

Abstract
Limited access to HIV drug resistance testing in low- and middle-income countries impedes clinical decision-making at the individual patient level. An efficient protocol to address this issue must be established to minimize negative therapeutic outcomes for HIV-1-infected individuals in such settings. This is an observational study to ascertain the potential of newer genomic sequencing platforms, such as the Illumina MiSeq instrument, to provide accurate HIV drug resistance genotypes for hundreds of samples simultaneously. Plasma samples were collected from Canadian patients during routine drug resistance testing (n = 759) and from a Ugandan study cohort (n = 349). Amplicons spanning HIV reverse transcriptase codons 90 to 234 were sequenced with both MiSeq sequencing and conventional Sanger sequencing methods. Sequences were evaluated for nucleotide concordance between methods, using coverage and mixture parameters for quality control. Consensus sequences were also analyzed for disparities in the identification of drug resistance mutations. Sanger and MiSeq sequencing was successful for 881 samples (80%) and 892 samples (81%), respectively, with 832 samples having results from both methods. Most failures were for samples with viral loads of <3.0 log10 HIV RNA copies/ml. Overall, 99.3% nucleotide concordance between methods was observed. MiSeq sequencing achieved 97.4% sensitivity and 99.3% specificity in detecting resistance mutations identified by Sanger sequencing. Findings suggest that the Illumina MiSeq platform can yield high-quality data with a high-multiplex “wide” sequencing approach. This strategy can be used for multiple HIV subtypes, demonstrating the potential for widespread individual testing and annual population surveillance in resource-limited settings.
INTRODUCTION
Advances in highly active antiretroviral therapy (HAART) in recent decades have resulted in sustained decreases in HIV-related morbidity and mortality rates. HIV-infected individuals who receive treatment now have nearly normal life expectancies, such that HIV is now considered a manageable chronic disease (1, 2); antiretroviral therapy (ART) not only provides benefits at the individual patient level but also results in a population-level advantage through HAART-induced suppression of HIV replication and the inherent prevention of onward transmission of the virus, termed “treatment as prevention” (3–6).
Drug resistance testing is an essential complement to HAART, enabling clinicians to identify patients infected with drug-resistant HIV and to prescribe the appropriate antiretroviral regimens (7). Lack of access to HIV drug resistance testing acts as a major barrier to long-term treatment success, either through prescription of ineffective regimens in the case of transmitted resistance or through decreased ability of physicians to identify causes of treatment failure (7–9). Cases of unsuppressed viremia allow continued transmission and can compromise the management of HIV on both the patient and population levels. These challenges are particularly acute in low- and middle-income countries (LMIC), where access to drug resistance testing is problematic due to limited resources and infrastructure. Currently, the majority of incident cases of clinically relevant HIV drug resistance involve nonnucleotide reverse transcriptase inhibitor (NNRTI) resistance; therefore, novel resistance testing methods for use in LMIC should, at a minimum, target the HIV reverse transcriptase (RT) region (10–13).
Sanger sequencing is the current standard methodology used for HIV drug resistance testing. Amplification and sequencing are performed with the genomic regions targeted by antiretroviral drugs, such as protease (PR), reverse transcriptase (RT), and integrase. This method of population sequencing generates a single consensus sequence per sample, which represents the predominant HIV quasispecies within the sample. Sanger consensus sequences can be analyzed for drug resistance mutations by various means, including but not limited to custom interpretive bioinformatic algorithms, linked genotype-phenotype information, and proprietary software included in commercial genotyping systems (e.g., Sierra [Stanford HIVdb Program], vircoTYPE HIV-1, or ViroSeq HIV-1 genotyping system) (14–16). However, Sanger sequencing is unable to reliably detect clinically relevant low-frequency drug-resistant variants (17–19).
In contrast, newer genomic sequencing (or next-generation sequencing [NGS]) platforms can be used to sequence diverse HIV quasispecies in order to detect rare resistant variants. In this deep sequencing approach, HIV RNA is PCR amplified and thousands of templates per sample are clonally sequenced. The depth of coverage obtained (typically several thousands to tens of thousands of reads per sample [20, 21]) can be used to detect low-frequency variants and to study within-host HIV evolution (22). Several studies have clearly demonstrated that the presence of low-frequency drug-resistant variants, particularly those with NNRTI resistance, can negatively affect treatment outcomes (20, 23–27).
Because the cost-per-base of NGS is substantially lower than that of Sanger sequencing, low-cost resistance testing could potentially be performed with those instruments. Furthermore, the greatly increased sequencing capacity of the newest NGS instruments offers additional benefits. Samples could be sequenced to greater depth, allowing more sensitive detection of rare variants (21). Alternatively, a correspondingly greater number of samples could be sequenced in a single run, a strategy that we refer to as “wide” sequencing. To our knowledge, however, NGS resistance testing methods have primarily been demonstrated using low sample numbers per run (21, 28–31).
In this proof-of-principle study, we describe a feasible, high-throughput sequencing method that uses the Illumina MiSeq system to produce high-quality sequences for hundreds of samples in parallel. This wide sequencing technique spreads the read coverage of deep sequencing, which typically is concentrated on a few samples, over a larger pool of amplicons. Some samples may be sequenced to a lesser depth than with deep sequencing approaches, but the coverage obtained for most successfully sequenced samples may still be sufficient to detect lower-frequency variants. This sequencing approach could be used for large-scale sequencing studies and HIV resistance surveillance in settings in which routine clinical testing is unavailable. Wide sequencing was evaluated for its overall accuracy, relative to Sanger sequencing, as well as its sensitivity and specificity in detecting resistance mutations.
MATERIALS AND METHODS
Plasma samples and RNA extraction.
Plasma samples were collected from Canadian patients (n = 759) or from participants in the Uganda AIDS Rural Treatment Outcomes (UARTO) study (n = 349) (32). Samples from Canadian patients were collected in 2013 and 2014, as part of routine physician-ordered genotypic HIV drug resistance testing at the British Columbia Centre for Excellence in HIV/AIDS (BCCfE) (Vancouver, Canada). Samples from the UARTO cohort consisted of baseline samples collected at the time of enrollment and samples collected for longitudinal virological monitoring following the initiation of ART, up to 7.5 years postinitiation (32). Plasma samples were stored at −20°C prior to extraction. Ethical approval was granted by the University of British Columbia Providence Health Care Research Ethics Board (protocols H13-00395 and H11-01642).
Plasma viral load (pVL) data were available for a majority subset of samples (n = 1,068). For those samples, the median HIV pVL values were 4.2 log10 copies/ml (interquartile range [IQR], 3.1 to 4.9 log10 copies/ml) and 5.2 log10 copies/ml (IQR, 4.6 to 5.5 log10 copies/ml) in the Canadian and UARTO samples, respectively, reflecting the differences in treatment experience in the two cohorts.
HIV RNA extraction was performed using the NucliSENS easyMAG system (bioMérieux, St. Laurent, Canada) or the Abbott m2000sp system (Abbott Molecular, Mississauga, Canada), according to the manufacturer's instructions. A laboratory clone, pNL4-3, resuspended in normal human plasma (n = 9), or a clinical isolate, POS08 (n = 5), was included in extraction runs as an internal control. Aliquots of nuclease-free water (n = 21) were included as negative controls for potential PCR contamination. A total of 1,108 patient samples were extracted.
RT PCR amplification of PR-RT and Sanger sequencing.
A two-step RT PCR, using Expand reverse transcriptase and the Expand High Fidelity PCR system (Roche Diagnostics, Laval, Canada), was used to generate HIV DNA fragments spanning the HIV protease (PR)-reverse transcriptase (RT) region, as described previously (33). Depending on the sample source, one of three PCR products was produced by nested PCR. Briefly, an amplicon spanning complete protease and RT codons 1 to 400 was produced for Canadian samples (34). In cases in which amplification of Canadian samples failed, backup amplification of a product covering protease and RT codons 1 to 240 was attempted (34). For UARTO samples, a smaller amplicon, spanning RT codons 35 to 234, was generated using PCR primers designed to target regions conserved among HIV-1 subtypes A, B, C, and D. PCR primers are listed in Table S1 in the supplemental material.
Amplified products were visualized on 0.5% agarose gels and sequenced bidirectionally on an ABI Prism 3730xl DNA analyzer (Life Technologies, Burlington, Canada), using the BigDye Terminator v3.1 cycle sequencing kit. Medians of >8 and >2 sequencing primers were used for the Canadian and UARTO samples, respectively, in order to obtain at least 2-fold coverage of the amplicons.
Sanger sequencing data processing.
ABI chromatograms were processed with in-house software (RECall) that automatically calls bases, trims primer sequences, and constructs consensus contigs (35). Briefly, chromatogram files were preprocessed by Phred to quantify the major and minor peaks at each position and to assign quality scores. Individual sequences were then aligned with the HIV HXB2 reference (GenBank accession number K03455.1), and a single contig was generated. Mixtures were called when the peak area of the minor base exceeded 20% of that of the major base across the majority of reads covering that position. The RECall default quality control criteria identified potential problematic bases and excluded any Sanger sequences that failed to meet RECall standards (35). Sanger sequences containing unknown bases (N) were excluded from the analysis, to allow complete comparisons of drug resistance profiles. All Sanger consensus sequences were then trimmed to a 435-bp region containing RT codons 90 to 234, to correspond to the region covered by the MiSeq sequences (as described below).
MiSeq library preparation and sequencing.
To minimize the potential variability introduced by the RNA extraction and reverse transcription steps, MiSeq library preparation began from the same first-round PCR products generated during the Sanger sequencing procedure. Since Sanger sequencing was performed as samples arrived at the BCCfE clinical laboratory, over a period of months, first-round PCR amplicons were stored at room temperature for up to 12 months prior to the initiation of this study, with >75% of samples being stored for no longer than 6 months. A fragment spanning RT codons 90 to 234 was amplified in a nested second PCR with primers incorporating Illumina indexing adaptors (see Table S1 in the supplemental material). A dual-index sequencing strategy was used in order to minimize the number of primers needed for such a large multiplex run. Briefly, 8-bp-long indices (or “barcodes”) are added to both the 5′ and 3′ ends of each amplified fragment. A unique pair of indices is used for each sample. Subsequent sequencing of each index pair unambiguously identifies a sequence read as belonging to a specific sample (36). A total of 24 i5 forward and 48 i7 reverse indices were used, allowing up to 1,152 samples to be sequenced simultaneously (see Table S2 in the supplemental material). Index tags were added in a third low-cycle PCR, as used in the Illumina Nextera XT indexing procedure. Indexed MiSeq amplicons were purified and normalized using Agencourt AMPure XP magnetic capture beads and were pooled into 12 DNA libraries (96 amplicons/library). Library concentrations were determined using the Invitrogen Quant-iT Picogreen dsDNA assay. The 12 libraries were pooled at equimolar concentrations (1 ng/μl) prior to MiSeq sequencing with a 2x250bp v2 kit. Illumina currently recommends a minimum of 5% PhiX spike-in, to avoid sequencing problems associated with low-diversity libraries; a more conservative 10% PhiX spike-in was used in this experiment. Thus, all 1,143 samples were sequenced in a single MiSeq run.
MiSeq data processing.
MiSeq short-read data were processed by an in-house pipeline using Bowtie2 and SAMtools (37, 38). Reads were initially mapped to the HXB2 reference; the mapped reads were used to generate sample-specific consensus sequences, to which the entire set of reads was subsequently remapped. Consensus sequence generation and remapping proceeded iteratively until >95% of all reads were mapped successfully or no improvement in the number of mapped reads was observed. Paired-end reads were then merged, with any differences in base calls in the overlapping portion being resolved using a quality score-informed algorithm. In brief, bases with quality scores of <15 were censored as unknown bases (N). Discordant bases were assigned as the base with the higher quality if the difference was ≥5; otherwise, the merged base was censored.
MiSeq consensus sequences spanning RT codons 90 to 234 were produced from the empirical raw nucleotide frequency distributions in the aligned and merged read data. An ambiguity threshold was used to allow minority bases present above the threshold to be included in consensus sequences as mixtures. A threshold of 20% was chosen in order to mimic the parameters of the RECall software used in Sanger sequence analysis. A depth-of-coverage parameter was used for quality control; sequences with low coverage (defined a priori as <100 times at any position) were rejected.
Analysis of concordance of nucleotide sequences and resistance interpretations.
Samples that were successfully amplified and sequenced by both Sanger and MiSeq sequencing methods were assessed for nucleotide concordance, calculated as the proportion of nucleotide agreement observed across all nucleotides sequenced. Any differences in nucleotide base calls, including differences in mixture calling, were considered mismatches. For technical replicates, a consensus sequence constructed from all available replicates was used to assess interassay sequencing variability. MiSeq controls were compared to their corresponding Sanger counterparts to determine variability between sequencing methods. Nucleotides that appeared in ≥20% of replicate sequences were included (as mixtures) in the consensus sequence. HIV subtyping of MiSeq sequences was performed by RIP, using a 90% confidence threshold and a 200-bp window size (39).
Agreement in resistance interpretations between methods was also evaluated by translating consensus sequences. Resistance mutations were defined according to the 2013 International Antiviral Society-USA (IAS-USA) list (40). Codons containing ambiguous nucleotides (mixtures) were translated to include all possible amino acids in drug resistance analyses.
RESULTS
Sanger and MiSeq sequencing success rates.
A total of 881 patient-derived (clinical) samples (80%) were successfully sequenced with the Sanger protocol; 576 (76%) of the Canadian samples and 305 (85%) of the samples from the UARTO cohort were successfully sequenced. Successfully sequenced pNL4-3 controls (7 of 9 replicates [78%]) were clonal, with 100% concordance between replicates, although two pNL4-3 failures were observed. Overall, 98.0% nucleotide concordance was observed among successful POS08 replicates (n = 5 [100%]), with three nucleotide mismatches being found in key resistance-associated positions. All differences between POS08 replicates were due to differences in mixture calls, all of which were compatible. It should be noted that both the Canadian and UARTO groups (primarily the former) contained samples from patients with low-level viremia (pVL of <1,000 copies/ml) or emerging virological failure, representing a reasonable cross-section of samples sent for HIV drug resistance testing. For example, 15% of samples had viral loads of 50 to 1,000 copies/ml, 18% had viral loads of 1,000 to 10,000 copies/ml, 31% had viral loads of 10,000 to 100,000 copies/ml, and 35% had viral loads of >100,000 copies/ml.
The MiSeq-reported quality metrics, taken from the averages across all sequence and index reads, were consistent with typical values observed in previous runs, i.e., cluster density of 1,086,000 clusters/mm2, 80.9% of bases with Q scores of ≥30, 77.4% of clusters passing filters, and a PhiX sequencing mean error rate of 1.6%. The median MiSeq coverage was ∼9,900 paired-end reads/sample (IQR, 5,300 to 13,400 paired-end reads/sample). Variance in sequence coverage across the amplicon was minimal, with the exception of a small drop in coverage between RT codons 167 and 171, corresponding to decreased sequencing quality at the end of the reverse R2 Illumina read (Fig. 1). The distributions of MiSeq read coverage values were comparable across all pVL strata.
FIG 1.
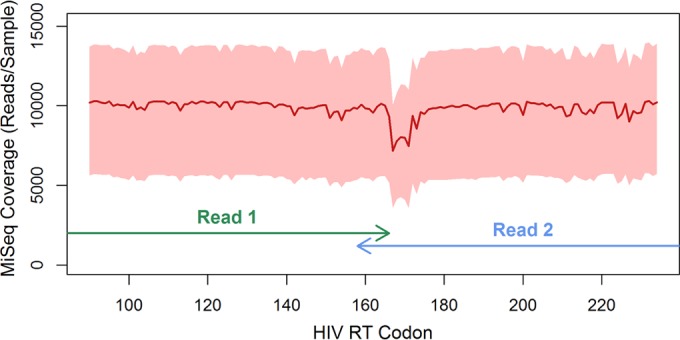
Distribution of MiSeq sequencing coverage. The depth of MiSeq sequencing coverage at each position across the sequenced amplicon (RT codons 90 to 234), after quality control filters were applied (n = 892 clinical samples), is shown. Red line, median coverage at each position; pink shading, interquartile range. Paired-end 2x250-bp sequencing kits were used. Arrows, positions covered by each read in the pair. The small drop in coverage observed around RT codon 170 reflects lower-quality bases at the end of read 2, which were discarded during data processing.
In total, 892 clinical samples (81%) were successfully sequenced by MiSeq sequencing, with 579 Canadian samples (76%) and 313 UARTO samples (87%) passing the predefined 100-fold coverage requirement. All pNL4-3 replicates and 4 POS08 samples (80%) were successfully sequenced, with 100% and 98.6% nucleotide concordance, respectively, being observed among replicates. A single nucleotide mismatch was found in a key resistance-associated position in one POS08 sample. Again, differences in POS08 replicate sequences were the result of mixture calls; no incompatible differences were identified.
The success rates of both Sanger and MiSeq sequencing were largely driven by plasma viral loads (pVLs), with neither method demonstrating a bias toward preferential amplification across pVL strata (Fig. 2A). Furthermore, the sequencing success rates among the cohorts were similar within each pVL stratum, suggesting that neither method preferentially amplified a particular subtype (see Fig. S1 in the supplemental material). The median pVL values for samples failing sequencing with the Sanger and MiSeq sequencing methods were 2.9 log10 copies/ml (IQR, 2.4 to 3.9 log10 copies/ml) and 2.7 log10 copies/ml (IQR, 2.4 to 3.7 log10 copies/ml), respectively. The median pVL for samples failing sequencing with both methods was 2.6 log10 copies/ml (IQR, 2.2 to 3.4 log10 copies/ml). Overall, sequencing by either Sanger or MiSeq sequencing was successful for >88% of samples with pVL values of >3.0 log10 copies/ml, with ∼85% being successfully sequenced by both methods. The duration of storage of the first-round PCR products at room temperature might have influenced MiSeq sequencing success rates. Success rates were marginally higher for samples stored for less than 6 months than for those stored for longer periods (94% versus 82% among Canadian samples with pVLs of >1,000 copies/ml).
FIG 2.

Success rates and concordance of Sanger and MiSeq sequencing methods, stratified by plasma viral loads. Overall, 881 samples (80%) and 892 samples (81%) were successfully sequenced by the Sanger and MiSeq methods, respectively, with 832 samples (75%) having sequences from both methods. (A) Sequencing success rates versus pVLs. Sequencing failure was driven largely by sample pVLs. Overall, 793 samples (88%) and 810 samples (90%) with pVLs of >3.0 log10 copies/ml were successfully sequenced by the Sanger and MiSeq sequencing methods, respectively. Numbers above the bars, total numbers of samples attempted in each category. (B) Sequencing concordance versus pVLs. Sequencing concordance was high across all pVL strata. Data points beyond 1.5 times the interquartile range from the box hinge are considered as outliers and were largely attributable to large numbers of mixed base calls in selected MiSeq sequences. Samples without viral load data (Unknown) also were successfully sequenced by both methods and yielded generally concordant results. Numbers above the boxes, total numbers of samples sequenced by both methods in each pVL category.
Sanger and MiSeq sequence concordance.
Overall, 832 clinical samples (75%) and 11 technical replicate samples (79%) were successfully sequenced by both Sanger and MiSeq sequencing. A total of 2,466 nucleotide mismatches were identified from 366,705 called bases in these samples (99.3% concordance) (Fig. 3). Sample mix-ups or cross-contaminations were not identified for samples with low (<98%) sequence concordance. The overwhelming majority (95.7%) of observed discordances were differences in mixture calls for which one method called a mixed base and the other called a component thereof. Neither method overcalled or undercalled mixtures relative to the other (Cohen's kappa = 0.72). It should be noted that use of the same first-round PCR products for both methods eliminates a source of potential random variation inherent in the RNA extraction and reverse transcription steps, due to random template sampling. Thus, the observed nucleotide differences between the methods are effectively limited to downstream PCR and sequencing errors. Comparisons of the laboratory clone pNL4-3 showed clonality for all sequences both within and between methods. As stated previously, the clinical isolate POS08 showed some variability between replicates among methods (∼98% concordance); however, high concordance between methods was observed when paired samples from the same first PCR product were compared (99.1% nucleotide concordance).
FIG 3.

Base calling differences for the Illumina MiSeq and ABI 3730xl Sanger sequencing methods. A change matrix displays the frequency of nucleotides detected by Sanger (rows) and MiSeq (columns) sequencing. Overall, 832 clinical samples and 11 technical replicate samples were successfully sequenced by both methods. A total of 2,466 mismatches were identified in 366,705 called bases in these samples. Concordant base calls are highlighted in green, partially discordant base calls (mixed bases detected by one method but not the other) are highlighted in yellow, and entirely discordant base calls are highlighted in red. Differences in mixture detection or calling accounted for >95% of all discordant bases. Rows for B, D, H, and V are not shown for Sanger base calls, as the RECall software does not call three-base mixtures.
MiSeq sequencing revealed that the PCR protocol was able to amplify successfully HIV RT from a variety of HIV subtypes. Phylogenetic analysis of MiSeq and HIV subtype reference sequences (downloaded from the Los Alamos HIV sequence database) displayed clustering by cohort (Fig. 4) consistent with the expected subtype prevalence, given the demographic characteristics of our cohort. Overall, Canadian samples were primarily subtype B (84%), with subtype C (12%) being the next most common. UARTO samples were primarily subtypes A (43%) and D (49%), with small numbers of recombinant sequences also being observed (see Table S3 in the supplemental material).
FIG 4.

Phylogenetic tree of samples successfully sequenced by MiSeq sequencing. A neighbor-joining tree constructed from MiSeq consensus sequences (n = 892 clinical samples) is depicted as a cladogram (with unscaled branch lengths). Mixed bases were called when minority bases exceeded 20% prevalence. Samples with <100-fold coverage were excluded. Sequences clustered by cohort and HIV subtype in agreement with expected prevalence rates. HIV-1 subtype consensus sequences (n = 16) (black tip labels) spanning RT codons 90 to 234 were included and represent subtypes A1, A2, B, C, D, F1, F2, G, and H, as well as recombinant viruses AE, AG, AB, BC, CD, BF, and BG (retrieved from http://www.hiv.lanl.gov). Canadian HIV RT sequences (blue tip labels) were primarily subtypes B and C, and UARTO HIV RT sequences (purple tip labels) were primarily subtypes D and A.
Resistance interpretations.
Drug resistance analysis of the 832 mutually successful clinical samples revealed that 155 Sanger and 156 MiSeq samples possessed one or more resistance mutations found on the 2013 IAS-USA list (see Table S4 in the supplemental material). MiSeq sequencing had 97.4% sensitivity and 99.3% specificity in detecting resistance mutations identified by Sanger sequencing. With the assumption that any one major drug resistance mutation confers a resistant phenotype, analysis of resistance by drug class revealed nearly identical interpretations with the two methods. Nucleotide reverse transcriptase inhibitor (NRTI) resistance mutations were detected in 62 samples (7.5%) and 63 samples (7.6%) sequenced by Sanger and MiSeq sequencing, respectively. Similarly, NNRTI resistance mutations were observed in 119 samples (14.3%) and 120 samples (14.4%) sequenced by Sanger and MiSeq sequencing, respectively. Sanger and MiSeq sequencing methods were 99.4% concordant in resistance interpretations for both NRTIs and NNRTIs. Given the relatively large depth of coverage obtained, it may be possible to identify resistance mutations with lower prevalences in the MiSeq data by lowering the threshold at which nucleotide mixtures are called. For example, 73 samples (8.8%) and 145 samples (17.4%) with NRTI and NNRTI mutations, respectively, at >5% frequency were identified (see Fig. S2 in the supplemental material); however, the appropriateness of the mixture-calling threshold and the clinical relevance of any low-frequency variants detected in this manner require further evaluation.
Sensitivity analysis of MiSeq coverage and mixture cutoff values determined a priori.
The suitability of the chosen minimum sequence coverage and mixture-calling thresholds was evaluated by recalculating the overall nucleotide concordance as these parameters were varied. Analysis of coverage and mixture cutoff parameters indicated that 100-fold minimum coverage and a 20% mixture threshold were suitable for obtaining reliable nucleotide concordance data (Fig. 5). Given our large sample size, lower coverage cutoff values appeared acceptable for obtaining dependable results. Nucleotide concordance began to decrease when minority bases were called at ≤15% or samples with <50-fold coverage were included. Sequencing failure rates increased as the minimum coverage threshold was increased; however, limited improvement in sequence concordance was observed. Although failure rates were substantially higher for samples with low pVL values, nucleotide concordance rates were comparable across pVL strata (Fig. 2B; see also Fig. S3 in the supplemental material). Receiver operating characteristic (ROC) curve analysis suggested that a minimum coverage cutoff value as low as 50 times would still yield acceptable sensitivity and specificity measurements for detection of resistance (see Table S5 in the supplemental material).
FIG 5.

Effects of MiSeq mixture-calling and minimum coverage thresholds on sequencing accuracy and success rates. (A) Raw nucleotide concordance between MiSeq and Sanger sequences derived from clinical samples versus minimum coverage. Concordance increased rapidly as the minimum coverage threshold was increased from 1-fold to 75-fold for all mixture-calling thresholds examined. Sequencing accuracy was highest at the 20% mixture cutoff value for all levels of coverage of >10 reads/sample. (B) Sequencing success versus minimum coverage. The number of successfully sequenced clinical samples decreased as the minimum coverage threshold was increased. Nucleotide concordance at the 20% mixture-calling threshold (A) and the number of samples successfully sequenced (B) are displayed for coverage cutoff values of 10, 100, 1,000, and 10,000 reads/sample.
DISCUSSION
The introduction of the large data-generating capacities of NGS platforms has markedly decreased the cost per base of DNA sequencing. At present, laboratories performing clinical HIV drug resistance genotyping do not generally benefit from these cost savings. Limited sample numbers, the requirement for rapid turnaround times, and a comparatively small genome means that the cost per sample of HIV resistance testing remains relatively unchanged.
The most common current application of NGS platforms in clinical HIV settings has been in deep sequencing of diverse virus populations, with the goal being to identify low-frequency variants that may influence treatment outcomes (20, 23–27). However, in settings in which large numbers of samples are routinely processed or requirements for turnaround times may be relaxed, it may be possible to perform cost-effective drug resistance genotyping by sequencing hundreds of samples in parallel. This high-multiplex wide sequencing would effectively spread the coverage conferred from deep sequencing across the samples. This concept has been proposed previously and partially evaluated with other platforms; however, it has not been demonstrated using such a large sample pool, and a systematic comparison of sequence concordance versus the current standard methodology (Sanger sequencing) has not been performed (29, 30).
In this proof-of-principle study, we demonstrated that high-multiplex sequencing with the Illumina MiSeq system could accurately replicate Sanger sequencing of HIV RT over a wide range of pVL inputs. For samples with >100-fold MiSeq coverage, consensus sequences (for which minority bases with >20% frequency were called as mixtures) were compared with Sanger sequences, and >99.3% nucleotide concordance was observed. The majority of discordances were due to differences in mixture calling; minimal incompatibilities were observed, and they did not substantially affect drug resistance interpretations. Amplification and sequencing success rates were comparable (∼80%) for the two methods, although it should be noted that these values likely represent underestimates of the success rates for the method implemented as a clinically validated assay, as PCR was not reattempted for failing samples. For example, the Sanger assay was able to amplify and to sequence successfully only ∼40% of samples with pVLs of <1,000 copies/ml in this experiment. In contrast, our laboratory typically achieves a >85% success rate for samples with low pVLs if samples that initially fail to produce a PCR product are retested with a backup protocol (34). Furthermore, the sequencing success rates for the internal controls (pNL4-3 and POS08) were comparable to those of the clinical samples for both the Sanger (86%) and MiSeq (93%) assays. Importantly, the MiSeq and Sanger methods were both able to amplify HIV RT from multiple subtypes. Taking into account the differences in the sizes of the amplicons generated (∼600 bp for UARTO samples and ∼1,700 bp for Canadian samples) and the pVL distributions in the cohorts, no obvious differences in success rates were observed with respect to the cohorts or HIV subtypes.
A previous study evaluated a pooled sequencing strategy for HIV drug resistance surveillance using the Roche 454 GS FLX system (30). The authors demonstrated that protease inhibitor resistance mutations and sequence polymorphisms, relative to the HXB2 reference sequence, observed by Sanger sequencing were also identified with the pooled sequencing approach. In addition, pooled pyrosequencing represented an estimated ∼35% costs savings, relative to Sanger sequencing, making it an attractive option for population resistance surveillance. However, as the individual samples in the pool were not uniquely barcoded, sequences from individual samples could not be distinguished, and thus no direct comparisons to Sanger sequencing could be performed on a per-sample basis. A similar study demonstrated that up to 48 samples could be multiplexed in a single Roche 454 GS Junior run, at a cost of approximately $20/sample; however, the accuracy relative to Sanger sequencing was not evaluated (29).
In contrast to the now-discontinued 454 system, this wide sequencing approach offers several advantages. First, the higher multiplexing density and the use of the MiSeq system rather than the 454 system result in further reductions in the cost of sequencing, down to approximately $1/sample. More importantly, unique barcoding of samples allows sequences from individual samples to be recovered, which allows individual-level clinical decisions to be made from pooled sequencing data. The relatively large depth of coverage (>9,900-fold) obtained for the majority of samples tested suggests that minority variant detection using the wide sequencing approach is possible, as the level of coverage achieved is comparable to that presented in earlier studies of HIV resistance testing by deep sequencing (20, 21). However, the clinical relevance of any minority variants identified by this method requires further investigation. Finally, increases in the availability of viral load testing in LMIC may offer the potential for even greater cost savings. In this study, RNA extraction was performed with two automated nucleic acid extractors that are used in the preparation of samples for pVL testing (the NucliSens easyMAG and Abbott m2000sp systems). With both instruments, more RNA is eluted than is required for the pVL assay, and this excess material is recoverable. Therefore, we envision a strategy in which laboratories performing HIV pVL testing could use this excess RNA to perform the initial RT PCR described here. The resulting heat-stable nonbiohazardous material could be shipped by regular mail to a centralized sequencing facility, where the nested PCR and MiSeq sequencing would be performed. Such a strategy would eliminate many of the logistical barriers to performing genotypic resistance testing in those settings. Therefore, wide sequencing also presents an opportunity to pursue a paradigm complementary to expanded testing from dried blood spots (41–43). Furthermore, the marginal cost increase of the RT PCR for the pVL testing laboratory (estimated as approximately $5/sample) is offset by the elimination of several costly sample-processing steps that are currently duplicated in pVL and drug resistance testing procedures (e.g., sample collection, accessioning, hazardous goods shipping, and RNA extraction). Thus, such an integrated strategy using centralized sequencing facilities (rather than individual local or regional laboratories) could represent a net cost savings for the health care system.
Several limitations to this wide sequencing approach should be addressed to validate this strategy. First, the requirements for a third (indexing) PCR and a bead-based normalization step introduced an incremental cost increase for MiSeq sample preparation (estimated as approximately $2/sample). It should be noted, however, that most of this cost could be eliminated by using barcoded primers in the second PCR, thus avoiding the indexing PCR step entirely. Most importantly, this proof-of-principle study evaluated only a small portion of HIV RT. All of the major NNRTI-associated resistance mutations on the IAS-USA 2013 list (40) were covered by the sequenced amplicon; however, important NRTI resistance mutations (notably, K65R), as well as all of the protease, were not. It would be possible to construct a longer amplicon covering RT codon 65, which could be sequenced using 2x300bp Illumina MiSeq kits. A second amplicon covering the protease could be prepared and sequenced in the same run. The sequence coverage of each sample would effectively be halved if the same multiplexing density was maintained, although we have demonstrated that this would have a minimal impact on sequencing accuracy. Sequencing additional amplicons, however, would marginally increase the preparation costs (only one additional second PCR would be required) and would create additional complexity.
Second, managing sample preparation for hundreds of samples in parallel is complicated, compared to conventional Sanger sequencing, and could increase the likelihood of sample mix-ups or cross-contaminations. For this reason, automated pipetting instruments are suggested, especially for the index PCR stage. However, the observation that MiSeq consensus sequences are highly concordant with those generated by Sanger sequencing may allow the existing quality control tools designed for Sanger sequencing to be repurposed for deep or wide sequencing approaches (35, 44).
Finally, consensus sequence generation and minority variant detection by wide sequencing require stringent bioinformatic pipelines for accurate analyses. Such tools might include optimization of mixture and depth-of-coverage cutoff parameters or further inclusion/exclusion criteria based on the number of observed mixtures per sample. The effect of detecting minority drug resistance variants on treatment outcomes is less clear. Briefly, protocols for population Sanger sequencing and minority variant detection have been established; if a mixture is detected above the threshold of about 20%, then it is considered clinically important (45). No definitive methods for NGS and minority variant detection in clinical settings have been established. These NGS tools need to be established and validated before this strategy is adapted to clinical practice. Unfortunately, the nature of the samples selected for this study did not allow validation analyses to be performed here. Clinical outcome analyses for this study were limited, in that only a few patients failed antiretroviral therapy and thus the significance of minority species could not be elucidated. Canadian samples represented an arbitrary cross-section of contemporaneous samples from treated patients, with limited subsequent follow-up monitoring. Treatment outcome data and MiSeq sequences were available for 212 antiretroviral-naive participants from the UARTO cohort for whom NNRTI-based HAART was initiated. However, we lacked the power to detect any meaningful association of minority resistant variants with virological outcomes, as over 95% of these individuals maintained pVLs below 50 RNA copies/ml over the course of follow-up monitoring.
Drug resistance testing provides physicians with clinically important information, enabling treatment decisions to be made on an individual basis while facilitating surveillance of HIV resistance transmission on a population level (9). In LMIC, access to HIV drug resistance testing continues to be limited. This has substantial clinical and epidemiological implications; patients often remain on suboptimal ART regimens, which enables the transmission of drug-resistant variants (10, 11). In this study, HIV drug resistance testing was attempted with 1,143 samples, representing approximately 20% of Canada's annual HIV drug resistance testing burden, in a single MiSeq run. HIV RT from multiple subtypes was successfully and accurately sequenced, which suggests that routine individual testing and/or annual population resistance surveillance in LMIC could be performed with this strategy. The potential to use surplus RNA from pVL testing presents the opportunity to further reduce costs and to reduce some of the logistical difficulties associated with resistance testing in LMIC.
Supplementary Material
ACKNOWLEDGMENTS
This study was funded by a Large-Scale Applied Research Project grant from Genome Canada to P.R.H. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We have no conflicts of interest to declare.
The work in this study was carried out through collaboration among all authors. We would also like to acknowledge the contribution of Peter Hunt to this study, namely, coordinating access to samples and data from the UARTO cohort. The wide sequencing strategy was designed by W.D., P.R.H., and C.J.B. D.R.B., J.N.M., A.R.M., and Y.B. provided UARTO samples and clinical data. BCCfE clinical staff members performed first-round PCR and Sanger sequencing for Canadian samples. H.R.L., G.Q.L., and A.K. performed first-round PCR and Sanger sequencing of UARTO samples. Laboratory work for MiSeq sequencing was done by H.R.L. The bioinformatic pipeline for MiSeq data processing was created by D.K. and A.F.Y.P. Data analysis was completed by H.R.L. and C.J.B. H.R.L. and C.J.B. wrote the manuscript, with contributions, review, and approval from all coauthors.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01490-15.
REFERENCES
- 1.Lima VD, Hogg RS, Harrigan PR, Moore D, Yip B, Wood E, Montaner JSG. 2007. Continued improvement in survival among HIV-infected individuals with newer forms of highly active antiretroviral therapy. AIDS 21:685–692. doi: 10.1097/QAD.0b013e32802ef30c. [DOI] [PubMed] [Google Scholar]
- 2.Samji H, Cescon A, Hogg RS, Modur SP, Althoff KN, Buchacz K, Burchell AN, Cohen M, Gebo KA, Gill MJ, Justice A, Kirk G, Klein MB, Korthuis PT, Martin J, Napravnik S, Rourke SB, Sterling TR, Silverberg MJ, Deeks S, Jacobson LP, Bosch RJ, Kitahata MM, Goedert JJ, Moore R, Gange SJ. 2013. Closing the gap: increases in life expectancy among treated HIV-positive individuals in the United States and Canada. PLoS One 8:e81355. doi: 10.1371/journal.pone.0081355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Montaner JSG, Hogg R, Wood E, Kerr T, Tyndall M, Levy AR, Harrigan PR. 2006. The case for expanding access to highly active antiretroviral therapy to curb the growth of the HIV epidemic. Lancet 368:531–536. doi: 10.1016/S0140-6736(06)69162-9. [DOI] [PubMed] [Google Scholar]
- 4.Montaner JSG, Lima VD, Barrios R, Yip B, Wood E, Kerr T, Shannon K, Harrigan PR, Hogg RS, Daly P, Kendall P. 2010. Association of highly active antiretroviral therapy coverage, population viral load, and yearly new HIV diagnoses in British Columbia, Canada: a population-based study. Lancet 376:532–539. doi: 10.1016/S0140-6736(10)60936-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Montaner JSG, Lima VD, Harrigan PR, Lourenço L, Yip B, Nosyk B, Wood E, Kerr T, Shannon K, Moore D, Hogg RS, Barrios R, Gilbert M, Krajden M, Gustafson R, Daly P, Kendall P. 2014. Expansion of HAART coverage is associated with sustained decreases in HIV/AIDS morbidity, mortality and HIV transmission: the “HIV Treatment as Prevention” experience in a Canadian setting. PLoS One 9:e87872. doi: 10.1371/journal.pone.0087872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen MS, Chen YQ, McCauley M, Gamble T, Hosseinipour MC, Kumarasamy N, Hakim JG, Kumwenda J, Grinsztejn B, Pilotto JHS, Godbole SV, Mehendale S, Chariyalertsak S, Santos BR, Mayer KH, Hoffman IF, Eshleman SH, Piwowar-Manning E, Wang L, Makhema J, Mills LA, de Bruyn G, Sanne I, Eron J, Gallant J, Havlir D, Swindells S, Ribaudo H, Elharrar V, Burns D, Taha TE, Nielsen-Saines K, Celentano D, Essex M, Fleming TR. 2011. Prevention of HIV-1 infection with early antiretroviral therapy. N Engl J Med 365:493–505. doi: 10.1056/NEJMoa1105243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baxter JD, Mayers DL, Wentworth DN, Neaton JD, Hoover ML, Winters MA, Mannheimer SB, Thompson MA, Abrams DI, Brizz BJ, Ioannidis JP, Merigan TC, CPCRA 046 Study Team for the Terry Beirn Community Programs for Clinical Research on AIDS. 2000. A randomized study of antiretroviral management based on plasma genotypic antiretroviral resistance testing in patients failing therapy. AIDS 14:F83–F93. doi: 10.1097/00002030-200006160-00001. [DOI] [PubMed] [Google Scholar]
- 8.DeGruttola V, Dix L, D'Aquila R, Holder D, Phillips A, Ait-Khaled M, Baxter J, Clevenbergh P, Hammer S, Harrigan R, Katzenstein D, Lanier R, Miller M, Para M, Yerly S, Zolopa A, Murray J, Patick A, Miller V, Castillo S, Pedneault L, Mellors J. 2000. The relation between baseline HIV drug resistance and response to antiretroviral therapy: re-analysis of retrospective and prospective studies using a standardized data analysis plan. Antivir Ther 5:41–48. [DOI] [PubMed] [Google Scholar]
- 9.Bennett DE, Bertagnolio S, Sutherland D, Gilks CF. 2008. The World Health Organization's global strategy for prevention and assessment of HIV drug resistance. Antivir Ther 13(Suppl 2):1–13. [PubMed] [Google Scholar]
- 10.Gupta RK, Jordan MR, Sultan BJ, Hill A, Davis DHJ, Gregson J, Sawyer AW, Hamers RL, Ndembi N, Pillay D, Bertagnolio S. 2012. Global trends in antiretroviral resistance in treatment-naïve individuals with HIV after rollout of antiretroviral treatment in resource-limited settings: a global collaborative study and meta-regression analysis. Lancet 380:1250–1258. doi: 10.1016/S0140-6736(12)61038-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamers RL, Wallis CL, Kityo C, Siwale M, Mandaliya K, Conradie F, Botes ME, Wellington M, Osibogun A, Sigaloff KCE, Nankya I, Schuurman R, Wit FW, Stevens WS, van Vugt M, de Wit TFR. 2011. HIV-1 drug resistance in antiretroviral-naive individuals in sub-Saharan Africa after rollout of antiretroviral therapy: a multicentre observational study. Lancet Infect Dis 11:750–759. doi: 10.1016/S1473-3099(11)70149-9. [DOI] [PubMed] [Google Scholar]
- 12.Gill VS, Lima VD, Zhang W, Wynhoven B, Yip B, Hogg RS, Montaner JSG, Harrigan PR. 2010. Improved virological outcomes in British Columbia concomitant with decreasing incidence of HIV type 1 drug resistance detection. Clin Infect Dis 50:98–105. doi: 10.1086/648729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rhee S-Y, Blanco JL, Jordan MR, Taylor J, Lemey P, Varghese V, Hamers RL, Bertagnolio S, de Wit TFR, Aghokeng AF, Albert J, Avi R, Avila-Rios S, Bessong PO, Brooks JI, Boucher CAB, Brumme ZL, Busch MP, Bussmann H, Chaix M-L, Chin BS, D'Aquin TT, De Gascun CF, Derache A, Descamps D, Deshpande AK, Djoko CF, Eshleman SH, Fleury H, Frange P, Fujisaki S, Harrigan PR, Hattori J, Holguin A, Hunt GM, Ichimura H, Kaleebu P, Katzenstein D, Kiertiburanakul S, Kim JH, Kim SS, Li Y, Lutsar I, Morris L, Ndembi N, Ng KP, Paranjape RS, Peeters M, Poljak M, Price MA, Ragonnet-Cronin ML, Reyes-Terán G, Rolland M, Sirivichayakul S, Smith DM, Soares MA, Soriano VV, Ssemwanga D, Stanojevic M, Stefani MA, Sugiura W, Sungkanuparph S, Tanuri A, Tee KK, Truong H-HM, van de Vijver DAMC, Vidal N, Yang C, Yang R, Yebra G, Ioannidis JPA, Vandamme A-M, Shafer RW. 2015. Geographic and temporal trends in the molecular epidemiology and genetic mechanisms of transmitted HIV-1 drug resistance: an individual-patient- and sequence-level meta-analysis. PLoS Med 12:e1001810. doi: 10.1371/journal.pmed.1001810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu TF, Shafer RW. 2006. Web resources for HIV type 1 genotypic-resistance test interpretation. Clin Infect Dis 42:1608–1618. doi: 10.1086/503914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vermeiren H, Van Craenenbroeck E, Alen P, Bacheler L, Picchio G, Lecocq P. 2007. Prediction of HIV-1 drug susceptibility phenotype from the viral genotype using linear regression modeling. J Virol Methods 145:47–55. doi: 10.1016/j.jviromet.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 16.Cunningham S, Ank B, Lewis D, Lu W, Wantman M, Dileanis J, Jackson JB, Palumbo P, Krogstad P, Eshleman SH. 2001. Performance of the Applied Biosystems ViroSeq human immunodeficiency virus type 1 (HIV-1) genotyping system for sequence-based analysis of HIV-1 in pediatric plasma samples. J Clin Microbiol 39:1254–1257. doi: 10.1128/JCM.39.4.1254-1257.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schuurman R, Brambilla D, de Groot T, Huang D, Land S, Bremer J, Benders I, Boucher CAB. 2002. Underestimation of HIV type 1 drug resistance mutations: results from the ENVA-2 genotyping proficiency program. AIDS Res Hum Retroviruses 18:243–248. doi: 10.1089/088922202753472801. [DOI] [PubMed] [Google Scholar]
- 18.Korn K, Reil H, Walter H, Schmidt B. 2003. Quality control trial for human immunodeficiency virus type 1 drug resistance testing using clinical samples reveals problems with detecting minority species and interpretation of test results. J Clin Microbiol 41:3559–3565. doi: 10.1128/JCM.41.8.3559-3565.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Church JD, Jones D, Flys T, Hoover D, Marlowe N, Chen S, Shi C, Eshleman JR, Guay LA, Jackson JB, Kumwenda N, Taha TE, Eshleman SH. 2006. Sensitivity of the ViroSeq HIV-1 genotyping system for detection of the K103N resistance mutation in HIV-1 subtypes A, C, and D. J Mol Diagn 8:430–432. doi: 10.2353/jmoldx.2006.050148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simen BB, Simons JF, Hullsiek KH, Novak RM, Macarthur RD, Baxter JD, Huang C, Lubeski C, Turenchalk GS, Braverman MS, Desany B, Rothberg JM, Egholm M, Kozal MJ. 2009. Low-abundance drug-resistant viral variants in chronically HIV-infected, antiretroviral treatment-naive patients significantly impact treatment outcomes. J Infect Dis 199:693–701. doi: 10.1086/596736. [DOI] [PubMed] [Google Scholar]
- 21.Ram D, Leshkowitz D, Gonzalez D, Forer R, Levy I, Chowers M, Lorber M, Hindiyeh M, Mendelson E, Mor O. 2015. Evaluation of GS Junior and MiSeq next-generation sequencing technologies as an alternative to Trugene population sequencing in the clinical HIV laboratory. J Virol Methods 212:12–16. doi: 10.1016/j.jviromet.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 22.Barzon L, Lavezzo E, Militello V, Toppo S, Palù G. 2011. Applications of next-generation sequencing technologies to diagnostic virology. Int J Mol Sci 12:7861–7884. doi: 10.3390/ijms12117861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson JA, Li JF, Wei X, Lipscomb J, Irlbeck D, Craig C, Smith A, Bennett DE, Monsour M, Sandstrom P, Lanier ER, Heneine W. 2008. Minority HIV-1 drug resistance mutations are present in antiretroviral treatment-naïve populations and associate with reduced treatment efficacy. PLoS Med 5:e158. doi: 10.1371/journal.pmed.0050158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson JA, Geretti AM. 2010. Low-frequency HIV-1 drug resistance mutations can be clinically significant but must be interpreted with caution. J Antimicrob Chemother 65:1322–1326. doi: 10.1093/jac/dkq139. [DOI] [PubMed] [Google Scholar]
- 25.Paredes R, Lalama CM, Ribaudo HJ, Schackman BR, Shikuma C, Giguel F, Meyer WA, Johnson VA, Fiscus SA, D'Aquila RT, Gulick RM, Kuritzkes DR. 2010. Pre-existing minority drug-resistant HIV-1 variants, adherence, and risk of antiretroviral treatment failure. J Infect Dis 201:662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li JZ, Kuritzkes DR. 2013. Clinical implications of HIV-1 minority variants. Clin Infect Dis 56:1667–1674. doi: 10.1093/cid/cit125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li JZ. 2014. HIV-1 drug-resistant minority variants: sweating the small stuff. J Infect Dis 209:639–641. doi: 10.1093/infdis/jit656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ekici H, Rao SD, Sönnerborg A, Ramprasad VL, Gupta R, Neogi U. 2014. Cost-efficient HIV-1 drug resistance surveillance using multiplexed high-throughput amplicon sequencing: implications for use in low- and middle-income countries. J Antimicrob Chemother 69:3349–3355. doi: 10.1093/jac/dku278. [DOI] [PubMed] [Google Scholar]
- 29.Dudley DM, Chin EN, Bimber BN, Sanabani SS, Tarosso LF, Costa PR, Sauer MM, Kallas EG, O'Connor DH. 2012. Low-cost ultra-wide genotyping using Roche/454 pyrosequencing for surveillance of HIV drug resistance. PLoS One 7:e36494. doi: 10.1371/journal.pone.0036494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ji H, Massé N, Tyler S, Liang B, Li Y, Merks H, Graham M, Sandstrom P, Brooks J. 2010. HIV drug resistance surveillance using pooled pyrosequencing. PLoS One 5:e9263. doi: 10.1371/journal.pone.0009263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gall A, Ferns B, Morris C, Watson S, Cotten M, Robinson M, Berry N, Pillay D, Kellam P. 2012. Universal amplification, next-generation sequencing, and assembly of HIV-1 genomes. J Clin Microbiol 50:3838–3844. doi: 10.1128/JCM.01516-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee GQ, Bangsberg DR, Muzoora C, Boum Y, Oyugi JH, Emenyonu N, Bennett J, Hunt PW, Knapp D, Brumme CJ, Harrigan PR, Martin JN. 2014. Prevalence and virologic consequences of transmitted HIV-1 drug resistance in Uganda. AIDS Res Hum Retroviruses 30:896–906. doi: 10.1089/aid.2014.0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alexander CS, Dong W, Chan K, Jahnke N, O'Shaughnessy MV, Mo T, Piaseczny MA, Montaner JS, Harrigan PR. 2001. HIV protease and reverse transcriptase variation and therapy outcome in antiretroviral-naive individuals from a large North American cohort. AIDS 15:601–607. doi: 10.1097/00002030-200103300-00009. [DOI] [PubMed] [Google Scholar]
- 34.Gonzalez-Serna A, Min JE, Woods C, Chan D, Lima VD, Montaner JSG, Harrigan PR, Swenson LC. 2014. Performance of HIV-1 drug resistance testing at low-level viremia and its ability to predict future virologic outcomes and viral evolution in treatment-naive individuals. Clin Infect Dis 58:1165–1173. doi: 10.1093/cid/ciu019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woods CK, Brumme CJ, Liu TF, Chui CKS, Chu AL, Wynhoven B, Hall TA, Trevino C, Shafer RW, Harrigan PR. 2012. Automating HIV drug resistance genotyping with RECall, a freely accessible sequence analysis tool. J Clin Microbiol 50:1936–1942. doi: 10.1128/JCM.06689-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. 2013. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siepel AC, Halpern AL, Macken C, Korber BT. 1995. A computer program designed to screen rapidly for HIV type 1 intersubtype recombinant sequences. AIDS Res Hum Retroviruses 11:1413–1416. doi: 10.1089/aid.1995.11.1413. [DOI] [PubMed] [Google Scholar]
- 40.Johnson VA, Calvez V, Gunthard HF, Paredes R, Pillay D, Shafer RW, Wensing AM, Richman DD. 2013. Update of the drug resistance mutations in HIV-1: March 2013. Top Antivir Med 21:6–14. [PMC free article] [PubMed] [Google Scholar]
- 41.Bertagnolio S, Soto-Ramirez L, Pilon R, Rodriguez R, Viveros M, Fuentes L, Harrigan PR, Mo T, Sutherland D, Sandstrom P. 2007. HIV-1 drug resistance surveillance using dried whole blood spots. Antivir Ther 12:107–113. [PubMed] [Google Scholar]
- 42.Hamers RL, Smit PW, Stevens W, Schuurman R, Rinke de Wit TF. 2009. Dried fluid spots for HIV type-1 viral load and resistance genotyping: a systematic review. Antivir Ther 14:619–629. [PubMed] [Google Scholar]
- 43.Steegen K, Luchters S, Demecheleer E, Dauwe K, Mandaliya K, Jaoko W, Plum J, Temmerman M, Verhofstede C. 2007. Feasibility of detecting human immunodeficiency virus type 1 drug resistance in DNA extracted from whole blood or dried blood spots. J Clin Microbiol 45:3342–3351. doi: 10.1128/JCM.00814-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DeLong AK, Wu M, Bennett D, Parkin N, Wu Z, Hogan JW, Kantor R. 2012. Sequence quality analysis tool for HIV type 1 protease and reverse transcriptase. AIDS Res Hum Retroviruses 28:894–901. doi: 10.1089/aid.2011.0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Larder BA, Kohli A, Kellam P, Kemp SD, Kronick M, Henfrey RD. 1993. Quantitative detection of HIV-1 drug resistance mutations by automated DNA sequencing. Nature 365:671–673. doi: 10.1038/365671a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.