in exon-proximal regions (Barash et al. 2013). A hypergeometric test was performed to identify features significantly overrepresented in JNK-dependent cassette exons compared with events not responsive to JNK inhibition. CELF2-binding sites were defined by AVISPA as UG-rich and CUG-rich motifs (see Barash et al. 2013 for complete description of feature set).
Supplementary Material
Acknowledgments
We thank the University of Pennsylvania Human Immunology Core for the human CD4+ cells, and the Wistar Mass Spectrometry Core for the mass spectrometry analysis. This work was supported by the National Institutes of Health through F31GM103255 to N.M.M., R01GM084034 and R01GM103383 to K.W.L., and R01GM052872 and R01HG004659 to X.-D.F. N.M.M. and K.W.L. conceived the project and wrote the manuscript. J.Q. and X.-D.F. performed RASL-seq analysis. M.R.G. and Y.B. developed and performed the AVISPA analysis. N.M.M., L.A., and M.J.M. performed all other experiments. All authors were involved in the design of experiments and analysis of data.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.267245.115.
References
- Allen SJ, Dawbarn D, Eckford SD, Wilcock GK, Ashcroft M, Colebrook SM, Feeney R, MacGowan SH. 1994. Cloning of a non-catalytic form of human trkB and distribution of messenger RNA for trkB in human brain. Neuroscience 60: 825–834. [DOI] [PubMed] [Google Scholar]
- Asaoka Y, Nishina H. 2010. Diverse physiological functions of MKK4 and MKK7 during early embryogenesis. J Biochem 148: 393–401. [DOI] [PubMed] [Google Scholar]
- Barash Y, Vaquero-Garcia J, González-Vallinas J, Xiong HY, Gao W, Lee LJ, Frey BJ. 2013. AVISPA: a Web tool for the prediction and analysis of alternative splicing. Genome Biol 14: R114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, et al. 2001. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci 98: 13681–13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blonska M, Pappu BP, Matsumoto R, Li H, Su B, Wang D, Lin X. 2007. The CARMA1–Bcl10 signaling complex selectively regulates JNK2 kinase in the T cell receptor-signaling pathway. Immunity 26: 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. 1994. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 369: 756–758. [DOI] [PubMed] [Google Scholar]
- Cheng C. 2006. A positive feedback loop couples Ras activation and CD44 alternative splicing. Genes Dev 20: 1715–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ, et al. 2013. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 152: 519–531. [DOI] [PubMed] [Google Scholar]
- DeSilva DR, Jones EA, Favata MF, Jaffee BD, Magolda RL, Trzaskos JM, Scherle PA. 1998. Inhibition of mitogen-activated protein kinase kinase blocks T cell proliferation but does not induce or prevent anergy. J Immunol 160: 4175–4181. [PubMed] [Google Scholar]
- Dhanasekaran DN, Reddy EP. 2008. JNK signaling in apoptosis. Oncogene 27: 6245–6251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Yang DD, Wysk M, Whitmarsh AJ, Davis RJ, Flavell RA. 1998. Defective T cell differentiation in the absence of Jnk1. Science 282: 2092–2095. [DOI] [PubMed] [Google Scholar]
- Dong C, Yang DD, Tournier C, Whitmarsh AJ, Xu J, Davis RJ, Flavell RA. 2000. JNK is required for effector T-cell function but not for T-cell activation. Nature 405: 91–94. [DOI] [PubMed] [Google Scholar]
- Esteban PF. 2006. A kinase-deficient TrkC receptor isoform activates Arf6-Rac1 signaling through the scaffold protein tamalin. J Cell Biol 173: 291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X-D, Ares M Jr. 2014. Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet 15: 689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazzara MR, Vaquero-Garcia J, Lynch KW, Barash Y. 2014. In silico to in vivo splicing analysis using splicing code models. Methods 67: 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habelhah H. 2001. Identification of new JNK substrate using ATP pocket mutant JNK and a corresponding ATP analogue. J Biol Chem 276: 18090–18095. [DOI] [PubMed] [Google Scholar]
- Haeusgen W, Herdegen T, Waetzig V. 2011. The bottleneck of JNK signaling: molecular and functional characteristics of MKK4 and MKK7. Eur J Cell Biol 90: 536–544. [DOI] [PubMed] [Google Scholar]
- Harris TE, Lawrence JC. 2003. TOR signaling. Sci STKE 2003: re15. [DOI] [PubMed] [Google Scholar]
- Heyd F, Lynch KW. 2010. Phosphorylation-dependent regulation of PSF by GSK3 controls CD45 alternative splicing. Mol Cell 40: 126–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho DT, Bardwell AJ, Grewal S, Iverson C, Bardwell L. 2006. Interacting JNK-docking sites in MKK7 promote binding and activation of JNK mitogen-activated protein kinases. J Biol Chem 281: 13169–13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchins EJ, Szaro BG. 2013. c-Jun N-terminal kinase phosphorylation of heterogeneous nuclear ribonucleoprotein K regulates vertebrate axon outgrowth via a posttranscriptional mechanism. J Neurosci 33: 14666–14680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip JY, Tong A, Pan Q, Topp JD, Blencowe BJ, Lynch KW. 2007. Global analysis of alternative splicing during T-cell activation. RNA 13: 563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens S, Burns K, Tschopp J, Beyaert R. 2002. Regulation of interleukin-1- and lipopolysaccharide-induced NF-κB activation by alternative splicing of MyD88. Curr Biol 12: 467–471. [DOI] [PubMed] [Google Scholar]
- Janssens S, Burns K, Vercammen E, Tschopp J, Beyaert R. 2003. MyD88S, a splice variant of MyD88, differentially modulates NF-κB- and AP-1-dependent gene expression. FEBS Lett 548: 103–107. [DOI] [PubMed] [Google Scholar]
- Jensen LE, Whitehead AS. 2001. IRAK1b, a novel alternative splice variant of interleukin-1 receptor-associated kinase (IRAK), mediates interleukin-1 signaling and has prolonged stability. J Biol Chem 276: 29037–29044. [DOI] [PubMed] [Google Scholar]
- Jin H-O, Seo S-K, Woo S-H, Kim E-S, Lee H-C, Yoo D-H, Choe T-B, Hong S-I, Kim J-I, Park I-C. 2009. SP600125 negatively regulates the mammalian target of rapamycin via ATF4-induced Redd1 expression. FEBS Lett 583: 123–127. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, Naziruddin B, Levy MF, Jackson A, McGlynn K. 2011. Calcineurin/nuclear factor of activated T cells and MAPK signaling induce TNF-α gene expression in pancreatic islet endocrine cells. J Biol Chem 286: 1025–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei K, Nimnual A, Zong WX, Kennedy NJ, Flavell RA, Thompson CB, Bar-Sagi D, Davis RJ. 2002. The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH2-terminal kinase. Mol Cell Biol 22: 4929–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Qiu J, Fu X-D. 2001. RASL-seq for massively parallel and quantitative analysis of gene expression. John Wiley & Sons, Inc, Hoboken, NJ, USA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch KW. 2004. Consequences of regulated pre-mRNA splicing in the immune system. Nat Rev Immunol 4: 931–940. [DOI] [PubMed] [Google Scholar]
- Lynch KW, Weiss A. 2000. A model system for activation-induced alternative splicing of CD45 pre-mRNA in T cells implicates protein kinase C and Ras. Mol Cell Biol 20: 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maimon A, Mogilevsky M, Shilo A, Golan-Gerstl R, Obiedat A, Ben-Hur V, Lebenthal-Loinger I, Stein I, Reich R, Beenstock J, et al. 2014. Mnk2 alternative splicing modulates the p38–MAPK pathway and impacts Ras-induced transformation. Cell Rep 7: 501–513. [DOI] [PubMed] [Google Scholar]
- Mallory MJ, Jackson J, Weber B, Chi A, Heyd F, Lynch KW. 2011. Signal- and development-dependent alternative splicing of LEF1 in T cells is controlled by CELF2. Mol Cell Biol 31: 2184–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallory MJ, Allon SJ, Qiu J, Gazzara MR, Tapescu I, Martinez NM, Fu X-D, Lynch KW. 2015. Induced transcription and stability of CELF2 mRNA drives widespread alternative splicing during T-cell signaling. Proc Natl Acad Sci 112: E2139–E2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez NM, Lynch KW. 2013. Control of alternative splicing in immune responses: many regulators, many predictions, much still to learn. Immunol Rev 253: 216–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez NM, Pan Q, Cole BS, Yarosh CA, Babcock GA, Heyd F, Zhu W, Ajith S, Blencowe BJ, Lynch KW. 2012. Alternative splicing networks regulated by signaling in human T cells. RNA 18: 1029–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda S, Moriguchi T, Koyasu S, Nishida E. 1998. T lymphocyte activation signals for interleukin-2 production involve activation of MKK6–p38 and MKK7–SAPK/JNK signaling pathways sensitive to cyclosporin A. J Biol Chem 273: 12378–12382. [DOI] [PubMed] [Google Scholar]
- Melton AA, Jackson J, Wang J, Lynch KW. 2007. Combinatorial control of signal-induced exon repression by hnRNP L and PSF. Mol Cell Biol 27: 6972–6984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middlemas DS, Lindberg RA, Hunter T. 1991. trkB, a neural receptor protein-tyrosine kinase: evidence for a full-length and two truncated receptors. Mol Cell Biol 11: 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR. 2010. Expansion of the eukaryotic proteome by alternative splicing. Nature 463: 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palko ME, Coppola V, Tessarollo L. 1999. Evidence for a role of truncated trkC receptor isoforms in mouse development. J Neurosci 19: 775–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelisch F, Blaustein M, Kornblihtt AR, Srebrow A. 2005. Cross-talk between signaling pathways regulates alternative splicing. J Biol Chem 280: 25461–25469. [DOI] [PubMed] [Google Scholar]
- Rauch J, Moran-Jones K, Albrecht V, Schwarzl T, Hunter K, Gires O, Kolch W. 2011. c-Myc regulates RNA splicing of the A-Raf kinase and its activation of the ERK pathway. Cancer Res 71: 4664–4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothrock C, Cannon B, Hahm B, Lynch KW. 2003. A conserved signal-responsive sequence mediates activation-induced alternative splicing of CD45. Mol Cell 12: 1317–1324. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Wada T, Kishimoto H, Irie-Sasaki J, Matsumoto G, Goto T, Yao Z, Wakeham A, Mak TW, Suzuki A, et al. 2001. The stress kinase mitogen-activated protein kinase kinase (MKK)7 is a negative regulator of antigen receptor and growth factor receptor-induced proliferation in hematopoietic cells. J Exp Med 194: 757–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao C, Yang B, Wu T, Huang J, Tang P, Zhou Y, Zhou J, Qiu J, Jiang L, Li H, et al. 2014. Mechanisms for U2AF to define 3′ splice sites and regulate alternative splicing in the human genome. Nat Struct Mol Biol 21: 997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RK, Xia Z, Bland CS, Kalsotra A, Scavuzzo MA, Curk T, Ule J, Li W, Cooper TA. 2014. Rbfox2-coordinated alternative splicing of Mef2d and Rock2 controls myoblast fusion during myogenesis. Mol Cell 55: 592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Garvin JE, Koretzky GA, Jordan MS. 2009. T cell activation. Annu Rev Immunol 27: 591–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su B, Jacinto E, Hibi M, Kallunki T, Karin M, Ben-Neriah Y. 1994. JNK is involved in signal integration during costimulation of T lymphocytes. Cell 77: 727–736. [DOI] [PubMed] [Google Scholar]
- Sveen A, Kilpinen S, Ruusulehto A, Lothe RA, Skotheim RI. 2015. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene. 10.1038/onc.2015.318. [DOI] [PubMed] [Google Scholar]
- Tournier C, Whitmarsh AJ, Cavanagh J, Barrett T, Davis RJ. 1997. Mitogen-activated protein kinase kinase 7 is an activator of the c-Jun NH2-terminal kinase. Proc Natl Acad Sci 94: 7337–7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai EY, Jain J, Pesavento PA, Rao A, Goldfeld AE. 1996a. Tumor necrosis factor α gene regulation in activated T cells involves ATF-2/Jun and NFATp. Mol Cell Biol 16: 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai EY, Yie J, Thanos D, Goldfeld AE. 1996b. Cell-type-specific regulation of the human tumor necrosis factor α gene in B cells and T cells by NFATp and ATF-2/JUN. Mol Cell Biol 16: 5232–5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoulfas P, Stephens RM, Kaplan DR, Parada LF. 1996. TrkC isoforms with inserts in the kinase domain show impaired signaling responses. J Biol Chem 271: 5691–5697. [DOI] [PubMed] [Google Scholar]
- Wan YY, Chi H, Xie M, Schneider MD, Flavell RA. 2006. The kinase TAK1 integrates antigen and cytokine receptor signaling for T cell development, survival and function. Nat Immunol 7: 851–858. [DOI] [PubMed] [Google Scholar]
- Wang X, Destrument A, Tournier C. 2007. Physiological roles of MKK4 and MKK7: insights from animal models. Biochim Biophys Acta 1773: 1349–1357. [DOI] [PubMed] [Google Scholar]
- Yang L, Dan HC, Sun M, Liu Q, Sun X-M, Feldman RI, Hamilton AD, Polokoff M, Nicosia SV, Herlyn M, et al. 2004. Akt/protein kinase B signaling inhibitor-2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res 64: 4394–4399. [DOI] [PubMed] [Google Scholar]
- Zhang T, Inesta-Vaquera F, Niepel M, Zhang J, Ficarro SB, Machleidt T, Xie T, Marto JA, Kim N, Sim T, et al. 2012. Discovery of potent and selective covalent inhibitors of JNK. Chem Biol 19: 140–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Collins SL, Lutz MA, Allen AN, Kole TP, Zarek PE, Powell JD. 2007. A role for mammalian target of rapamycin in regulating T cell activation versus anergy. J Immunol 178: 2163–2170. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Qiu J, Liu W, Zhou Y, Plocinik RM, Li H, Hu Q, Ghosh G, Adams JA, Rosenfeld MG, et al. 2012. The Akt–SRPK–SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol Cell 47: 422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.