

Abstract
Over the last 10 years, Mycobacterium abscessus group strains have emerged as important human pathogens, which are associated with significantly higher fatality rates than any other rapidly growing mycobacteria. These opportunistic pathogens are widespread in the environment and can cause a wide range of clinical diseases, including skin, soft tissue, central nervous system, and disseminated infections; by far, the most difficult to treat is the pulmonary form. Infections with M. abscessus are often multidrug-resistant (MDR) and require prolonged treatment with various regimens and, many times, result in high mortality despite maximal therapy. We report here the evaluation of diverse mouse infection models for their ability to produce a progressive high level of infection with M. abscessus. The nude (nu/nu), SCID (severe combined immunodeficiency), gamma interferon knockout (GKO), and granulocyte-macrophage colony-stimulating factor (GMCSF) knockout mice fulfilled the criteria for an optimal model for compound screening. Thus, we set out to assess the antimycobacterial activity of clarithromycin, clofazimine, bedaquiline, and clofazimine-bedaquiline combinations against M. abscessus-infected GKO and SCID murine infection models. Treatment of GKO and SCID mice with a combination of clofazimine and bedaquiline was the most effective in decreasing the M. abscessus organ burden.
INTRODUCTION
Over the last 10 years, Mycobacterium abscessus group strains (including M. abscessus subsp. abscessus and massiliense) have emerged as important human pathogens and are associated with significantly higher fatality rates than any other rapidly growing mycobacteria (RGM) (1, 2). It is becoming increasingly clear that the incidence of RGM species may be increasing. This increase exceeds that which can be explained due to improved recognition, typing, and diagnosis (3). Recent reports suggest that, in many areas of the United States, the prevalence of nontuberculosis mycobacteria (NTM) pulmonary disease exceeds that of tuberculosis (4).
NTM are ubiquitous in the environment and are responsible for colonization, infection, and pseudo-outbreaks in health care settings throughout the world (5, 6). RGM cause serious, life-threatening chronic lung disease and are responsible for disseminated and often fatal infections (6, 7). Infections are typically caused by contaminated materials and invasive procedures involving catheters, nonsterile surgical procedures, or injections and implantations of foreign bodies. Exposure risks linked to shower heads and Jacuzzi baths have also been commonly reported (8). NTM typically cause opportunistic infections in patients with pulmonary diseases, such as interferon-gamma (IFN-γ)–interleukin-12 (IL-12) axis deficits, chronic obstructive disease, and bronchiectasis, as well as in immunocompetent postmenopausal Caucasian women and those with cystic fibrosis (CF) (7–9). M. abscessus infection in CF patients is particularly problematic, as it results in enhanced pulmonary destruction and is often impossible to treat, with failure rates as high as 60% despite maximal therapy (9).
RGM infections provide many obstacles to establishing effective antibiotic therapy, including a hydrophobic cell wall that can potentially protect the organism against lipophilic antibiotics. Many are oligotrophic and require a minimal two carbon sources and limited access to metal ions, allowing for survival and persistence in the environment (10). One important issue regarding RGM is antibiotic inactivation with multiple antibiotic resistance mechanisms, including beta-lactamases, aminoglycoside phosphotransferases, and aminoglycoside acetyltransferases (11, 12). Additional impediments include erythromycin ribosomal methylase (erm) genes found in almost all M. abscessus subsp. abscessus isolates, which result in methylation of the 23S rRNA, rendering the bacteria resistant against macrolides—a mainstay of RGM treatment regimens (6, 11, 12).
As an additional impediment, in vitro susceptibility testing does not always correlate with in vivo clinical outcomes in NTM patients (13). NTM exhibit a significant ability to form biofilms in the environment and hypothetically in vivo (14), critically complicating in vitro susceptibility testing. Testing methods may, therefore, represent a true measure of a compound's activity only against planktonic bacilli and not against biofilms (13–15). As a result of these multiple resistance mechanisms paired with incorrect MICs, very few antibiotics have good in vivo activity against RGM (13–16).
It has been challenging to develop an animal model for screening compounds against RGM, such as M. abscessus, because of their opportunistic nature and relative avirulence. Prior studies have revealed that most immunocompetent models, which serve as excellent models for Mycobacterium tuberculosis, such as C57BL/6 mice, guinea pigs, and an obese mouse model (leptin deficient [OBOB]), when infected with M. abscessus are rapidly cleared by host immunity (17). In spite of this, we have continued to evaluate diverse mouse infection models to assess the progression of the infection, granuloma formation, and therapeutic activity against M. abscessus.
Our studies focused primarily around the evaluation of beige, Nos2−/−, Cybb−/−, TNFR−/−, C3HeB/FeJ, GKO−/−, and MyD88−/− mice, which have been shown to lack major innate and acquired antimycobacterial protective immunity (18). Upon mycobacterial alveolar cell infection, innate immunity is induced by an adaptor protein (MyD88), used by almost all Toll-like receptors to activate NF-κB, and releases inflammatory mediators (19). Mice with M. abscessus infection that have the MyD88 gene knocked out and a loss of NF-κB activation were shown to be still capable of bacterial clearance. Additionally, Cybb knockout mice, a mouse model of chronic granulomatous disease (CGD), have a recessive disorder characterized by a defective phagocyte respiratory burst oxidase required for mycobacterial intracellular killing, but the lack of this oxidase does not allow for RGM survival (20). Moreover, targeting more essential genes for the cytokines required for clearance of M. tuberculosis and even Mycobacterium avium, such as the tumor necrosis factor receptor (TNFR) and IFN-γ, two cytokines essential for acquired immune removal of more virulent pathogens when knocked out, allowed for early RGM persistence but eventually resulted in clearance (21). The beige model has a dominant Th2 immunity that allows for M. avium growth (22); however, RGM are also removed by this model. Lastly, futile attempts to infect M. abscessus in the nitric oxide synthase 2 (NOS2) knockout required for intracellular bacterial killing and the C3HeB/FeJ mice (also known as the Kramnik mouse model, which develop necrotic granulomas upon M. tuberculosis infection) resulted in M. abscessus elimination (23).
Thus, we set out to assess the antimycobacterial activity of clarithromycin, clofazimine, bedaquiline, and a clofazimine-bedaquiline combination against M. abscessus in mouse models that fulfilled the criteria of developing high bacterial levels of infection and developing necrotizing granulomas. The GKO and SCID models proved to be an acceptable preclinical model for acute antimycobacterial drug treatment. In the GKO and SCID mouse models infected with M. abscessus, the clofazimine-bedaquiline combination proved to be the best treatment regimen evaluated.
MATERIALS AND METHODS
Mice.
Six- to eight-week-old, specific-pathogen-free female SCID/beige mice (C.B-Igh-1b/GbmsTac-Prkdcscid-Lystbg N7), referred to here as SCID, were purchased from Taconic Laboratories (Hudson, NY). Beige, Nos2 knockout, Cybb knockout, TNFR knockout, C3HeB/FeJ, MyD88 knockout, nude, granulocyte-macrophage colony-stimulating factor (GMCSF) knockout, and IFN-γ knockout (GKO) mice were obtained from Jackson Laboratories (Bar Harbor, ME). Mice were maintained in the biosafety level 3 facilities at Colorado State University and were given sterile water, chow, bedding, and enrichment for the duration of the experiments. Sentinel animals were routinely tested to determine the specific-pathogen-free nature of the mouse colonies. All experimental protocols were approved by the Animal Care and Use Committee of Colorado State University.
Animal infection.
Mice were infected with a tail vein injection of 100 μl containing 1 × 106 CFU of M. abscessus 103 with a rough colony morphology and positive for biofilm formation (a gift from Mary Jackson, Colorado State University). The M. abscessus 103 inoculum was prepared by thawing the bacterial vial, sonicating for 10 to 15 s, and vortexing to remove any clumps that formed during freezing. Thereafter, the mycobacterial suspension was obtained from the vial with a 1-ml tuberculin syringe fitted with a 26.5-gauge needle and expelled back into the vial. This procedure was repeated back and forth into the vial 20 times without removing the needle to mix the suspension and break up any small clumps of bacilli. The following day, three mice were euthanized and their lungs, spleens, and livers were harvested to determine the bacterial burden baseline. Organs were homogenized in phosphate-buffered saline (PBS), and serial dilutions were plated on nutrient 7H11 agar and Mueller-Hinton agar for 1 week at 30°C, when CFU were enumerated.
MICs.
The MICs for the test compounds against M. abscessus 103 are shown in Table 1. Briefly, the MIC testing was done in duplicate by broth microdilution. M. abscessus 103 isolates were grown in a 96-well plate, and the expected absorbance after 2 to 3 days was (optical density [OD]) 0.08 to 0.1 (0.5 McFarland standard). Compounds were stored as 1.28 mg/ml stocks in dimethyl sulfoxide (DMSO), and a test range of 64 to 0.06 μg/ml was utilized and assayed for ODs on day 7 using the alamarBlue method as described and recommended by the Clinical and Laboratory Standards Institute (24). The MIC is the lowest drug concentration that inhibits 90% (MIC90) of the M. abscessus isolates. Minimum bactericidal concentrations (MBCs) were assayed by plating the MIC well-diluted contents on 7H11 and Mueller-Hinton agar plates (0 to 7) in quadruplicate (4 plates per well), agar plates were incubated in the incubator, and the number of CFU was calculated. The MBC was defined as the lowest drug concentration that killed ≤99.99% of the initial population.
TABLE 1.
MICs for test compounds against M. abscessus 103
| Drug | MIC (μg/ml) |
|---|---|
| CLR | 0.75 (0.125) ± 0.02 |
| AMI | 24 ± 0.01 |
| CIP | 0.25 ± 0.02 |
| BDQ | 1 ± 0.01 |
| CLF | 1.3 ± 0.3 |
CLR, clarithromycin; AMI, amikacin; CIP, ciprofloxacin; CLF, clofazimine; BDQ, bedaquiline. MICs are averages ± standard errors from testing done in duplicate.
Model development for M. abscessus.
Mice (beige, Nos2 knockout, Cybb knockout, TNFR knockout, C3HeB/FeJ, GKO knockout, MyD88 knockout, SCID, nude, and GMCSF knockout [41]) were infected with a tail vein injection of 100 μl containing 1 × 106 CFU of M. abscessus 103 from a cystic fibrosis patient (a gift from Mary Jackson, Colorado State University). The following day, three mice were euthanized and whole organs (lungs, spleens, and livers) were harvested to determine baseline bacterial burden. Organs were homogenized in PBS, and serial dilutions were plated on nutrient 7H11 agar and Mueller-Hinton agar plates for 1 week at 30°C. Then, CFU were enumerated. Each mouse model experiment was completed in triplicate.
Acute experimental antibiotic treatment.
The initial trial was conducted in GKO−/− mice. Mice were intravenously infected with 1 × 106 CFU of M. abscessus 103. Starting at day 2 after infection, mice were dosed with an antibiotic for nine consecutive days via oral gavage of 200 μl. The trials was conducted with the following experimental groups by gavage: the control (saline), clarithromycin at 250 mg/kg of body weight diluted in 0.5% methyl cellulose, clofazimine at 20 mg/kg diluted in 0.05% agarose, bedaquiline at 30 mg/kg diluted in 20% 2-hydroxypropyl-beta-cyclodextrin, bedaquiline at 30 mg/kg and clofazimine at 20 mg/kg, ciprofloxacin at 100 mg/kg, and amikacin at 150 mg/kg daily by subcutaneous injection. Bedaquiline was kindly provided by Johnson and Johnson, whereas the other reagents were obtained from Sigma-Aldrich (St. Louis, MO). For the group receiving clofazimine and bedaquiline, each antibiotic was administered separately and interspersed by at least 6 h to avoid any interference. At day 10 after infection (including 8 days of antibiotic treatment), antibiotic administration was terminated. The group receiving clarithromycin was euthanized at day 11 in order to allow antibiotic clearance during a 24-h period. For the groups receiving clofazimine, bedaquiline or the combination of these drugs, mice were euthanized 5 days after antibiotic cessation in order to allow sufficient time for antibiotic clearance. Experimental groups of mouse lungs, spleens, and livers were harvested from the control (saline) or one of the antibiotic treatment groups. To minimize antibiotic carry-over, organs were homogenized in 10% bovine serum albumin (BSA; Sigma) instead of PBS. Antibiotics such as bedaquiline and clofazimine are known to bind strongly to proteins such as BSA; thus, its inclusion in the homogenizing buffer is a way to remove the drugs (25). Finally, homogenates were plated on nutrient 7H11 agar and Mueller-Hinton agar plates containing 0.4% charcoal in order to chelate any remaining antibiotics. Plates were incubated for 14 days at 32°C, and then CFU were enumerated. Results shown in this study are representative of two experiments using GKO−/− and SCID mice. In addition, initial studies were conducted to evaluate the potential for carryover in our organ homogenates as previously described (26). The results of these initial studies using 10% BSA and 0.4% charcoal plates with various dilutions of drug-treated tissue homogenates confirmed a lack of carryover.
The subsequent trials were conducted in SCID mice. Mice were intravenously infected with 1 × 106 CFU of M. abscessus 103. Starting at day 2 after infection, mice were dosed for nine consecutive days with 200 μl of each antibiotic via oral gavage. The trial was conducted with the following groups by gavage: the control (saline), clarithromycin at 250 mg/kg diluted in 0.5% methyl cellulose, clofazimine at 20 mg/kg diluted in 0.05% agarose, bedaquiline at 30 mg/kg diluted in 20% 2-hydroxypropyl-beta-cyclodextrin, and bedaquiline at 30 mg/kg and clofazimine 20 mg/kg. The experimental group outline was the same as described for the GKO−/− study.
Histology.
Organs were perfused with 4% formaldehyde. Paraffin-embedded tissues were stained with hematoxylin and eosin and stained for acid-fast bacilli (17).
Statistical analysis.
Bacterial burdens in the untreated control and drug-treated animal organs were analyzed with GraphPad Prism version 4 (GraphPad Software, San Diego, CA), using analysis of variance (ANOVA) and Dunnett and Tukey multiple comparison tests. Data are presented using the mean values (n = 5) plus or minus the standard error of the mean (SEM). Significance was considered below a P value of <0.050.
RESULTS
Course of M. abscessus infection in mouse models.
We previously published our findings showing that the immune response of C57BL/6 mice and guinea pigs infected with M. abscessus results in eventual clearance of these organisms (17). Hence, a series of diverse mouse infection models were evaluated for their ability to establish a progressive high level of infection. The course of the experimental infection is shown in Fig. 1. In each model, beige, Nos2−/−, Cybb−/−, TNFR−/−, C3HeB/FeJ, GKO−/−, MyD88−/−, SCID, nude, and GMCSF−/− mice were exposed to an intravenous infection with approximately 1 × 106 CFU of the clinical strain M. abscessus 103. The rationale for the use of an intravenous infection rather than a pulmonary infection route is due to technical problems associated with aerosol exposure of RGM. Attempts to develop a pulmonary model exposing mice to M. abscessus at 1.0 × 10 11 CFU resulted in a low level of infection in SCID and GMCSF−/− mice (our unpublished data). Moreover, further attempts to deliver higher bacterial CFU of 1.0 × 1012 were technically impossible due to the inoculum becoming a solid paste that cannot be aerosolized. Mice infected with M. abscessus 103 were evaluated for bacterial loads in the lungs (Fig. 1A), spleens (Fig. 1B), and livers (Fig. 1C) after 1, 20, and 40 days of infection.
FIG 1.
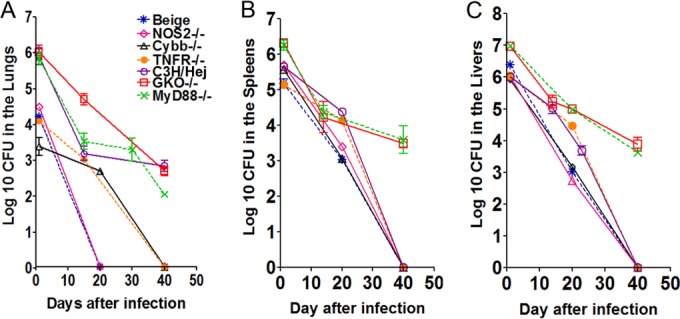
M. abscessus infection in mouse models. Bacterial counts in the lungs (A), spleens (B), and livers (C) of beige, Nos2−/−, Cybb−/−, TNFR−/−, C3H/HeJ, GKO−/−, and MyD88−/− mice infected with 1 × 106 CFU of M. abscessus 103 are shown. CFU were determined at days 1, 20, and 40 after infection by plating serial dilutions of organ homogenates on nutrient 7H11 and TSA agar and counting CFU after 7 days incubation at 32°C. (A) The beige, Nos2−/−, Cybb−/−, and TNFR−/− mice showed complete clearance after 40 days of infection. After 40 days of infection, the C3H/HeJ, GKO−/−, and MyD88−/− mice showed about 2 to 2.5 log10 CFU in the lungs. (B and C) Complete clearance of bacterial numbers in the spleens and livers was evident in the case of the beige, Nos2−/−, Cybb−/−, TNFR−/−, and C3H/HeJ animals after 40 days of infection. After 40 days of infection GKO−/− and MyD88−/− mice showed 3.5 log10 CFU in the spleen and liver. Results represent the average of three experiments (n = 5 mice per experiment) on bacterial load in each group and are expressed as log10 CFU (± SEM).
The beige, Nos2−/−, Cybb−/−, and TNFR−/− mice showed an early innate immune clearance of about 2 to 2.6 log10 in the lungs over the first day of infection (Fig. 1A), resulting in a complete clearance at 20 days in the beige and Nos2−/− animals. Similarly, the Cybb−/− and TNFR−/− mice showed decreased bacterial burdens resulting in clearance of bacilli in the lungs after 40 days of infection (Fig. 1A). The C3HeB/FeJ, GKO−/−, and MyD88−/−, mice had about 2.0 to 2.5 log10 bacilli persisting in the lungs after 40 days of infection.
Complete clearance of bacterial numbers in the spleens and livers was evident in the case of the beige, Nos2−/−, Cybb−/−, TNFR−/−, and C3HeB/FeJ animals after 40 days of infection (Fig. 1B and C). However, in the GKO−/− and MyD88−/− mouse models, about 3.5 log10 remained in the spleen and liver after 40 days of infection.
Figure 2 demonstrates the experimental infection in the SCID, nude, and GMCSF−/− mouse models. The SCID, nude, and GMCSF−/− mice were exposed to an intravenous infection with approximately 1 × 106 CFU of the clinical strain M. abscessus 103. The SCID and GMCSF−/− mice showed the highest increase in bacterial burden in the lungs, spleens, and livers over 40 days of infection compared to those of the nude mouse model (Fig. 1A, B, and C).
FIG 2.
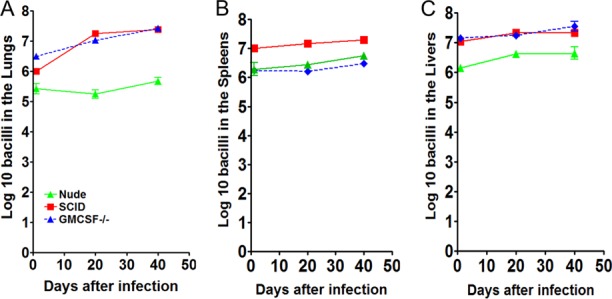
Progressive M. abscessus infection in mouse models. Bacterial counts in the lungs (A), spleens (B), and livers (C) of SCID, nude, and GMCSF−/− mice infected with 1 × 106 CFU of M. abscessus 103. CFU were determined at day 1, 20, and 40 after infection by plating serial dilutions of organ homogenates on nutrient 7H11 and TSA agar and counting CFU after 7 days incubation at 32°C. (A, B, C) The SCID, nude, and GMCSF−/− mice showed a high level of progressive infection after 40 days of infection. Results represent the average of three experiments (n = 5 mice per experiment) on bacterial load in each group and are expressed as log10 CFU (± SEM).
Development of pathology in SCID, nude, and GMCSF−/− animals.
An optimal model for antimycobacterial treatment must consider the type of granulomas that are formed during infection and not only mimic the bacterial burden. Therefore, we evaluated the changes in lung pathology in SCID, GMCSF−/−, and nude mice shown in Fig. 3. As early as day 20 after infection, SCID (Fig. 3A), GMCSF−/− (Fig. 3B), and nude (Fig. 3C) mice demonstrated the influx of monocytes and the thickening of the interstitium indicative of inflammation. Large numbers of foamy cells were present in SCID (Fig. 3A, arrows) and GMCSF−/− (Fig. 3B, arrows) mice. The lungs of SCID, GMCSF−/−, and nude mice infected with M. abscessus 103 exhibited progressive lesion development, and by day 40, lung tissue was composed of notable granulomas and inflammation (Fig. 3A, B, and C). The SCID, nude, and GMCSF−/− mouse models with more than 1.0 × 106 CFU in the lungs, spleens, and livers represent levels of bacteria present in an NTM-infected patient with cavitary disease (27). NTM patients generally have two types of pulmonary granulomas, nonnecrotizing and necrotizing (28, 29). The SCID model was the only model to show nonnecrotizing and necrotizing granulomas as well as a large numbers of foamy cells (Fig. 3A, B, and C). The nude and GMCSF−/− mice only demonstrated nonnecrotizing granulomas after 40 days of infection.
FIG 3.

Pulmonary pathology in SCID, nude, and GMCSF−/− mice. Representative photomicrographs of hematoxylin- and eosin-stained slides from the lungs of control or vaccinated mice. Changes in lung pathology in SCID, GMCSF−/−, and nude mice are shown in Fig. 3. As early as day 20 after infection, SCID (A), GMCSF−/− (B), and nude (C) mice demonstrated an influx of monocytes and thickening of the interstitium indicative of inflammation. Large numbers of foamy cells were present in SCID (A, arrow) and GMCSF−/− (B, arrow). (A, B, C) The lungs of SCID, GMCSF−/−, and nude mice infected with M. abscessus 103 exhibited progressive lesion development, and by day 40, lung tissue was composed of notable granulomas and inflammation. The SCID mouse model was the only model that demonstrate both non-necrotizing and necrotizing granulomas and large numbers of foamy cells. Magnifications: -, 1×, –, 5×, and —, 10×.
Antimycobacterial treatment in the acute GKO−/− mouse infection model.
The GKO−/− mouse model lacks IFN-γ, which is protective against mycobacterial infections. Humans with an IFN-γ–IL-12 axis deficit have increased susceptibility to NTM infection (7). The deficit of this model is, of course, that the immune system of these animals can slowly clear M. abscessus, but the benefit is that the bacilli are capable of persisting, albeit at lower levels. Groups of GKO−/− mice were exposed to an intravenous infection with approximately 1 × 106 CFU of the clinical strain M. abscessus 103. The GKO−/− mice were treated starting on day 2 for 8 days with saline, clarithromycin, ciprofloxacin, amikacin, clofazimine, bedaquiline, and a combination of bedaquiline and clofazimine. Experimental groups of mice were evaluated for bacterial burden on days 1, 5, and 15 after infection.
The course of the experimental infection is shown in Fig. 4. The numbers of bacteria in the untreated mice decreased over time due to immune clearance. Clarithromycin treatment for 5 days resulted in significantly reduced bacterial burdens in the lungs (P < 0.05). However, treatment with clarithromycin for 8 days did not show any significant differences in any of the organs assayed compared to those of the the M. abscessus-infected control. This may have occurred due to the reduced bacterial burden of the control animals over time due to immune clearance. The clofazimine-treated group compared to the untreated control group demonstrated statistically significant reductions in the bacterial burdens in the lung (P < 0.050) (Fig. 4A), spleen (P < 0.050) (Fig. 4B), and liver (P < 0.050) (Fig. 4C) after 8 days of treatment.
FIG 4.

Acute GKO−/− mouse treatment model. Bacterial counts in the lungs (A), spleens (B), and livers (C) of GKO−/− mice infected with 1 × 106 CFU of M. abscessus 103. The GKO−/− mice were treated starting on day 2 for 9 days with saline, 250 mg/kg clarithromycin, 100 mg/kg ciprofloxacin, and 150 mg/kg amikacin subcutaneous injection, 20 mg/kg clofazimine, 30 mg/kg bedaquiline, or a combination of 30 mg/kg bedaquiline and 20 mg/kg clofazimine. Experimental groups of mice were evaluated for bacterial burden on days 1, 5, and 15 after infection by plating serial dilutions of organ homogenates on nutrient 7H11 and TSA agar and counting CFU after 14 days incubation at 32°C. The log10 CFU reduction after 8 days of treatment with bedaquiline compared to that of no treatment was 1.03 in the lung, 1.09 in the spleen, and 1.06 in the liver. After 8 days of treatment with the clofazimine and bedaquiline combination, the most significant reductions in bacterial numbers were observed in the lung (P < 0.001), spleen (P < 0.001), and liver (P < 0.001) compared to all the other compounds assayed. Log10 CFU reduction was afforded by clofazimine and bedaquiline combination treatment—1.17 in the lung, 1.52 in the spleen, and 1.60 in the liver. Results represent the average of two experiments (n = 5 mice per experiment) on bacterial load in each group and are expressed as log10 CFU (± SEM) cells. *, P < 0.050 by ANOVA and the Dunnett and Tukey multiple comparison tests.
The bedaquiline group, after 4 days of treatment (day 5 bacterial CFU), compared to the untreated control group yielded significantly reduced bacterial burdens in the spleen (P < 0.001) and liver (P < 0.001). After 8 days of treatment, the bedaquiline group compared to the control group achieved significant reductions in all three organs—lung (P < 0.01), spleen (P < 0.001), and liver (P < 0.001). Bedaquiline (8 days treatment) significantly reduced the bacterial burden in the lung (P < 0.050), spleen (P < 0.001), and liver (P < 0.001) compared to clarithromycin according to the results obtained after treatment. In addition, bedaquiline was more effective in reducing the bacterial numbers in the lung (P < 0.050), spleen (P < 0.001), and liver (P < 0.001) than the ciprofloxacin and amikacin treatments.
More importantly, combination treatment with clofazimine and bedaquiline (treatment for 4 days) compared to the untreated control yielded reduced bacterial burdens in not only the spleen (P < 0.001) and liver (P < 0.001) but also the lung (P < 0.050). After 8 days, treatment with a combination of clofazimine and bedaquiline resulted in the most significant reductions in bacterial numbers in the lung (P < 0.001), spleen (P < 0.001), and liver (P < 0.001) compared to the untreated control group. In addition, the combination was also significantly better than all of the other treatments evaluated (P < 0.050).
Eight days of ciprofloxacin treatment compared to treatment with the saline control did not result in a significant reduction in bacterial burden in any of the organs assayed. However, eight days of amikacin treatment resulted in significant reductions in bacterial burdens in the spleen (P < 0.010) and liver (P < 0.001) but not in the lungs. Treatment with amikacin (15 days) reduced the bacterial burden in the spleen (P < 0.010) and liver (P < 0.001) compared to treatment with ciprofloxacin.
Acute antimycobacterial treatment in SCID mice.
The SCID mouse model lacks functional lymphocytes and NK cells. However, studies in these animals have shown the response of normal pulmonary macrophages to IFN-γ and IL-12 but not to tumor necrosis factor alpha (TNF-α) during Mycobacterium bovis bacillus Calmette-Guérin infection (30). Humans with such immunocompromised conditions as SCID, AIDS, and genetic deficiencies in type 1 cytokines IFN- γ and IL-12 or their receptors are especially susceptible to intracellular infection by NTM (7). Optimization of the SCID model showed strong reproducibility and robust treatment data over more than 20 intravenous infections using clarithromycin treatment.
To confirm the results obtained from M. abscessus-infected GKO−/− mice treated with clofazimine and bedaquiline, we repeated the experiment in SCID mice, which develop a high level of bacterial burden. Groups of SCID mice were exposed to an intravenous infection with approximately 1 × 106 CFU of the clinical strain M. abscessus 103. Starting on day 2, the SCID mice were treated for 8 days with saline, clarithromycin, clofazimine, bedaquiline, and a combination of bedaquiline and clofazimine. Experimental groups of mice were evaluated for bacterial burden on day 1 and 15 after infection. Eight days of clarithromycin treatment caused significant reductions in bacterial burdens in the lung (P < 0.010), spleen (P < 0.050), and liver (P < 0.010) compared to that of the untreated controls. Treatment with the single drug clofazimine or bedaquiline for 8 days resulted in reduced bacterial burdens in the lung (P < 0.050), spleen (P < 0.050), and liver (P < 0.010) compared to those of the untreated controls. The combination treatment of clofazimine and bedaquiline for 8 days yielded significant reductions in the bacterial burdens in the lung (P < 0.050) (Fig. 5A), spleen (P < 0.050) (Fig. 5B), and liver (P < 0.050) (Fig. 5C) compared to those of the the saline group and all of the other compounds assayed. Figure 5D demonstrates the lung pathology after infections with M. abscessus 103 and treatment with saline, clarithromycin, clofazimine, bedaquiline, and a combination of bedaquiline and clofazimine. The saline control demonstrated the influx of mononuclear cells and inflammation. It was evident that some foci of cellular aggregates remained in the clofazimine-treated and bedaquiline-treated mice, while the combination of bedaquiline and clofazimine resulted in less inflammation and nominal cellular masses.
FIG 5.

Acute SCID treatment mouse model. Bacterial counts in the lungs (A), spleens (B), and livers (C) of SCID mice infected with 1 × 106 CFU of M. abscessus 103. The GKO−/− mice were treated starting on day 2 for 9 days with saline, 250 mg/kg clarithromycin, 20 mg/kg clofazimine, 30 mg/kg bedaquiline, or a combination of 30 mg/kg bedaquiline and 20 mg/kg clofazimine. Experimental groups of mice were evaluated for bacterial burden on days 1 and 15 after infection by plating serial dilutions of organ homogenates on nutrient 7H11 and TSA agar and counting CFU after 14 days incubation at 32°C. The combination treatment of clofazimine and bedaquiline for 8 days yielded significant reductions in the bacterial burden in the lung (P < 0.050), spleen (P < 0.001), and liver (P < 0.001) compared to those of the saline and clarithromycin control. Results represent the average of two experiments (n = 5 mice per experiment) on bacterial load in each group and is expressed as Log10 CFU (± SEM) cells. *, P < 0.050, by ANOVA and the Dunnett and Tukey multiple comparison tests. (D) Representative photomicrographs of hematoxylin and eosin stained slides from the lungs of control or treated mice. As early as 15 days after infection and treatment with bedaquiline and clofazimine, reduced granulomatous lesions and reduced thinking of the interstitium indicative of reduced inflammation were observed in these mice. (D) Lung pathology after infection with M. abscessus 103 and treatment with saline, clarithromycin, clofazimine, bedaquiline, or a combination of bedaquiline and clofazimine. The saline demonstrated the influx of mononuclear cells and inflammation. It was evident that some foci of cellular aggregates (D, arrows) remained in the clofazimine- and bedaquiline-treated mice while the combination of bedaquiline and clofazimine resulted in less inflammation and nominal cellular masses. Magnification, 1×.
DISCUSSION
The results of this study show that most of the mouse models with major deficits in innate or acquired immunity can still clear a high level of M. abscessus infection. The immune systems of the following mice were capable of clearing M. abscessus: the beige mice with a dominant Th2 immunity; the Nos2−/− mice lacking NO2; Cybb−/− mice, which are devoid of the superoxide-generating enzyme that forms the reactive oxygen species (ROS) associated with chronic granulomatous disease; TNFR−/− mice, which lack the tumor necrosis factor receptor; C3HeB/FeJ mice capable of forming tubercular granulomas and GKO−/− deficient in IFN-γ; and the MyD88 knockout mice, with an allele that encodes a deletion of exon 3 of the myeloid differentiation primary response deleteriously impacting innate and acquired immunity causing increased susceptibility to bacterial pathogens (19–23). After 40 days of infection, the C3HeB/FeJ, GKO−/−, and MyD88−/− mice had bacterial loads of about 2.0 to 2.5 log10 persisting in the lungs. After 40 days of infection in the GKO−/− and MyD88−/− animal models, about 3.5 log10 bacilli remained in the spleen and liver. With regard to M. tuberculosis infection, C3HeB/FeJ mice dominantly expressed necrotic granulomas in the lungs allowing for bacterial replication, while fewer tubercular granulomas were present in the spleens and livers, leading to better bacterial clearance (23).
Our prior studies (17) demonstrated that immunocompetent C57/BL6 mice and guinea pigs are also capable of rapid removal of M. abscessus infection. In addition, a leptin deficient OBOB knockout obese mouse model that lacks leptin—a satiety hormone whose absence causes insatiable hunger in these animals—is capable of M. abscessus clearance (17). One major but poorly understood NTM patient cohort is a group of white Caucasian postmenopausal women who are thin; thus, low leptin levels have been proposed as a factor for increased susceptibility to NTM simply due to the fact that leptin has a positive feedback regulation on production of IFN-γ (3).
The cure rate for chronic lung and disseminated infections due to NTM is dismal despite prolonged treatment with various multidrug regimens (6, 7). Treatment of M. abscessus consists of macrolides, quinolones, amikacin, and other antibiotics. Except for modest susceptibility to ethambutol and rifampin, M. abscessus group strains are not susceptible to the typical antituberculosis drugs and are in fact considered the NTM with the greatest antibiotic resistance (11, 12). Pulmonary disease caused by M. abscessus is treated based on routine susceptibility testing. In general, multidrug regimens for M. abscessus include clarithromycin, 1,000 mg/day, with 1 or more of the parenteral medications (amikacin, cefoxitin, or imipenem) and may cause symptomatic improvement and disease regression (11).
Studies using an intravenous infection of M. abscessus in nude mice have been reported, which detail a static high level of bacterial burden (31). In the latter study, nude mice infected with 106 to 108 CFU of M. abscessus ATCC 19977 reference strain were treated for 2 months with bedaquiline, and this did not result in significantly reduced bacterial burdens in lungs and spleens (31). The M. abscessus ATCC 19977 MICs showed clarithromycin inducible resistance and 0.5 μg/ml bedaquiline in agar and 0.06 μg/ml in brain heart infusion (BHI) broth (31). Our study differed in many ways from the latter study, as we infected GKO−/− and SCID mice with a clinical M. abscessus 103 strain (derived from a cystic fibrosis patient) at a consistent 1.0 × 106 CFU/mouse dose (32). Moreover, our M. abscessus 103 strain MICs were 0.75 μg/ml for clarithromycin and 1.0 μg/ml for bedaquiline. Thus, these combined studies add insight into the importance of strain MIC differences and treatment outcomes in anti-nontuberculosis mouse models.
An intratracheal infection in GMCSF−/− mice (33) meets the requirements for a preclinical antimycobacterial M. abscessus model. In our hands, the nude model produced lower levels of bacterial burden, which remained static compared to the progressive growth present in the SCID and GMCSF−/− mice. Additional models, such as the embryonic zebrafish test system, have been developed to assay M. abscessus (34) for rapid compound screening.
The advantage of mouse models with severe immunodeficiencies is that after M. abscessus infection, a progressively high level of infection results, allowing for detection of significant differences between the M. abscessus control and the drug treated groups. This is not surprising, since NTM thrive in humans as an opportunistic infection. Our studies demonstrate that M. abscessus infection of SCID, nude, and GMCSF−/− mice results in a sustained high level of infection, exponentially dividing bacteria, reproducible bacterial loads, and organ histopathology, which are suitable for antimycobacterial studies. Another benefit of the three preclinical models after 40 days of M. abscessus infection was the influx of foamy cells to the lungs, typical in human NTM disease (28, 29). Another characteristic of pulmonary NTM infection in humans is the development of non-necrotic and necrotizing granulomas (28, 29), both of which could only be reproduced in the SCID mice model. It is very likely, though, that by changing parameters, such as the virulence of the M. abscessus strain, bacterial burden, and duration of disease, the GMCSF−/− and nude models could potentially produce necrotizing granulomas as well. SCID mice lack mature T and B lymphocytes, nude mice have an absent thymus, resulting in reduced numbers of T cells, and GMCSF−/− mice have a deficit of this growth factor for monocytes and leukocytes. The ability of SCID, GMCSF−/−, and nude mice to support sustained NTM growth points to the strong role of T cells and GMCSF-dependent cell phenotypes being required for protective immunity against NTM.
Additional studies were completed in GKO−/− and SCID mice to evaluate the antimycobacterial activity of clarithromycin, ciprofloxacin, amikacin, clofazimine, bedaquiline, and a combination of bedaquiline and clofazimine against M. abscessus. The first trial in GKO−/− mice consisted of an 8-day treatment with these aforementioned drugs. The reduction in the bacterial load of the clarithromycin-treated GKO mice was limited in all of the organs assayed compared to the M. abscessus-infected control. The lack of reduction in bacterial burden after clarithromycin treatment of GKO−/− mice infected with M. abscessus support earlier reports using nude mice (31) as well as a retrospective clinical analysis (28). Amikacin treatment for 8 days resulted in significantly reduced bacterial burdens in the spleen and liver but not in lungs, supporting the findings of reports on a chronic GMSCF−/− model (33) and contradicting the findings of other reports on a chronic nude model (31). Ciprofloxacin treatment for 5 and 8 days did not result in a significant reduction in bacterial burden in any of the organs assayed. However, clofazimine treatment demonstrated statistically significant reductions in the bacterial burdens in lung, spleen, and liver after 8 days of treatment. Five days of bedaquiline treatment yielded significantly reduced bacterial burdens in spleen and liver. Furthermore, eight days of bedaquiline treatment achieved significant reductions in all three organs. More importantly, the addition of clofazimine to bedaquiline led to reduced bacterial burdens in not only the spleen and liver but also the lung after 5 days of treatment. After 8 days of treatment with the clofazimine and bedaquiline combination, the most significant reductions in bacterial numbers were observed in the lung, spleen, and liver, which were superior to all of the other compounds assayed.
To confirm our results in the GKO−/− models, we used SCID mice exposed to an intravenous infection with approximately 1 × 106 CFU of the clinical strain M. abscessus 103. The SCID mice showed consistent bacterial burden and granuloma development (necrotizing and non-necrotizing) and lacked clinical signs of compound toxicity. SCID mice were treated starting on day 2 for 8 days with the same drug panel as described above (saline, clarithromycin, clofazimine, bedaquiline, or a combination of bedaquiline and clofazimine). Experimental groups of mice were evaluated for bacterial burden on day 1 and day 15 after infection. The combination treatment of clofazimine and bedaquiline for 8 days yielded significant reductions in the bacterial burdens in the lung, spleen, and liver compared to those of either the saline control or the clarithromycin-positive control.
The lung pathology after infections with M. abscessus 103 and drug treatments was also evaluated. The saline control demonstrated an influx of mononuclear cells and signs of inflammation. It was evident that some foci of cellular aggregates remained in the clofazimine- and bedaquiline-treated mice, while the combination of bedaquiline and clofazimine resulted in less inflammation and nominal cellular aggregates. These results support the reduced bacterial burden present in bedaquiline- and clofazimine-treated animals.
Studies using multiple animal models have shown that bedaquiline is efficacious against M. tuberculosis, Mycobacterium leprae, and to some extent M. avium (25, 35–37). Bedaquiline potently inhibits the mycobacterial enzyme complex ATP synthase, thus interfering with energy production and homeostasis. As a consequence, it is highly active against drug-sensitive and drug-resistant isolates of M. tuberculosis (25). The mechanism of clofazimine is not entirely known but has been shown to affect the electron transport of M. tuberculosis (38). It is impressively active in vitro against multidrug-resistant strains of M. tuberculosis and is currently extensively evaluated clinically against MDR-tuberculosis patients (38). Although the efficacy of bedaquiline and clofazimine showed some promise against M. abscessus in a mouse model, the combination treatment has some complications, such as cardiovascular safety and dual cross-resistance, that require more study (39, 40).
In conclusion, NTM infections are becoming an emerging problem worldwide. To deal with the increasing morbidity and mortality of these pathogens, multiple laboratories are focused on developing new preclinical models to screen new or repurposed compounds to combat the emergence of these pathogens. Our studies support the use of the SCID, GMCSF−/−, and nude preclinical mouse models for antibacterial treatment against Mycobacterium abscessus. Moreover, by using the GKO−/− and SCID acute infection models, the combination of clofazimine and bedaquiline was found to significantly reduce the bacterial loads in major organs compared to the saline control. The in vivo activity of the combination of clofazimine and bedaquiline against M. abscessus merits further study.
ACKNOWLEDGMENTS
This work was supported by funding provided by the National Institutes of Health (grant numbers R21AIO99534-02 and AIO99534-02 and NIH/NIAID task order HHSN272201000009I/HHSN27200001, subtask 6, principal investigator [PI], Anne J. Lenaerts, co-PI, Diane J. Ordway) (program officers, Christine Sizemore and Andre McBride).
REFERENCES
- 1.Wallace RJ Jr, Swenson JM, Silcox VA, Good RC, Tschen JA, Stone MS. 1983. Spectrum of disease due to rapidly growing mycobacteria. Rev Infect Dis 5:657–679. doi: 10.1093/clinids/5.4.657. [DOI] [PubMed] [Google Scholar]
- 2.Woods GL, Bergmann JS, Witebsky FG, Fahle GA, Wanger A, Boulet B, Plaunt M, Brown BA, Wallace RJ Jr. 1999. Multisite reproducibility of results obtained by the broth microdilution method for susceptibility testing of Mycobacterium abscessus, Mycobacterium chelonae, and Mycobacterium fortuitum. J Clin Microbiol 37:1676–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Epson E, Cassidy M, Marshall-Olson A, Hedberg K, Winthrop KL. 2012. Patients with nontuberculous mycobacteria: comparison of updated and previous diagnostic criteria for lung disease. Diagn Microbiol Infect Dis 74:98–100. doi: 10.1016/j.diagmicrobio.2012.05.035. [DOI] [PubMed] [Google Scholar]
- 4.Shang S, Gibbs S, Henao-Tamayo M, Shanley CA, McDonnell G, Duarte RS, Ordway DJ, Jackson M. 2011. Increased virulence of an epidemic strain of Mycobacterium massiliense in mice. PLoS One 6:e24726. doi: 10.1371/journal.pone.0024726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Falkinham JD., III 1996. Epidemiology of infection by nontuberculous mycobacteria. Clin Microbiol Rev 9:177–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orme IM, Ordway DJ. 2014. Host response to nontuberculous mycobacterial infections of current clinical importance. Infect Immun 82:3516–3522. doi: 10.1128/IAI.01606-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan ED, Bai X, Kartalija M, Orme IM, Ordway DJ. 2010. Host immune response to rapidly growing mycobacteria, an emerging cause of chronic lung disease. Am J Respir Cell Mol Biol 43:387–393. doi: 10.1165/rcmb.2009-0276TR. [DOI] [PubMed] [Google Scholar]
- 8.Glazer CS, Martyny JW, Lee B, Sanchez TL, Sells TM, Newman LS, Murphy J, Heifets L, Rose CS. 2007. Nontuberculous mycobacteria in aerosol droplets and bulk water samples from therapy pools and hot tubs. J Occup Environ Hyg 4:831–840. doi: 10.1080/15459620701634403. [DOI] [PubMed] [Google Scholar]
- 9.Bar-On O, Mussaffi H, Mei-Zahav M, Prais D, Steuer G, Stafler P, Hananya S, Blau H. 2015. Increasing nontuberculous mycobacteria infection in cystic fibrosis. J Cyst Fibros 14:53–62. doi: 10.1016/j.jcf.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 10.Falkinham JO. 2010. Impact of human activities on the ecology of nontuberculous mycobacteria. Future Microbiol 5:951–960. doi: 10.2217/fmb.10.53. [DOI] [PubMed] [Google Scholar]
- 11.Brown-Elliott BA, Wallace RJ Jr. 2002. Clinical and taxonomic status of pathogenic nonpigmented or late-pigmenting rapidly growing mycobacteria. Clin Microbiol Rev 15:716–746. doi: 10.1128/CMR.15.4.716-746.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Ingen J, Boeree MJ, van Soolingen D, Mouton JW. 2012. Resistance mechanisms and drug susceptibility testing of nontuberculous mycobacteria. Drug Resist Updat 15:149–161. doi: 10.1016/j.drup.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 13.Greendyke R, Byrd TF. 2008. Differential antibiotic susceptibility of Mycobacterium abscessus variants in biofilms and macrophages compared to that of planktonic bacteria. Antimicrob Agents Chemother 52:2019–2026. doi: 10.1128/AAC.00986-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams MM, Yakrus MA, Arduino MJ, Cooksey RC, Crane CB, Banerjee SN, Hilborn ED, Donlan RM. 2009. Structural analysis of biofilm formation by rapidly and slowly growing nontuberculous mycobacteria. Appl Environ Microbiol 75:2091–2098. doi: 10.1128/AEM.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maurer FP, Bruderer VL, Ritter C, Castelberg C, Bloemberg GV, Bottger EC. 2014. Lack of antimicrobial bactericidal activity in Mycobacterium abscessus. Antimicrob Agents Chemother 58:3828–3836. doi: 10.1128/AAC.02448-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daley CL, Griffith DE. 2002. Pulmonary disease caused by rapidly growing mycobacteria. Clin Chest Med 23:623–632. doi: 10.1016/S0272-5231(02)00021-7. [DOI] [PubMed] [Google Scholar]
- 17.Ordway D, Henao-Tamayo M, Smith E, Shanley C, Harton M, Troudt J, Bai X, Basaraba RJ, Orme IM, Chan ED. 2008. Animal model of Mycobacterium abscessus lung infection. J Leukoc Biol 83:1502–1511. doi: 10.1189/jlb.1007696. [DOI] [PubMed] [Google Scholar]
- 18.Cooper AM. 2009. Cell-mediated immune responses in tuberculosis. Annu Rev Immunol 27:393–422. doi: 10.1146/annurev.immunol.021908.132703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sugawara I, Yamada H, Mizuno S, Takeda K, Akira S. 2003. Mycobacterial infection in MyD88-deficient mice. Microbiol Immunol 47:841–847. doi: 10.1111/j.1348-0421.2003.tb03450.x. [DOI] [PubMed] [Google Scholar]
- 20.Deffert C, Schäppi MG, Pache JC, Cachat J, Vesin D, Bisig R, Ma Mulone X, Kelkka T, Holmdahl R, Garcia I, Olleros ML, Krause KH. 2014. Bacillus Calmette-Guerin infection in NADPH oxidase deficiency: defective mycobacterial sequestration and granuloma formation. PLoS Pathog 10:e1004325. doi: 10.1371/journal.ppat.1004325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haug M, Awuh JA, Steigedal M, Frengen Kojen J, Marstad J, Nordrum IS, Halaas Ø, Flo TH. 2013. Dynamics of immune effector mechanisms during infection with Mycobacterium avium in C57BL/6 mice. Immunology 140:232–243. doi: 10.1111/imm.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bermudez LE, Parker A, Petrofsky M. 1999. Apoptosis of Mycobacterium avium-infected macrophages is mediated by both tumour necrosis factor (TNF) and Fas, and involves the activation of caspases. Clin Exp Immunol 116:94–99. doi: 10.1046/j.1365-2249.1999.00852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henao-Tamayo M, Obregon-Henao A, Creissen E, Shanley C, Orme I, Ordway DJ. 2015. Differential Mycobacterium bovis BCG vaccine-derived efficacy in C3Heb/FeJ and C3H/HeOuJ mice exposed to a clinical strain of Mycobacterium tuberculosis. Clin Vaccine Immunol 22:91–98. doi: 10.1128/CVI.00466-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clinical and Laboratory Standards Institute. 2011. Susceptibility testing of mycobacteria, nocardiae, and other aerobic actinomycetes; approved standard—–2nd ed CLSI document M24-A2. Clinical and Laboratory Standards Institute, Wayne, PA. [PubMed] [Google Scholar]
- 25.Lenaerts AJ, Hoff D, Aly S, Ehlers S, Andries K, Cantarero L, Orme IM, Basaraba RJ. 2007. Location of persisting mycobacteria in a guinea pig model of tuberculosis revealed by r207910. Antimicrob Agents Chemother 51:3338–3345. doi: 10.1128/AAC.00276-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gangadharam PR, Reddy MV. 1995. Carryover of clofazimine into culture media. Antimicrob Agents Chemother 39:1388–1389. doi: 10.1128/AAC.39.6.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koh WJ, Hong G, Kim K, Ahn S, Han J. 2012. Pulmonary sequestration infected with nontuberculous mycobacteria: a report of two cases and literature review. Asian Pac J Trop Med 5:917–919. doi: 10.1016/S1995-7645(12)60172-2. [DOI] [PubMed] [Google Scholar]
- 28.Jeong Y, Lee KS, Koh WJ, Han J, Kim TS, Kwon OJ. 2004. Nontuberculous mycobacterial pulmonary infection in immunocompetent patients: comparison of thin-section CT and histopathologic findings. Radiology 231:880–886. doi: 10.1148/radiol.2313030833. [DOI] [PubMed] [Google Scholar]
- 29.Lammas DA, De Heer E, Edger JD, Novelli V, Ben-Smith A, Baretto R, Drysdale J, Binch P, MacLennan C, Kumararatne DS, Panchalingam S, Ottenhoff TH, Casanova JL, Emile JF. 2002. Heterogeneity in the granulomatous response to mycobacterial infection in patients with defined genetic mutations in the interleukin 12-dependent interferon-gamma production pathway. Int J Exp Pathol 83:1–20. doi: 10.1046/j.1365-2613.2002.00216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xing Z, Zganiacz A, Wang J, Sharma SK. 2001. Enhanced protection against fatal mycobacterial infection in SCID beige mice by reshaping innate immunity with IFN-γ transgene. J Immunol 167:375–383. doi: 10.4049/jimmunol.167.1.375. [DOI] [PubMed] [Google Scholar]
- 31.Lerat I, Cambau E, Roth Dit Bettoni R, Gaillard JL, Jarlier V, Truffot C, Veziris N. 2014. In vivo evaluation of antibiotic activity against Mycobacterium abscessus. J Infect Dis 209:905–912. doi: 10.1093/infdis/jit614. [DOI] [PubMed] [Google Scholar]
- 32.Saito H, Tasaka H. 1969. Comparison of the pathogenicity for mice of Mycobacterium fortuitum and Mycobacterium abscessus. J Bacteriol 99:851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Groote MA, Johnson L, Podell B, Brooks E, Basaraba R, Gonzalez-Juarrero M. 2014. GM-CSF knockout mice for preclinical testing of agents with antimicrobial activity against Mycobacterium abscessus. J Antimicrob Chemother 69:1057–1064. doi: 10.1093/jac/dkt451. [DOI] [PubMed] [Google Scholar]
- 34.Bernut A, Le Moigne V, Lesne T, Lutfalla G, Herrmann JL, Kremer L. 2014. In vivo assessment of drug efficacy against Mycobacterium abscessus using the embryonic zebrafish test system. Antimicrob Agents Chemother 58:4054–4063. doi: 10.1128/AAC.00142-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shang S, Shanley CA, Caraway ML, Orme EA, Henao-Tamayo M, Hascall-Dove L, Ackart D, Lenaerts AJ, Basaraba RJ, Orme IM, Ordway DJ. 2011. Activities of TMC207, rifampin, and pyrazinamide against Mycobacterium tuberculosis infection in guinea pigs. Antimicrob Agents Chemother 55:124–131. doi: 10.1128/AAC.00978-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Veziris N, Ibrahim M, Lounis N, Chauffour A, Truffot-Pernot C, Andries K, Jarlier V. 2009. A once-weekly R207910-containing regimen exceeds activity of the standard daily regimen in murine tuberculosis. Am J Respir Crit Care Med 179:75–79. doi: 10.1164/rccm.200711-1736OC. [DOI] [PubMed] [Google Scholar]
- 37.Locher CP, Jones SM, Hanzelka BL, Perola E, Shoen CM, Cynamon MH, Ngwane AH, Wiid IJ, van Helden PD, Betoudji F, Nuermberger EL, Thomson JA. 2015. A novel inhibitor of gyrase B is a potent drug candidate for treatment of tuberculosis and nontuberculosis mycobacterial infections. Antimicrob Agents Chemother 55:1455–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cholo MC, Steel HC, Fourie PB, Germishuizen WA, Anderson R. 2012. Clofazimine: current status and future prospects. J Antimicrob Chemother 70:290–298. [DOI] [PubMed] [Google Scholar]
- 39.Andries K, Villellas C, Coeck N, Thys K, Gevers T, Vranckx L, Lounis N, de Jong BC, Koul A. 2014. Acquired resistance of Mycobacterium tuberculosis to bedaquiline. PLoS One 9:e102135. doi: 10.1371/journal.pone.0102135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nessar R, Cambau E, Reyrat JM, Murray A, Gicquel B. 2012. Mycobacterium abscessus: a new antibiotic nightmare. J Antimicrob Chemother 67:810–818. doi: 10.1093/jac/dkr578. [DOI] [PubMed] [Google Scholar]
- 41.Willinger T, Rongvaux A, Takizawa H, Yancopoulos GD, Valenzuela DM, Murphy AJ, Auerbach W, Eynon EE, Stevens S, Manz MG, Flavell RA. 2011. Human IL-3/GM-CSF knock-in mice support human alveolar macrophage development and human immune responses in the lung. Proc Natl Acad Sci U S A 108:2390–2395. doi: 10.1073/pnas.1019682108. [DOI] [PMC free article] [PubMed] [Google Scholar]