

Abstract
The widespread use of oxyimino-cephalosporin antibiotics drives the evolution of the CTX-M family of β-lactamases that hydrolyze these drugs and confer antibiotic resistance. Clinically isolated CTX-M enzymes carrying the P167S or D240G active site-associated adaptive mutation have a broadened substrate profile that includes the oxyimino-cephalosporin antibiotic ceftazidime. The D240G substitution is known to reduce the stability of CTX-M-14 β-lactamase, and the P167S substitution is shown here to also destabilize the enzyme. Proteins are marginally stable entities, and second-site mutations that stabilize the enzyme can offset a loss in stability caused by mutations that enhance enzyme activity. Therefore, the evolution of antibiotic resistance enzymes can be dependent on the acquisition of stabilizing mutations. The A77V substitution is present in CTX-M extended-spectrum β-lactamases (ESBLs) from a number of clinical isolates, suggesting that it may be important in the evolution of antibiotic resistance in this family of β-lactamases. In this study, the effects of the A77V substitution in the CTX-M-14 model enzyme were characterized with regard to the kinetic parameters for antibiotic hydrolysis as well as enzyme expression levels in vivo and protein stability in vitro. The A77V substitution has little effect on the kinetics of oxyimino-cephalosporin hydrolysis, but it stabilizes the CTX-M enzyme and compensates for the loss of stability resulting from the P167S and D240G mutations. The acquisition of global stabilizing mutations, such as A77V, is an important feature in β-lactamase evolution and a common mechanism in protein evolution.
INTRODUCTION
The introduction of new antibiotics into clinical use fuels the evolution of enzymes causing drug resistance. These enzymes gain the ability to inactivate antibiotics more efficiently by acquiring point mutations in and around their active sites that increase catalytic activity (1). However, the introduction of new gain-of-function mutations within highly organized active sites often comes at a stability cost to the enzymes (2–6). Therefore, enzymes can absorb only a limited number of destabilizing mutations before they unfold and lose function (7). This limitation in the evolution of catalytic efficiency is often overcome by the acquisition of global stabilizing mutations that offset the incremental loss in stability due to the primary mutation. This compensation mechanism allows the enzymes to continue a mutational trajectory, resulting in increased resistance.
The role of global stabilizing mutations in enzyme evolution has been widely studied (4, 8, 9). In recent decades, the selection of mutant β-lactamase enzymes with expanded substrate specificity has occurred for the plasmid-mediated TEM- and SHV-type β-lactamases to give rise to extended-spectrum β-lactamases (ESBLs) (10, 11). These enzymes arise due to mutations resulting in substitutions near the active site that increase hydrolysis of oxyimino-cephalosporins such as cefotaxime and ceftazidime. The substitutions that increase hydrolysis and create new interactions with the substrate or transition state often remove interactions that would otherwise stabilize the enzyme in the absence of substrate (3, 5, 12, 13). These variant enzymes depend strongly on stabilizing mutations that correct the stability defect associated with the original mutations (12, 14). For example, the M182T mutation has been identified in many TEM-1 β-lactamase variants from clinical isolates, and it compensates for stability and folding defects caused by various mutations in the enzyme that increase oxyimino-cephalosporin hydrolysis (15). Additional mutations such as A184V, T265M, R275Q, and N276D work similarly to M182T to stabilize TEM-1 and thereby increase its evolvability (4). Most second-site stabilizing mutations are found to be associated with functional mutations that alter the substrate specificity or the function of the enzyme.
The introduction of oxyimino-cephalosporin antibiotics, such as cefotaxime and ceftazidime, has resulted in the selection of new β-lactamase genes from the environment encoding enzymes with high cephalosporinase activity (16, 17). In this regard, the CTX-M enzymes, named for their enhanced ability to hydrolyze cefotaxime, were derived from Kluyvera species and have rapidly replaced the TEM ESBL β-lactamases in prevalence in Gram-negative pathogens (17–19). Currently, more than 140 different CTX-M variants have been identified in clinical isolates, and their genetic sequences have been deposited in databases (http://www.lahey.org/Studies/other.asp). These variants can be divided into five subfamilies (CTX-M-1, -2, -8, -9, and -25) based on amino acid sequence identity. CTX-M enzymes belonging to different subfamilies have ≤90% amino acid sequence similarity, and members within a subfamily have ≥95% sequence similarity, differing only by point mutations (16, 20).
CTX-M enzymes have the ability to hydrolyze cefotaxime, but ceftazidime is a poor substrate (20). However, the recently identified D240G and P167S mutations broaden the substrate profile of the CTX-M enzymes to include ceftazidime (21–25). CTX-M variants carrying the D240G substitution have been identified in the CTX-M-1, -2, -9, and -25 subfamilies (Table 1). The D240G substitution is located at the bottom of the B3 β strand within the active site (Fig. 1). This substitution increases flexibility, enlarging the active site cavity to accommodate the bulkier ceftazidime molecule (25, 26). The D240G substitution also decreases protein stability, indicating that there is a trade-off for increased ceftazidime hydrolysis (25). The P167S substitution has been identified in variants belonging to the CTX-M-1 and -9 subfamilies (Table 1) and is located within the omega loop (Fig. 1). This substitution increases the flexibility of the omega loop and alters its interactions with ceftazidime, making it a more favorable substrate (23, 27). The effect of the P167S substitution on protein stability has not been reported. Previous studies indicate that the D240G and P167S substitutions cause increased catalytic efficiency for ceftazidime hydrolysis, resulting in an increased MIC. On the other hand, the increase in ceftazidimase activity is associated with a decrease in the catalytic efficiency and MIC for cefotaxime (21–25, 28). The A77V mutation, found in several members of the CTX-M family also in combination with P167S or D240G, can partially restore cefotaximase activity (16, 21).
TABLE 1.
CTX-M variants from clinical isolates that contain the mutations presented in this studya
| Substitution(s) | CTX-M-1 | CTX-M-2 | CTX-M-9 | CTX-M-25 |
|---|---|---|---|---|
| A77V | CTX-M-1, -23, -60, -61 | CTX-M-51, -65, -87, -90 | CTX-M-91 | |
| P167S | CTX-M-62 | CTX-M-19, -99 | ||
| D240G | CTX-M-15, -28, -29, -33, -71, -82, -88, -96, -101, -107, -108, -109, -117 | CTX-M-43, -131 | CTX-M-16, -27, -93, -102, -121 | CTX-M-41, -94, -100 |
| A77V/P167S | CTX-M-52 | |||
| A77V/D240G | CTX-M-32, -53, -55/57, -69, -79, -114 | CTX-M-98, -105 | CTX-M-25 |
The subfamily to which the CTX-M variant belongs is in bold. The CTX-M variants listed contain substitutions in addition to those listed in the “Substitution(s)” column.
FIG 1.
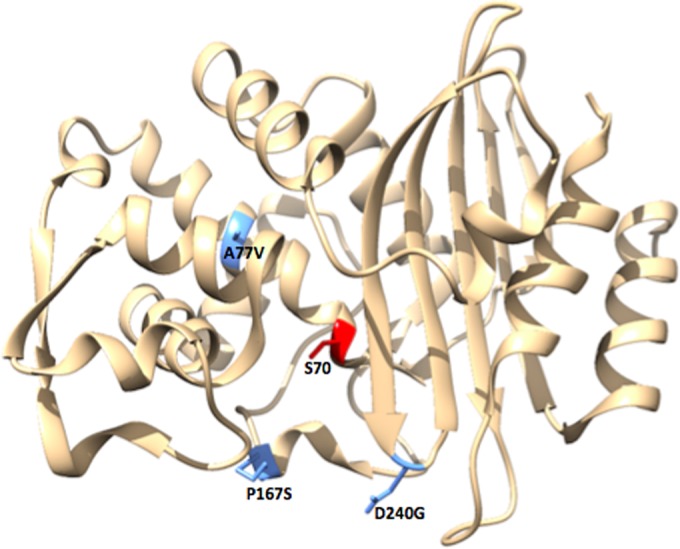
Variable amino acid positions in CTX-M-14 β-lactamase. The structure of CTX-M-14 β-lactamase is shown in tan. Amino acid residues at positions 77, 167, and 240 are highlighted in blue. The catalytic serine (S70) is highlighted in red. The figure was generated using UCSF Chimera (46).
Here, the A77V mutation is identified as a global stabilizing substitution in the CTX-M class of enzymes. The A77V mutation occurs in combination with the P167S and D240G mutations in several clinical isolates (Table 1). Our studies confirm that the active site mutations P167S and D240G allow CTX-M β-lactamases to effectively hydrolyze ceftazidime and that the D240G substitution destabilizes the enzyme. In addition, it is shown that the P167S substitution destabilizes the enzyme. We hypothesize that A77V acts as a stabilizing mutation that offsets the defects caused by the P167S and D240G mutations. This allows the enzyme to retain its stability and in vivo protein levels while acquiring the ability to hydrolyze ceftazidime. The A77V, P167S, and D240G substitutions were introduced individually and in combination within the CTX-M-14 model enzyme and examined for growth competition/fitness, thermostability, in vivo protein levels, and kinetic parameters of the purified proteins. The results indicate that the A77V substitution does not alter substrate specificity with respect to oxyimino-cephalosporins but increases enzyme stability and, when present with either P167S or D240G, increases protein expression levels and the fitness of Escherichia coli in the presence of cefotaxime and ceftazidime.
MATERIALS AND METHODS
Bacterial strains and plasmids.
The E. coli strain XL1-Blue {recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F9 proAB lacIqZΔM15 Tn10 (Tetr)]} (Stratagene, Inc., La Jolla, CA) was used as the host for the construction of the CTX-M-14 mutants. The CTX-M-14-pTP123 plasmid was used for site-directed mutagenesis, MIC determinations, competition experiments, immunoblotting, and protein purification (29). This plasmid was constructed by insertion of the CTX-M-14 gene into the previously constructed pTP123 plasmid, which encodes chloramphenicol resistance (30). The resulting plasmid contains the blaCTX-M-14 gene under the transcriptional control of the Ptrc promoter. This promoter provides low-level basal expression of CTX-M-14 β-lactamase in the absence of isopropyl-β-d-thiogalactopyranoside (IPTG) and high-level expression in the presence of IPTG (29, 30). The basal expression of CTX-M-14 β-lactamase in the absence of IPTG was used for the MIC determinations, competition experiments, and immunoblotting, while IPTG induction of high-level CTX-M-14 expression was used for protein purification experiments. The CTX-M-14 wild-type and mutant proteins of interest were expressed in E. coli RB791 (strain W3110 lacIqL8) for protein purification (31). The E. coli RB791 strain was also used as a host for MIC determination and immunoblot analysis.
In order to obtain sufficient quantities of enzyme for circular dichroism (CD) thermal denaturation experiments, the CTX-M-14 P167S and A77V/P167S mutants were also expressed using the pET28a plasmid. For this purpose, NdeI and SacI restriction sites were used to introduce the blaCTX-M-14 genes with the P167S and A77V/P167S mutations into the pET28a vector using a Gibson Assembly kit (New England BioLabs, Ipswich, MA). The proteins expressed from the pET28a vector contained an N-terminal His tag, and their signal sequence was excluded during the cloning process. The E. coli strain BL21(DE3) [fhuA2 (lon) ompT gal (λ DE3) (dcm) ΔhsdS λ DE3 = λ sBamHIo ΔEcoRI-B int::(lacI::PlacUV5::T7 gene1) i21 Δnin5] was used as the host for protein expression from the CTX-M-14-pET28a plasmids (32).
Construction of CTX-M-14 mutant enzymes by site-directed mutagenesis.
All CTX-M-14 β-lactamase mutants were created in the CTX-M-14-pTP123 plasmid described above. Site-directed mutagenesis was performed using Phusion DNA polymerase (New England BioLabs) according to the manufacturer's protocol. The forward and reverse primers used to introduce mutations into the blaCTX-M-14 gene are listed in Table 2. Primers were obtained from Integrated DNA Technologies (Coralville, IA). DNA sequencing was performed by Lone Star Labs (Houston, TX) to verify the sequence of the entire gene of all site-directed mutants.
TABLE 2.
Primers used for site-directed mutagenesis to create the mutant CTX-M β-lactamases in this study
| Primer | Sequence |
|---|---|
| A76E-F | 5′-AGTAAAGTTATGGAGGCCGCGGCG-3′ |
| A77V-F | 5′-CAGTAAAGTTATGGCGAACGCGGCGGTGCTTAAG-3′ |
| A77V-R | 5′-CTTAAGCACCGCCGCGTTCGCCATAACTTTACTG-3′ |
| D240G-F | 5′-CCGGCAGCGGCGGCTACGGCACCAC-3′ |
| D240G-R | 5′-GTGGTGCCGTAGCCGCCGCTGCCGG-3′ |
| P167S-F | 5′- CTGGATCGCACTGAAAGCACGCTGAATACCGCC-3′ |
| P167S-R | 5′-GGCGGTATTCAGCGTGCTTTCAGTGCGATCCAG-3′ |
MICs.
The MICs for ampicillin, cephalothin, cefotaxime, and ceftazidime were determined using broth microdilution. The CTX-M-14-pTP123 plasmid encoding the wild-type or mutant CTX-M-14 enzyme was transformed into E. coli RB791 cells. The cultures were grown overnight at 37°C in 10 ml LB containing 12.5 μg/ml chloramphenicol. The overnight cultures were diluted 1:100 (optical density at 600 nm [OD600] of ∼0.03), and 1 μl was used to inoculate 0.5 ml LB containing 12.5 μg/ml chloramphenicol and increasing concentrations of ampicillin, cephalothin, cefotaxime, or ceftazidime. IPTG was not used in order to maintain low expression levels of CTX-M-14 to increase the sensitivity of the MIC experiments. The concentrations of ampicillin tested were 0, 31, 47, 63, 94, 125, 188, 250, 375, and 500 μg/ml. The concentrations of cephalothin tested were 0, 312, 468, 625, 938, 1,250, 1,875, 2,500, 3,750, and 5,000 μg/ml. The concentrations of cefotaxime tested were 0, 1, 2, 3, 4, 5, 6, and 7 μg/ml. The concentrations of ceftazidime tested were 0, 0.25, 0.5, 1, 2, 3, 4, and 5 μg/ml. The cultures were incubated at 37°C overnight with shaking for ∼18 h. The MIC was reported as the concentration of antibiotic at which there was no bacterial growth.
Competition assays.
To compare the fitness of E. coli encoding two mutant enzymes in culture, competition experiments were performed as previously described (9, 33). E. coli B Ara+ and Ara− strains were transformed with the CTX-M-14-pTP123 plasmid carrying blaCTX-M-14 with the P167S, A77V/P167S, D240G, and A77V/D240G mutations and grown at 37°C overnight in 10 ml LB supplemented with 12.5 μg/ml chloramphenicol (34). The competing variants were mixed 1:1 and then diluted 1:100 into 20 ml LB with chloramphenicol and increasing concentrations of cefotaxime or ceftazidime. IPTG induction of the CTX-M-14 gene was not used in order to maintain low-level expression of the CTX-M-14 β-lactamase variants to increase the sensitivity of the competition experiments. The competition experiments were carried out at 37°C overnight. The overnight cultures were diluted to 1 × 103 cells/ml before 100 μl was spread onto tetrazolium arabinose (TA) agar plates and incubated at 37°C overnight. Ara− cells form red colonies and Ara+ cells form white colonies on TA agar plates. The percentage of red to white colonies was determined for each concentration of cefotaxime and ceftazidime. All experiments were done in duplicate and repeated in the opposite strain; i.e., competitions were performed after reversing the Ara+ and Ara− strain backgrounds for each mutant.
Purification of the wild-type and mutant CTX-M-14 enzymes.
The CTX-M-14-pTP123 plasmid with genes encoding the wild-type or mutant CTX-M-14 enzyme was introduced into E. coli RB791 cells and grown at 37°C in LB containing 12.5 μg/ml chloramphenicol. High-level expression of CTX-M-14 was induced with 0.2 mM IPTG at a culture density of OD600 of ∼0.9, and the mixture was shaken for an additional 16 h at 23°C. The culture was centrifuged at 4,000 rpm for 15 min at 4°C. Bacterial pellets were resuspended in a periplasmic protein extraction solution (10 mM Tris-HCl [pH 8.0], 20% [wt/vol] sucrose) at a volume of 20 ml/g of bacterial pellet, and the mixture was shaken for 1 h at 4°C. An equal volume of water was added to the resuspended cells, and the mixture was shaken for an additional hour. The mixture was then spun at 9,000 rpm for 30 min, and the supernatant (pH 7.0) was collected and passed through an SP Sepharose Fast Flow (SPFF) cation-exchange column (GE Healthcare) equilibrated with 10 mM morpholineethanesulfonic acid (MES) (pH 6.0) several times to bind the contaminating protein. The pH of the supernatant was lowered to 6.0 using a 1 M MES buffer solution to place a positive charge on the CTX-M protein. The supernatant was passed through the SPFF column once more to bind the positively charged CTX-M protein. The β-lactamase enzyme was eluted using an NaCl gradient (10 mM MES, 2.5 M NaCl [pH 6.0]), and fractions with >90% purity (as determined by SDS-PAGE) were collected and concentrated.
In order to obtain sufficient quantities of enzyme for the CD thermal denaturation experiments, the CTX-M-14 P167S and A77V/P167S mutants were expressed using the pET28a plasmid. The CTX-M-14-pET28a plasmids containing the mutants were introduced into E. coli BL21(DE3) cells and grown at 37°C in LB containing 25 μg/ml kanamycin. For each mutant, the culture was induced with a final concentration of 0.2 mM IPTG at a culture density of OD600 of ∼0.9, and the mixture was shaken for an additional 16 h at 23°C. The culture was centrifuged at 7,000 rpm for 15 min at 4°C. Bacterial pellets were resuspended in 30 ml of lysis buffer (25 mM sodium phosphate [pH 7.2], 300 mM NaCl, 40 μM MgCl2, 10 μg/ml DNase I [Sigma], and 1 EDTA-free protease inhibitor cocktail tablet [Roche]). A French press was used at 1,250 lb/in2 to release protein from the cell. The mixture was then spun at 10,000 × g for 45 min, and the supernatant was collected and passed through a HisTrap FF column (GE Healthcare) equilibrated with 25 mM phosphate buffer [pH 7.2] and 300 mM NaCl. The protein was eluted using an imidazole gradient, and fractions with >90% purity (as determined by SDS-PAGE) were collected and concentrated. The N-terminal His tag was removed by incubation of 0.5 mg of TEV protease with 25 mg of CTX-M protein at 4°C overnight. The TEV protease was removed from the cleaved CTX-M protein using Ni Sepharose high-performance beads (GE Healthcare). The CTX-M protein was run on an SDS-PAGE gel to confirm cleavage of the His tag.
Determination of steady-state kinetic parameters.
The wild-type and mutant CTX-M-14 β-lactamase enzymes were purified to >90% homogeneity, and the in vitro kinetic parameters for β-lactam substrate hydrolysis were determined as previously described (35, 36). Experiments were performed at 30°C in 50 mM sodium phosphate buffer [pH 7.0]. Bovine serum albumin (BSA) was added to the buffer at a concentration of 0.1 mg/ml to stabilize the β-lactamase enzyme, which is used at a low concentration for this assay. However, BSA was not added for the ampicillin reaction due to an overlap in absorbance. The substrate hydrolysis was monitored using a DU800 spectrophotometer at wavelengths of 235 nm, 262 nm, 264 nm, 260 nm, and 482 nm for ampicillin, cephalothin, cefotaxime, ceftazidime, and nitrocefin, respectively. The initial velocities were measured and fit to the Michaelis-Menten equation, v = Vmax[S]/(Km + [S]), using GraphPad Prism 6 to determine kcat and Km. When the CTX-M-14 wild-type or mutant enzyme had a high Km value that prevented determination of Vmax, the catalytic efficiency (kcat/Km) was estimated using the equation v = kcat/Km [E][S], where [S] ≪ Km. The initial velocities were measured at least in duplicate and averaged to determine the kinetic parameters. The standard deviations between the two runs were determined for the kcat and Km values. The standard deviations reported for kcat/Km are the sums of the percent standard deviations of kcat and Km.
Determination of thermal stabilities.
CD was used to determine the thermal stability of the CTX-M-14 wild-type and mutant β-lactamases as previously described (4). The thermal stability was tested by increasing the sample temperature from 30°C to 70°C at a rate of 0.01°C/s, while the signal was measured at 222 nm with a Jasco J-815 CD spectropolarimeter (Jasco, Essex, United Kingdom) coupled to a Peltier PFD-425S/15 instrument to control the temperature. The heat-induced unfolding transitions were monitored for 75 μg of enzyme in 500 μl of 50 mM phosphate buffer [pH 7.0]. The melting temperature (Tm) or the temperature midpoint of protein unfolding was determined by fitting the transition from the folded to unfolded state to a single Boltzmann model for a two-state unfolding mechanism. The data were fit to these equations with GraphPad Prism 6.
Immunoblot analysis.
Immunoblot analysis was performed to determine the in vivo expression levels of the wild-type and mutant CTX-M β-lactamases as previously described (9). E. coli RB791 cells containing the CTX-M-14-pTP123 plasmid with genes encoding the wild-type or mutant CTX-M β-lactamases were grown overnight at 37°C in LB supplemented with 12.5 μg/ml chloramphenicol. The overnight cultures were diluted 100-fold and grown at 37°C to an OD600 of ∼0.8. Then 2 ml of the culture was pelleted by centrifugation, and the supernatant was decanted. IPTG was not added in order to maintain low levels of CTX-M-14 expression to increase the sensitivity of the immunoblot experiment. The cell pellet was resuspended in 100 μl of periplasmic bacterial protein extraction solution (10 mM Tris-HCl [pH 8.0], 20% [wt/vol] sucrose) and incubated at 4°C for 30 min. Next, 100 μl of cold water was added to each sample to release the periplasmic contents of the cells. The samples were again incubated at 4°C for 30 min. The cells were then pelleted by centrifugation, and the supernatant was collected. Next, 15 μl of each sample was incubated at 90°C for 10 min in 4× SDS loading buffer and fractionated on a 12.5% SDS-PAGE gel. A Bio-Rad semidry transfer apparatus was used to transfer the proteins to a nitrocellulose membrane (Whatman Protran). The membrane was blocked with 5% milk before being probed with a rabbit polyclonal anti-CTX-M-14 antibody. The immunoblot was developed using a donkey anti-rabbit secondary antibody conjugated with horseradish peroxidase (GE Healthcare) and the SuperSignal West Pico chemiluminescent substrate (Thermo Scientific). Quantification was performed using densitometry with ImageJ 64-bit software.
RESULTS
Antibiotic susceptibility and fitness of CTX-M-14 β-lactamase and mutant enzymes.
MICs for several antibiotics were determined for E. coli strains expressing the A77V, D240G, P167S, A77V/D240G, and A77V/P167S β-lactamase variants in order to examine the fitness advantage conferred by the A77V substitution (Table 3). E. coli isolates expressing the wild-type CTX-M-14 showed high resistance to ampicillin and cephalothin (250 μg/ml and 938 μg/ml, respectively), as well as a relatively high MIC for cefotaxime (4 μg/ml) and a low MIC for ceftazidime (0.5 μg/ml). This resistance profile is characteristic of the CTX-M class of β-lactamases (16, 20, 35). The A77V mutant exhibited MICs similar to those of the wild-type enzyme to all antibiotics with the exception of cephalothin (468 μg/ml). Consistent with earlier reports in a number of clinical isolates (21), the E. coli strains containing the P167S and D240G mutant enzymes displayed an increase in the MIC for ceftazidime but a decrease in the MIC for cefotaxime (Table 3). The presence of A77V in combination with P167S or D240G restored the MICs for cefotaxime to greater than wild-type levels and further increased the MICs for ceftazidime, indicating that A77V improves the fitness of the P167S and D240G mutants.
TABLE 3.
MICs for E. coli RB791-containing CTX-M-14 mutants, the wild type, and the empty vector control
| Plasmid or WT or mutant β-lactamase | MIC (μg/ml)a |
|||
|---|---|---|---|---|
| AMP | CEF | CTX | CAZ | |
| pTP123 | <4 | 8 | <0.05 | 0.19 |
| CTX-M-14 | 250 | 938 | 4 | 0.5 |
| A77V | 250 | 468 | 3 | 0.5 |
| P167S | 125 | 312 | 2 | 3 |
| D240G | 63 | 312 | 2 | 1 |
| A77V/P167S | 375 | 468 | 7 | 5 |
| A77V/D240G | 188 | 468 | 6 | 3 |
AMP, ampicillin; CEF, cephalothin; CTX, cefotaxime; CAZ, ceftazidime.
Competition experiments were performed to further assess the fitness advantage for E. coli when the A77V mutation was introduced into the P167S and D240G mutant enzymes (see Materials and Methods). E. coli expressing the A77V/P167S mutant enzyme outcompeted E. coli expressing the P167S mutant enzyme in the presence of increasing concentrations of either cefotaxime or ceftazidime (Fig. 2A). Similarly, E. coli expressing the A77V/D240G mutant enzyme outcompeted E. coli expressing the D240G mutant enzyme as concentrations of either cefotaxime or ceftazidime were increased (Fig. 2B). These results conclusively demonstrate that the A77V mutation provides an in vivo fitness advantage when found in combination with the D240G or P167S functional mutations and explain the presence of the double mutant enzymes in naturally occurring variants. The results of the competition experiments are in congruence with the higher MICs that are observed for cefotaxime and ceftazidime when the P167S or D240G single mutant enzymes gain the A77V substitution (Table 3). Taken together, these results are consistent with the A77V substitution either enhancing the catalytic efficiency of P167S and D240G for cefotaxime and ceftazidime hydrolysis and/or increasing the in vivo expression levels of these enzymes.
FIG 2.

Competition experiments between E. coli B cells expressing single mutant and double mutant variants of CTX-M-14. (A) Competition experiments between P167S and A77V/P167S in the presence of increasing concentrations of cefotaxime (left panel) and ceftazidime (right panel). (B) Competition experiments between D240G and A77V/D240G with increasing concentrations of cefotaxime (left panel) and ceftazidime (right panel).
Enzyme kinetic parameters for CTX-M-14 β-lactamase and the mutant enzymes.
Enzyme kinetic parameters were determined to understand the effect of the A77V substitution on catalytic efficiency. Although the A77V substitution is distant from the active site, it may work by altering the active site via long-range structural changes. To investigate this further, the A77V, D240G, P167S, A77V/D240G, and A77V/P167S enzymes were expressed and purified for kinetic analysis with ampicillin, cephalothin, cefotaxime, ceftazidime, and nitrocefin as substrates (Table 4). The A77V mutation alone increased the Km values for ampicillin and cephalothin compared to those for the CTX-M-14 enzyme. Although the A77V substitution decreases the kcat/Km values for ampicillin and cephalothin by 2- to 3-fold, it maintains high levels of resistance to these β-lactams in vivo (Table 3). This supports the hypothesis that the A77V substitution acts by stabilizing the CTX-M-14 enzyme and increases the steady-state enzyme levels, compensating for the reduced kcat/Km values for hydrolysis of these drugs.
TABLE 4.
Enzyme kinetic parameters for CTX-M-14 β-lactamase and mutant enzymes
| WT or mutant β-lactamase | Parameter | Valuea |
||||
|---|---|---|---|---|---|---|
| AMP | CEF | CTX | CAZ | Nitrocefin | ||
| CTX-M-14 | kcat (s−1) | 140 ± 13.2 | 1,626 ± 147.0 | 203 ± 13.4 | NDb | 304 ± 52.4 |
| Km (μM) | ≤12 | 93 ± 8.0 | 72 ± 6.7 | >500 | 22 ± 12.0 | |
| kcat/Km (s−1 μM−1) | ≥12.0 | 17.5 ± 2.2 | 2.8 ± 0.3 | 0.001 ± 0.0007 | 13.8 ± 7.9 | |
| A77V | kcat (s−1) | 100 ± 3.8 | 1,354 ± 15.8 | 99 ± 4.9 | ND | 487 ± 36.1 |
| Km (μM) | 22 ± 10.3 | 187 ± 17.7 | 47.8 ± 9.1 | >500 | 17 ± 4.0 | |
| kcat/Km (s−1 μM−1) | 4.5 ± 2.1 | 7.2 ± 0.7 | 2.1 ± 0.4 | 0.001 ± 0.00004 | 28.6 ± 7.1 | |
| P167S | kcat (s−1) | 29 ± 3.5 | 681 ± 36.8 | 297 ± 29.7 | ND | 225 ± 4.5 |
| Km (μM) | ≤12 | 32 ± 0.8 | 37 ± 6.3 | >500 | 15 ± 1.8 | |
| kcat/Km (s−1 μM−1) | ≥2.4 | 21.1 ± 1.3 | 8.0 ± 1.6 | 0.011 ± 0.0002 | 15 ± 1.8 | |
| D240G | kcat (s−1) | 644 ± 12.9 | 471 ± 41.7 | 321 ± 46.2 | ND | 186 ± 5.7 |
| Km (μM) | 75 ± 12.3 | 47 ± 10.9 | 52 ± 8.6 | >500 | 24 ± 4.7 | |
| kcat/Km (s−1 μM−1) | 8.6 ± 1.4 | 10.0 ± 2.5 | 6.2 ± 1.4 | 0.013 ± 0.0007 | 7.8 ± 1.5 | |
| A77V/P167S | kcat (s−1) | 60 ± 4.9 | 789 ± 117 | 396 ± 96.0 | ND | 145 ± 4.6 |
| Km (μM) | ≤12 | 22 ± 0.01 | 46 ± 4.6 | >500 | 9.4 ± 0.3 | |
| kcat/Km (s−1 μM−1) | ≥5.0 | 35.9 ± 5.3 | 8.6 ± 2.3 | 0.018 ± 0.0003 | 15.4 ± 0.7 | |
| A77V/D240G | kcat (s−1) | 57 ± 6.3 | 317 ± 14.9 | 194 ± 17.5 | ND | 213 ± 0.1 |
| Km (μM) | 31 ± 8.3 | 38 ± 0.2 | 32 ± 7.3 | >500 | 15 ± 0.1 | |
| kcat/Km (s−1 μM−1) | 1.8 ± 0.5 | 8.4 ± 0.4 | 6.1 ± 1.5 | 0.019 ± 0.0008 | 14.2 ± 0.1 | |
AMP, ampicillin; CEF, cephalothin; CTX, cefotaxime; CAZ, ceftazidime.
ND, not determined.
The D240G, P167S, A77V/D240G, and A77V/P167S mutant enzymes displayed 10-fold higher kcat/Km values for ceftazidime hydrolysis than the wild-type CTX-M-14 and A77V mutant enzymes. This finding is consistent with the increased MICs for D240G, P167S, A77V/D240G, and A77V/P167S to ceftazidime. The kcat/Km values for the enzymes carrying the D240G and A77V/D240G mutations are similar in relation to cefotaxime hydrolysis (∼6.0 s−1 μM−1). Similarly, the kcat/Km values for enzymes carrying the P167S and A77V/P167S mutations are both ∼8.0 s−1 μM−1 for cefotaxime hydrolysis. However, the MICs for the A77V/D240G and A77V/P167S double mutant enzymes (6.0 μg/ml and 7.0 μg/ml, respectively) are higher than the MICs for the D240G and P167S single mutant enzymes (2.0 μg/ml) (Table 3). The competition experiments clearly establish the significance of the difference in MICs observed between the double and single mutant enzymes. In both cases, the double mutant enzymes outcompete the single mutant enzymes at higher concentrations of cefotaxime or ceftazidime. Taken together, the results indicate that the A77V substitution does not alter the substrate specificity of the enzymes with respect to oxyimino-cephalosporin hydrolysis.
Stability of CTX-M variants.
To determine the stabilizing effects of the A77V substitution on the CTX-M-14 enzyme, the purified A77V, D240G, P167S, A77V/D240G, and A77V/P167S enzymes were thermally denatured. Table 5 shows the melting temperature (Tm) of each variant as determined from denaturation curves generated by measuring the α-helix ellipticity by circular dichroism while increasing temperature (4, 37). The Tm for wild-type CTX-M-14 was 54.0°C. The A77V substitution stabilized the CTX-M-14 enzyme by increasing the Tm by 3.3°C. Thus, the A77V substitution alone acts as a stabilizing substitution. The D240G substitution only slightly destabilized the CTX-M-14 enzyme (−0.4°C), which might explain its frequent occurrence in almost all CTX-M subfamilies (21, 24). The difference in the stability of D240G versus that of the wild type is consistent with that observed previously (25). The A77V substitution was able to compensate for this loss and further stabilize the A77V/D240G variant, increasing the Tm of the enzyme by 3.2°C, significantly above that of wild-type CTX-M-14. Similarly, the P167S substitution destabilizes the wild-type enzyme (−2.2°C), but the A77V substitution is able to partially compensate for this loss, raising the Tm of the P167S enzyme by 0.5°C. These results indicate that A77V is a stabilizing substitution and suggest that it acts as a suppressor of stability defects.
TABLE 5.
Thermal stabilities of CTX-M variants
| WT or mutant β-lactamase | Tm (°C) | ΔTm (°C) |
|---|---|---|
| CTX-M-14 | 54.0 ± 0.09 | |
| A77V | 57.3 ± 0.14 | +3.3 |
| P167S | 51.8 ± 0.16 | −2.2 |
| D240G | 53.6 ± 0.08 | −0.4 |
| A77V/P167S | 52.3 ± 0.12 | −1.7 |
| A77V/D240G | 56.8 ± 0.08 | +2.8 |
Steady-state protein expression levels of CTX-M-14 and mutant enzymes in vivo.
One way in which the β-lactamase enzymes can increase their resistance to β-lactam antibiotics is by increasing the in vivo steady-state amount of enzyme (4, 9). The stability of an enzyme correlates closely with the steady-state expression levels. Enzymes that are less stable are prone to proteolysis by periplasmic proteases (8, 15). In contrast, more stable enzymes will escape the periplasmic proteases, resulting in greater amounts of enzyme protein and a corresponding increased antibiotic resistance. Because the A77V mutation increases the overall stability of the enzyme, it is expected to increase the in vivo steady-state enzyme levels.
The in vivo expression levels of wild-type CTX-M-14 and the mutant enzymes were examined using immunoblot analysis (Fig. 3). The A77V mutant enzyme displayed an 80% increase in protein levels compared to those for the wild-type enzyme. In addition, the A77V/D240G double mutant enzyme displayed a 200% increase in protein levels compared to those for the D240G single mutant enzyme. Similarly, the A77V/P167S double mutant enzyme displayed a 70% increase in protein levels compared to those for the P167S single mutant enzyme (Fig. 3A). The expression data are consistent with the thermal stability data and demonstrate that the A77V substitution is able to rescue the reduced stability and expression levels of the D240G and P167S mutant enzymes. The increases in the protein expression levels of the double mutant enzymes also explain their higher MICs for cefotaxime and ceftazidime and their ability to outcompete the single mutant enzymes in the competition experiments.
FIG 3.
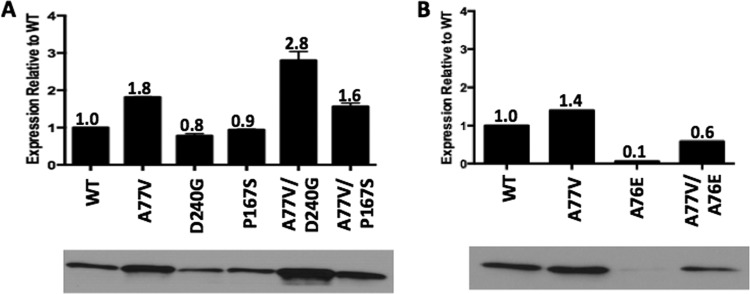
Steady-state expression levels of wild-type (WT) and mutant CTX-M-14 β-lactamases. (A) Immunoblot analysis showing protein expression levels in the periplasmic fraction of E. coli cultures carrying blaCTX-M-14 with the A77V, D240G, P167S, A77V/D240G, and A77V/P167S mutations on the CTX-M-14-pTP123 plasmid. (B) Immunoblot analysis showing protein expression levels of the A76E and A77V/A76E CTX-M β-lactamases. The density of the bands was quantified using ImageJ 64-bit software and plotted relative to that of the WT enzyme.
To determine if the A77V substitution can rescue the expression of CTX-M enzymes carrying mutations other than D240G and P167S, the A76E mutation was studied. The A76E mutant CTX-M enzyme is not found in nature. The substitution was chosen for study due to its location and its ability to disrupt the hydrophobic core of the CTX-M enzyme. The A77V/A76E double mutant enzyme displayed a 50% increase in protein levels compared to the A76E single mutant enzyme as determined by immunoblot analysis (Fig. 3B). This demonstrates that the A77V substitution can compensate for multiple mutations and suggests that the A77V substitution increases the fitness of CTX-M enzymes by increasing stability and steady-state protein levels in vivo. Since the A77V substitution is able to rescue the protein expression of CTX-M enzymes carrying various deleterious mutations, in the future, it may be found associated with new mutations that further expand the functionality of these enzymes.
DISCUSSION
In a continuously changing selective environment, gene products do not always perform optimally. The acquisition of amino acid substitutions due to mutations can change the properties of proteins to provide an altered function and a selective advantage for an organism (38, 39). Most proteins, however, are marginally stable entities and can become unstable in the face of amino acid substitutions (7, 13, 40). Therefore, stability itself imposes limits on the evolution of protein function. Mutations resulting in substitutions that increase overall protein stability overcome this limitation and allow proteins to continually evolve (3, 4, 7).
As shown in Table 1, the mutations A77V, A77V/D240G, and A77V/P167S have been identified independently in multiple subfamilies, particularly subfamilies 1 and 9, of clinically isolated CTX-M ESBLs that confer resistance to oxyimino-cephalosporin antibiotics. Roughly 25 to 30% of the CTX-M isolates containing D240G or P167S also contain A77V. Previous studies demonstrated that the P167S and D240G substitutions broaden the substrate profile of the CTX-M enzymes to include ceftazidime and that the D240G substitution destabilizes the enzyme (21, 25, 27). This study confirms the previous findings and in addition demonstrates that the P167S substitution destabilizes the CTX-M enzyme. Furthermore, A77V functions as a suppressor mutation in CTX-M enzymes due to its ability to compensate for defects caused by the D240G and P167S mutations.
Stabilizing mutations have been well characterized in the TEM-1 β-lactamase family of resistance enzymes (4, 5, 15). Similar to the TEM-1 system, the CTX-M β-lactamase enzymes exhibit increased catalytic activity and altered substrate specificity due to mutations in and near the active site, which come at a cost in terms of stability (21, 22, 24, 25, 41). Mutations that stabilize the protein can arise and enhance in vivo activity by increasing enzyme expression levels (7, 15). The thermodynamic stability of β-lactamases is directly proportional to the ratio of folded to unfolded protein. Unfolded periplasmic proteins are highly susceptible to the action of nonspecific periplasmic proteases in E. coli (8). A stabilized enzyme with a lower fraction of unfolded protein might escape the action of these proteases, increasing the enzyme expression levels and enhancing the fitness of E. coli with genes encoding that particular variant. Several studies have shown that stabilizing mutations can result in increased quantities of active enzyme that will, in turn, cause increased antibiotic resistance in the organism (9, 15, 40). The A77V mutation stabilizes the wild-type CTX-M-14 β-lactamase and both of the previously described P167S and D240G enzymes that exhibit increased ceftazidime hydrolysis, resulting in increased resistance to oxyimino-cephalosporin antibiotics.
The A77V/D240G and A77V/P167S double mutant enzymes hydrolyze cefotaxime and ceftazidime more efficiently than the wild-type enzyme. However, this increased resistance comes at a cost of catalytic efficiency against older β-lactam antibiotics. Specifically, the A77V/P167S mutant enzyme exhibits lower catalytic efficiency for ampicillin, and the A77V/D240G mutant enzyme exhibits lower catalytic efficiency for both ampicillin and cephalothin hydrolysis. This type of “specificity trade-off” has been observed among TEM-1 ESBLs and suggests that the changes in the active site that improve ceftazidime hydrolysis decrease interactions important for the binding and hydrolysis of ampicillin and cephalothin (5). Nonetheless, a decrease in catalytic efficiency against these substrates is not reflected by a corresponding decrease in resistance values for E. coli containing the mutant. This is presumably due to the increased levels of active enzyme in the periplasmic space of the bacterium carrying variants with these substitutions.
Stabilizing mutations have been shown to play a central role in the evolution of β-lactamases and other enzymes (5, 8, 15, 42–44). Amino acid substitutions in the active site of CTX-M enzymes may change their activity and substrate specificity at the cost of stability. The A77V substitution is a stabilizing mutation that compensates for a loss in stability associated with substitutions that alter catalytic activity. The A77V substitution increases the overall thermal stability of the enzyme, allowing it to absorb a greater number of deleterious destabilizing mutations and increasing its access to alternate evolutionary paths (45). These results suggest that the effects of drug resistance mutations on protein stability are an important determining force in the evolution of these enzymes.
ACKNOWLEDGMENTS
This work was supported by NIH grant R01 AI32956 to T.P. M.P.P. was supported in part by award number T32 GM088129 from the National Institute of General Medical Sciences.
We thank Hiram Gilbert for comments on the manuscript. We also thank Paul Leonard, Todd Link, and the Center for Biomolecular Structure and Function at the University of Texas MD Anderson Cancer Center for use of the circular dichroism instrument for thermal stability measurements.
REFERENCES
- 1.Bush K. 2013. Proliferation and significance of clinically relevant β-lactamases. Ann N Y Acad Sci 1277:84–90. doi: 10.1111/nyas.12023. [DOI] [PubMed] [Google Scholar]
- 2.Fromer M, Shifman JM. 2009. Tradeoff between stability and multispecificity in the design of promiscuous proteins. PLoS Comput Biol 5:e1000627. doi: 10.1371/journal.pcbi.1000627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tokuriki N, Tawfik DS. 2009. Stability effects of mutations and protein evolvability. Curr Opin Struct Biol 19:596–604. doi: 10.1016/j.sbi.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Brown NG, Pennington JM, Huang W, Ayvaz T, Palzkill T. 2010. Multiple global suppressors of protein stability defects facilitate the evolution of extended-spectrum TEM β-lactamases. J Mol Biol 404:832–846. doi: 10.1016/j.jmb.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X, Minasov G, Shoichet BK. 2002. Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J Mol Biol 320:85–95. doi: 10.1016/S0022-2836(02)00400-X. [DOI] [PubMed] [Google Scholar]
- 6.Orencia MC, Yoon JS, Ness JE, Stemmer WPC, Stevens RC. 2001. Predicting the emergence of antibiotic resistance by directed evolution and structural analysis. Nat Struct Biol 8:238–242. doi: 10.1038/84981. [DOI] [PubMed] [Google Scholar]
- 7.Bloom JD, Labthavikul ST, Otey CR, Arnold FH. 2006. Protein stability promotes evolvability. Proc Natl Acad Sci U S A 103:5869–5874. doi: 10.1073/pnas.0510098103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang W, Palzkill T. 1997. A natural polymorphism in beta-lactamase is a global suppressor. Proc Natl Acad Sci U S A 94:8801–8806. doi: 10.1073/pnas.94.16.8801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marciano DC, Pennington JM, Wang X, Wang J, Chen Y, Thomas VL, Shoichet BK, Palzkill T. 2008. Genetic and structural characterization of an L201P global suppressor substitution in TEM-1 beta-lactamase. J Mol Biol 384:151–164. doi: 10.1016/j.jmb.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tzouvelekis LS, Bonomo RA. 1999. SHV-type beta-lactamases. Curr Pharm Des 5:847–864. [PubMed] [Google Scholar]
- 11.Knox JR. 1995. Extended-spectrum and inhibitor-resistant TEM-type beta-lactamases: mutations, specificity, and three-dimensional structure. Antimicrob Agents Chemother 39:2593–2601. doi: 10.1128/AAC.39.12.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomas VL, McReynolds AC, Shoichet BK. 2010. Structural bases for stability-function tradeoffs in antibiotic resistance. J Mol Biol 396:47–59. doi: 10.1016/j.jmb.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shoichet BK, Baase WA, Kuroki R, Matthews BW. 1995. A relationship between protein stability and protein function. Proc Natl Acad Sci U S A 92:452–456. doi: 10.1073/pnas.92.2.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomas VL, Golemi-Kotra D, Kim C, Vakulenko SB, Mobashery S, Shoichet BK. 2005. Structural consequences of the inhibitor-resistant Ser130Gly substitution in TEM beta-lactamase. Biochemistry 44:9330–9338. doi: 10.1021/bi0502700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sideraki V, Huang W, Palzkill T, Gilbert HF. 2001. A secondary drug resistance mutation of TEM-1 beta-lactamase that suppresses misfolding and aggregation. Proc Natl Acad Sci U S A 98:283–288. doi: 10.1073/pnas.98.1.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.D'Andrea MM, Arena F, Pallecchi L, Rossolini GM. 2013. CTX-M-type β-lactamases: a successful story of antibiotic resistance. Int J Med Microbiol 303:305–317. doi: 10.1016/j.ijmm.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 17.Cantón R, González-Alba JM, Galán JC. 2012. CTX-M enzymes: origin and diffusion. Front Microbiol 3:110. doi: 10.3389/fmicb.2012.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rossolini GM, D'Andrea MM, Mugnaioli C. 2008. The spread of CTX-M-type extended-spectrum beta-lactamases. Clin Microbiol Infect 14(Suppl 1):33–41. doi: 10.1111/j.1469-0691.2007.01867.x. [DOI] [PubMed] [Google Scholar]
- 19.Bae IK, Lee BH, Hwang HY, Jeong SH, Hong SG, Chang CL, Kwak H-S, Kim HJ, Youn H. 2006. A novel ceftazidime-hydrolysing extended-spectrum beta-lactamase, CTX-M-54, with a single amino acid substitution at position 167 in the omega loop. J Antimicrob Chemother 58:315–319. doi: 10.1093/jac/dkl252. [DOI] [PubMed] [Google Scholar]
- 20.Bonnet R. 2004. Growing group of extended-spectrum β-lactamases: the CTX-M enzymes. Antimicrob Agents Chemother 48:1–14. doi: 10.1128/AAC.48.1.1-14.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Novais A, Cantón R, Coque TM, Moya A, Baquero F, Galán JC. 2008. Mutational events in cefotaximase extended-spectrum beta-lactamases of the CTX-M-1 cluster involved in ceftazidime resistance. Antimicrob Agents Chemother 52:2377–2382. doi: 10.1128/AAC.01658-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kimura S, Ishii Y, Tateda K, Yamaguchi K. 2007. Predictive analysis of ceftazidime hydrolysis in CTX-M-type β-lactamase family members with a mutational substitution at position 167. Int J Antimicrob Agents 29:326–331. doi: 10.1016/j.ijantimicag.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 23.Kimura S, Ishiguro M, Ishii Y, Alba J. 2004. Role of a mutation at position 167 of CTX-M-19 in ceftazidime hydrolysis. Antimicrob Agents Chemother 48:1454–1460. doi: 10.1128/AAC.48.5.1454-1460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonnet R, Recule C, Baraduc R, Chanal C, Sirot D, De Champs C, Sirot J. 2003. Effect of D240G substitution in a novel ESBL CTX-M-27. J Antimicrob Chemother 52:29–35. doi: 10.1093/jac/dkg256. [DOI] [PubMed] [Google Scholar]
- 25.Chen Y, Delmas J, Sirot J, Shoichet B, Bonnet R. 2005. Atomic resolution structures of CTX-M beta-lactamases: extended spectrum activities from increased mobility and decreased stability. J Mol Biol 348:349–362. doi: 10.1016/j.jmb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 26.Delmas J, Chen Y, Prati F, Robin F, Shoichet BK, Bonnet R. 2008. Structure and dynamics of CTX-M enzymes reveal insights into substrate accommodation by extended-spectrum beta-lactamases. J Mol Biol 375:192–201. doi: 10.1016/j.jmb.2007.10.026. [DOI] [PubMed] [Google Scholar]
- 27.Poirel L, Naas T, Thomas ILE, Karim A, Bingen E, Nordmann P, Debre R. 2001. CTX-M-type extended-spectrum β-lactamase that hydrolyzes ceftazidime through a single amino acid substitution in the omega loop. Antimicrob Agents Chemother 45:3355–3361. doi: 10.1128/AAC.45.12.3355-3361.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delmas J, Robin F, Carvalho F, Mongaret C, Bonnet R. 2006. Prediction of the evolution of ceftazidime resistance in extended-spectrum β-lactamase CTX-M-9. Antimicrob Agents Chemother 50:731–738. doi: 10.1128/AAC.50.2.731-738.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adamski CJ, Cardenas AM, Brown NG, Horton LB, Sankaran B, Prasad BVV, Gilbert HF, Palzkill T. 2015. Molecular basis for the catalytic specificity of the CTX-M extended-spectrum β-lactamases. Biochemistry 54:447–457. doi: 10.1021/bi501195g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petrosino J, Rudgers G, Gilbert H, Palzkill T. 1999. Contributions of aspartate 49 and phenylalanine 142 residues of a tight binding inhibitory protein of β-lactamases. J Biol Chem 274:2394–2400. doi: 10.1074/jbc.274.4.2394. [DOI] [PubMed] [Google Scholar]
- 31.Amann E, Brosius J, Ptashne M. 1983. Vectors bearing a hybrid trp-lac promoter useful for regulated expression of cloned genes in Escherichia coli. Gene 25:167–178. doi: 10.1016/0378-1119(83)90222-6. [DOI] [PubMed] [Google Scholar]
- 32.Studier FW, Moffatt BA. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol 189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- 33.Majiduddin FK, Palzkill T. 2003. An analysis of why highly similar enzymes evolve differently. Genetics 466:457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lenski RE, Simpson SC, Nguyen TT. 1994. Genetic analysis of a plasmid-encoded, host genotype-specific enhancement of bacterial fitness. J Bacteriol 176:3140–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ishii Y, Galleni M, Ma L, Frère J-M, Yamaguchi K. 2007. Biochemical characterisation of the CTX-M-14 beta-lactamase. Int J Antimicrob Agents 29:159–164. doi: 10.1016/j.ijantimicag.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 36.Brown NG, Palzkill T. 2010. Identification and characterization of beta-lactamase inhibitor protein-II (BLIP-II) interactions with beta-lactamases using phage display. Protein Eng Des Sel 23:469–478. doi: 10.1093/protein/gzq017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deng Z, Huang W, Bakkalbasi E, Brown NG, Adamski CJ, Rice K, Muzny D. 2012. Deep sequencing of systematic combinatorial libraries reveals β-lactamase sequence constraints at high resolution. J Mol Biol 424:150–167. doi: 10.1016/j.jmb.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Romero PA, Arnold FH. 2009. Exploring protein fitness landscapes by directed evolution. Nat Rev Mol Cell Biol 10:866–876. doi: 10.1038/nrm2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Serrano L, Day AG, Fersht AR. 1993. Step-wise mutation of barnase to binase A procedure for engineering increased stability of proteins and an experimental analysis of the evolution of protein stability. J Mol Biol 233:305–312. [DOI] [PubMed] [Google Scholar]
- 40.Teilum K, Olsen JG, Kragelund BB. 2011. Protein stability, flexibility and function. Biochim Biophys Acta 1814:969–976. doi: 10.1016/j.bbapap.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 41.Celenza G, Luzi C, Aschi M, Segatore B, Setacci D, Pellegrini C, Forcella C, Amicosante G, Perilli M. 2008. Natural D240G Toho-1 mutant conferring resistance to ceftazidime: biochemical characterization of CTX-M-43. J Antimicrob Chemother 62:991–997. doi: 10.1093/jac/dkn339. [DOI] [PubMed] [Google Scholar]
- 42.Shortle D, Lin B. 1985. Genetic analysis of staphylococcal nuclease: identification of three intragenic “global” suppressors of nuclease-minus mutations. Genetics 110:539–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blance SJ, Williams NL, Preston ZA, Bishara J, Smyth MS, Maxwell A. 2000. Temperature-sensitive suppressor mutations of the Escherichia coli DNA gyrase B protein. Protein Sci 9:1035–1037. doi: 10.1110/ps.9.5.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fane B, Villafanej R, Mitraki A, King J. 1991. Identification of global suppressors for temperature-sensitive folding mutations of the P22 tailspike protein. J Biol Chem 261:11640–11648. [PubMed] [Google Scholar]
- 45.Weinreich DM, Delaney NF, Depristo MA, Hartl DL. 2006. Darwinian evolution can follow only very few mutational paths to fitter proteins. Science 312:111–114. doi: 10.1126/science.1123539. [DOI] [PubMed] [Google Scholar]
- 46.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]