

Abstract
Elbasvir is an investigational NS5A inhibitor with in vitro activity against multiple HCV genotypes. Antiviral activity of elbasvir was measured in replicons derived from wild-type or resistant variants of genotypes 1a, 1b, and 3. The barrier to resistance was assessed by the number of resistant colonies selected by exposure to various elbasvir concentrations. In a phase 1b dose-escalating study, virologic responses were determined in 48 noncirrhotic adult men with chronic genotype 1 or 3 infections randomized to placebo or elbasvir from 5 to 50 mg (genotype 1) or 10 to 100 mg (genotype 3) once daily for 5 days. The NS5A gene was sequenced from plasma specimens obtained before, during, and after treatment. Elbasvir suppressed the emergence of resistance-associated variants (RAVs) in vitro in a dose-dependent manner. Variants selected by exposure to high elbasvir concentrations typically encoded multiple amino acid substitutions (most commonly involving loci 30, 31, and 93), conferring high-level elbasvir resistance. In the monotherapy study, patients with genotype 1b had greater reductions in HCV RNA levels than patients with genotype 1a at all elbasvir doses; responses in patients with genotype 3 were generally less pronounced than for genotype 1, particularly at lower elbasvir doses. M28T, Q30R, L31V, and Y93H in genotype 1a, L31V and Y93H in genotype 1b, and A30K, L31F, and Y93H in genotype 3 were the predominant RAVs selected by elbasvir monotherapy. Virologic findings in patients were consistent with the preclinical observations. NS5A-RAVs emerged most often at amino acid positions 28, 30, 31, and 93 in both the laboratory and clinical trial. (The MK-8742 P002 trial has been registered at ClinicalTrials.gov under identifier NCT01532973.)
INTRODUCTION
Elbasvir (MK-8742) is a small-molecule inhibitor of nonstructural protein 5A (NS5A) of hepatitis C virus (HCV) being developed as a component of treatment regimens for chronic HCV infection (1–4). Elbasvir possesses activity against genotype 1a, 1b, and 3 in vitro, including against some viral variants resistant to other NS5A inhibitors (4). In a phase 1b dose escalation study, elbasvir once daily for 5 days resulted in mean reductions in HCV RNA levels of 3.7 to 5.1 log10 IU/ml in patients with genotype 1a or 1b infections given 5 to 50 mg/day and of ∼3 log10 IU/ml in patients with genotype 3 given 50 or 100 mg/day (3). In later phase 2 trials, treatment with elbasvir combined with grazoprevir (MK-5172, an investigational once-daily NS3/4A protease inhibitor) with or without ribavirin for 12 weeks produced rates of sustained virological response at week 12 (SVR12) of 87 to 98% for patients with genotype 1 infections, including in historically difficult-to-treat subgroups (1, 2).
Drug resistance poses a challenge to interferon-sparing regimens for chronic HCV infection (5–12). Variants associated with decreased drug susceptibility exist at low levels in most patients before any exposure to direct-acting antiviral agents. Preexistent resistance-associated variants (RAVs) may then emerge as a dominant species under selective drug pressure during or after direct-acting antiviral therapy. The current report compares and contrasts RAVs identified in preclinical studies with baseline and emerging variants encountered during a phase 1b dose-escalating trial using elbasvir monotherapy for 5 days in patients chronically infected by HCV genotypes 1 and 3 (3).
MATERIALS AND METHODS
Preclinical studies.
HCV replicons were used to determine the effective concentrations (EC) of elbasvir necessary to inhibit HCV RNA levels by a specified percentage (50% or 90%) for genotypes 1a, 1b, and 3 variants compared to no treatment (13). Replicons maintained in 0.5 mg/ml of G418 (HyClone, GE Healthcare Bio-Sciences, Pittsburgh, PA, USA) to select replicating cells were seeded on 384-well plates in Dulbecco's modified Eagle medium (DMEM) containing 5% fetal calf serum. Twofold dilutions of elbasvir concentrations from 1 μM down to 0.002 μM were added to the medium the next day in the presence of 0.5% dimethyl sulfoxide (DMSO). After 72 h of incubation, cells were harvested and subjected to real-time reverse transcriptase PCR (RT-PCR) (14). For each variant, threshold cycle numbers were plotted against the log of elbasvir concentrations and fitted to a sigmoid dose-response curve using Prism (GraphPad Software, San Diego, CA, USA) to obtain the EC50 and EC90 (the drug concentrations needed to achieve 50% and 90% inhibition, respectively, relative to a DMSO control without drug). For reference, the steady-state minimum concentration (Cmin) for once-daily 50 mg elbasvir dosing is about 22 nM (3).
To select cell lines with decreased elbasvir susceptibility, subconfluent monolayers of replicon cells were cultured in the presence of various drug concentrations at multiples of EC90. Plates were prepared at 2 × 105 cells per 60-mm plate and passaged only once at a 1:10 ratio when the cells reached 95% confluence. Colonies surviving selection were first counted and then pooled and expanded for analysis. The colony count in the presence of elbasvir was divided by the number of cells seeded to calculate resistance frequency. Total cellular RNA was isolated from pooled colonies and amplified by RT-PCR. The RT-PCR products were purified with a QIAquick PCR purification kit (Qiagen, Germantown, MD, USA), and the full-length NS5A gene was sequenced. Additionally, the RT-PCR products were cloned into the TOPO TA vector (Invitrogen), and the plasmid DNA from bacterial colonies was sequenced to look for linked variations. The replicative capacity (“fitness”) of RAVs was evaluated in a replicon colony formation assay (13).
Phase 1b randomized clinical trial design.
MK-8742 P002 was a randomized, double-blind, placebo-controlled, sequential dose-escalating phase 1b study of elbasvir monotherapy to assess safety, pharmacokinetics, and viral responses in adult men with chronic HCV-1 or HCV-3 infection (3). Patients between 18 and 60 years of age (up to 65 years old at the discretion of the investigator) with HCV RNA levels of >100,000 IU/ml were eligible. Elbasvir doses were 5, 10, and 50 mg once daily for patients infected with genotype 1a or 1b, and 10, 50, and 100 mg once daily for patients infected with genotype 3. A total of 6 patients were to be enrolled at each dosing level, including 5 patients to receive elbasvir and 1 patient to receive a matching placebo orally for 5 consecutive days, starting at the lowest dose for the infecting genotype. Doses were escalated stepwise once adequate safety data had been reviewed from the previous dosing group. Viral load and resistance testing was to be performed daily and every other day, respectively, for the first 10 days of the study and then at 2 weeks, 3 weeks, 1 month, and 2 months after the last dose of elbasvir. The study was conducted in accordance with good clinical practice guidelines. All participants provided written informed consent.
Viral quantification, sequencing, and resistance analyses.
In the phase 1b study (3), HCV RNA levels were measured in plasma specimens obtained at baseline, during and at the end of elbasvir monotherapy, and at periodic follow-up visits by the TaqMan 2.0 assay (Roche Diagnostics, Branchburg, NJ, USA) with lower limits of quantification and detection of 25 and 9.3 IU/ml, respectively. Blood samples were also collected for viral resistance testing at prespecified time points, including prior to the first elbasvir dose, near the nadir of the HCV RNA level, and up to 2 months after the last elbasvir dose provided that the HCV RNA level remained >1,000 IU/ml.
The full-length NS5A gene was amplified from plasma samples using RT-PCR followed by population and selective clonal sequencing (15). Due to the sensitivity of the assay, resistance analyses were routinely performed only on samples with HCV RNA levels of >1,000 IU/ml. The resultant amino acid sequences were compared to genotype 1a (H77; GenBank no. NC_004102), genotype 1b (con1; GenBank no. AJ238799), or genotype 3 (S52; GenBank no. GU814263) referents. The limit of variant detection in population sequencing was presumed to be ∼20 to 25% of the viral quasispecies (16). Polymorphisms identified in ≥10% of patients were selected for more detailed analysis. For clonal sequencing, amino acids 1 to 448 of NS5A were amplified by RT-PCR, and the resultant amplicons were cloned into a TOPO TA vector (Invitrogen). Approximately 40 clones were sequenced at each time point. Polymorphisms detected in more than a single clone were included in the analysis.
In phenotypic analyses to determine the antiviral potency of elbasvir against detected variants, genotype-specific replicons were constructed to incorporate NS5A polymorphisms. The shift (n-fold) in elbasvir EC for each variant replicon was expressed relative to the EC for the corresponding wild-type replicon. The fitness of resistant variants was evaluated by comparing colony counts to a wild-type referent.
RESULTS
Preclinical results. (i) Activity of elbasvir against genotype 1a and 1b replicons.
In the 3-day replicon assay, the EC90 of elbasvir was 0.006 nM for both the wild-type 1a_H77 and 1b_con1 replicons (Table 1). The elbasvir EC90 against genotype 1a shifted 13-fold to 0.08 nM when 40% human serum was added to the assay medium in addition to the standard 5% fetal calf serum. To assess elbasvir activity against a range of genetically diverse clinical isolates, more than 200 independent genotype 1 sequences collected from clinical databases were subjected to phylogenetic analysis. The full-length NS5A sequences from 5 genotype 1a and 4 genotype 1b patients were then synthesized and cloned into a con1 subgenomic replicon. The EC90 of elbasvir ranged from 0.01 to 0.02 nM for genotype 1a and 0.01 to 0.03 nM for genotype 1b.
TABLE 1.
Activity of elbasvir against HCV genotype 1 NS5A variant repliconsa
| Sequence identificationb | Effective inhibitory concn (nM)c |
|
|---|---|---|
| 50% | 90% | |
| 1a_H77 | 0.004 ± 0.002 | 0.006 ± 0.002 |
| 1a_H77 in 40% NHS | 0.040 ± 0.013 | 0.082 ± 0.027 |
| 1a DQ889262 | 0.009 ± 0.007 | 0.019 ± 0.012 |
| 1a DQ889305 | 0.005 ± 0.000 | 0.011 ± 0.004 |
| 1a DQ889320 | 0.003 ± 0.000 | 0.009 ± 0.005 |
| 1a EU155348 | 0.003 ± 0.002 | 0.007 ± 0.001 |
| 1a EU155380 | 0.006 ± 0.001 | 0.015 ± 0.004 |
| 1b_con1 | 0.003 ± 0.001 | 0.006 ± 0.004 |
| 1b AF033358 | 0.005 ± 0.003 | 0.012 ± 0.006 |
| 1b AJ132996 | 0.009 ± 0.005 | 0.019 ± 0.011 |
| 1b B9016 | 0.014 ± 0.006 | 0.029 ± 0.009 |
| 1b EU482860 | 0.010 ± 0.004 | 0.026 ± 0.013 |
A convenience sample of genotype 1 isolates available in the public domain was used to test susceptibility to elbasvir in the replicon assay. The table summarizes the results from cell lines that were able to replicate in cell culture.
Numbers are GenBank accession numbers except for 1b B9016. H77 and con1 are wild-type referents for genotypes 1a and 1b, respectively. NHS, normal human serum.
Values are averages and ranges from duplicate independent experiments.
(ii) Activity of elbasvir against genotype 3 replicons.
To assess the potency of elbasvir against genotype 3, a subgenomic replicon harboring the genotype 3 NS5A sequence was first constructed by cloning the full-length NS5A gene from a patient isolate (GenBank accession no. NC_009824) into a con1 replicon background, yielding a cell line with an elbasvir EC90 of 0.12 nM (Table 2). Subsequently, more than 100 independent genotype 3 sequences collected from clinical databases were subjected to phylogenetic analysis, from which 11 unique and nonclustered sequences were selected to ensure genetic diversity and cloned into con1 and JFH replicons for further evaluation. In all cases, replicons containing the JFH backbone yielded higher colony counts than the con1 constructs. Whereas con1 and JFH replicon backbones differentially impacted replication fitness, the potency of elbasvir remained nearly constant irrespective of the replicon backbone. The elbasvir EC90 stayed in the subnanomolar range for all the tested variants.
TABLE 2.
Activity of elbasvir against HCV genotype 3 repliconsa
| Sequence identificationb | Con1 backbone |
JFH backbone |
||
|---|---|---|---|---|
| No. of replicon colonies | Meanc EC90 ± SD (nM) | No. of replicon colonies | Meanc EC90 ± SD (nM) | |
| Parental replicon | 300 | 0.006 ± 0.004 | TMTC | 0.019 ± 0.010 |
| GU814823 (S52) | 9 | 0.042 ± 0.011 | 503 | 0.065 ± 0.026 |
| GQ356207 | 3 | 0.016 ± 0.008 | 472 | 0.035 ± 0.027 |
| AM493639 | 0 | ND | 0 | ND |
| EU826291 | 21 | 0.005 ± 0.003 | TMTC | 0.096 ± 0.117 |
| EU826299 | 0 | ND | 0 | ND |
| GQ300882 | 0 | ND | 482 | 0.613 ± 0.855 |
| GQ356215 | 0 | ND | 242 | 0.016 |
| HM042077 | 2 | 0.011 ± 0.010 | 598 | 0.034 ± 0.037 |
| HM042078 | 4 | 0.007 ± 0.006 | 412 | 0.031 ± 0.022 |
| HQ912953 | 2 | 0.007 ± 0.004 | 51 | 0.016 |
| AF320799 | 0 | 0.119 | 43 | 0.387 ± 0.380 |
| NC_009824 | ND | 0.120 ± 0.060 | ND | ND |
EC, effective (inhibitory) concentration; TMTC, too many to count; ND, not done.
Numbers are GenBank accession numbers. Parental replicons of con1 (genotype 1b) and JFH (genotype 2a) were used as controls.
Average from ≥3 independent experiments.
(iii) Resistance selection in genotype 1 and genotype 3 replicons.
To examine the resistant barrier for elbasvir, genotype 1a, 1b, and 3 replicons were treated with elbasvir in the presence of G418. Surviving colonies were counted, and pooled colonies were subjected to phenotypic and genotypic analysis (Table 3). Elbasvir demonstrated dose-dependent suppression of resistant genotype 1a replicons, illustrated by the reductions in colony counts at higher doses. The numbers of surviving colonies were 204, 56, and 4, respectively, in the presence of 0.06 nM, 0.6 nM, and 6 nM elbasvir (ranging from 10× to 1,000× EC90 against the control genotype 1a replicon). Cells emerged as loosely interconnected colonies at 1× EC90 which were too numerous to count. Replicons selected by treatment with high-dose elbasvir were highly resistant to elbasvir, as measured by fold shifts in EC90 from values determined in cells treated with DMSO in the absence of drug. The resistance frequency was estimated to be 0.002% at 6 nM elbasvir. Population sequencing of the full length of the NS5A gene identified Y93N as the only substitution selected by exposure to 0.6 nM elbasvir. After exposure to 6 nM elbasvir, Q30D was detected in all colonies, usually associated with Y93N. Clonal analysis confirmed the Q30D-Y93N linkage in cells treated with 6 nM elbasvir. Y93N conferred high-level elbasvir resistance in vitro.
TABLE 3.
In vitro selection of RAVs in genotype 1a, 1b, and 3a replicons after exposure to varying concentrations of elbasvira
| Replicon cell type | Elbasvir treatment (×EC90)b | Recovered colony counts | Elbasvir susceptibility |
Observed amino acid substitution(s)e | ||
|---|---|---|---|---|---|---|
| EC50 (nM)c | EC90 (nM)c | Fold shift in EC90d | ||||
| Genotype 1a_H77 | 1,000 | 4 | 135 | 526 | 90,000 | Q30D, Q30D-Y93N |
| 100 | 56 | 5 | 15 | 3,000 | Y93N | |
| 10 | 204 | 2 | 11 | 2,000 | Not detected | |
| 1 | TMTC | <2 | <2 | <300 | Not detected | |
| DMSO | NA | 0.008 | 0.015 | <3 | Not detected | |
| Genotype 1b_con1 | 1,000 | 3 | 27 | 120 | 20,000 | Y93H, L31F-Y93H-V121I |
| 100 | 5 | NA | NA | NA | NA | |
| 10 | 38 | 0.6 | 12 | 2,000 | Y93H, V121I, Y93H-V121I | |
| 1 | 122 | 0.2 | 1 | 200 | Y93H | |
| DMSO | NA | 0.008 | 0.011 | <3 | Not detected | |
| Genotype 3_NC_009824f | 1,000 | 15 | 328 | 959 | 10,000 | E92K, Y93H |
| 100 | 23 | 245 | 518 | 5,000 | E92K, Y93H | |
| 10 | TMTC | 122 | 704 | 7,000 | E92K, Y93H | |
| 1 | TMTC | 157 | 725 | 7,000 | Y93H | |
| DMSO | NA | 0.2 | 0.8 | 6 | Not detected | |
EC, effective (inhibitory) concentration; TMTC, too many to count (as the result of a lawn of cells); NA, not available (because cells failed to expand).
Elbasvir concentration (in multiples of EC90) used to select RAVs.
Values represent single experiments.
Calculated as the EC90 for selected cell line/EC90 for the corresponding wild-type genotype 1a_H77 or genotype 1b_con1 referent (EC90 = 0.006 nM for both wild-type genotype 1 replicons).
Based on population sequencing. Because the lower limit of reliable detection is >20% with population sequencing (16), the failure to detect polymorphisms by this method does not necessarily imply that minority variants were not present in low concentrations.
Replicons were constructed using genotype 3 NS5A sequences in a con1 background (EC90 = 0.12 nM for the wild-type genotype 3 replicon).
For genotype 1b replicons, dose-dependent suppression of resistance was also observed. The respective numbers of surviving colonies selected were 122, 38, 5, and 3 in the presence of 0.006 nM, 0.06 nM, 0.6 nM, and 6 nM elbasvir (ranging from 1× to 1,000× EC90 for the control genotype 1b replicon). Compared with genotype 1a, fewer colonies emerged in genotype 1b at 1×, 10×, and 100× EC90 under identical experimental conditions. Y93H was the majority variation in the population treated with 0.006 nM elbasvir. Y93H and V121I were detected in cells treated with 0.06 nM elbasvir, and Y93H-V121I linkage was confirmed with clonal analysis. Cells exposed to 0.6 nM elbasvir failed to expand and thus were not available for testing. L31F, Y93H, and V121I were detected in cells treated with 6 nM elbasvir, and L31F-Y93H-V121I linkage was identified in 80% of tested clones. Y93H was associated with high-level elbasvir resistance.
A subgenomic replicon cell line harboring the genotype 3 NS5A sequence from a patient isolate (NC_009824) cloned into a con1 background with an elbasvir EC90 of 0.12 nM was used for the genotype 3 resistance selection experiments. As for genotype 1a and genotype 1b, genotype 3 replicons were treated with elbasvir at multiples of the EC90. Innumerable colonies grew in the presence of 0.12 nM and 1.2 nM elbasvir (corresponding to 1× and 10× EC90). Population sequencing detected low levels of the Y93H and E92K variants. Increasing the elbasvir concentrations to 12 nM and 120 nM (corresponding to 100× and 1,000× EC90) significantly reduced the number of resistant colonies to 23 and 15, respectively. Analysis of 40 individual clones did not identify a linkage between the Y93H and E92K variations. Y93H conferred high-level elbasvir resistance. Establishing a genotype 3 replicon with E92K alone was not successful despite many attempts, suggesting that the isolated E92K substitution rendered the variant unfit.
(iv) Activity of elbasvir against variants selected by other NS5A inhibitors.
NS5A RAVs previously identified in clinical trials with other NS5A inhibitors were included among the resistant variants selected in our experiments. Substitutions arose principally at amino acid residues 28 (genotype 1a and 1b), 30 (genotype 1a), 31 (genotypes 1a and 1b), and 93 (genotypes 1a, 1b, and 3) (17). In replicons containing individual NS5A substitutions, the elbasvir EC90 against 9 of the 13 genotype 1a variants tested had increases in EC90 of >10× (range, 12× to 1,333×), whereas EC90s for only 2 of 6 tested genotype 1b variants increased by >10× over that for wild type [L31F (44×) and Y93H (67×)] (Table 4). All 3 tested genotype 3a variants had increases in EC90 of >20×.
TABLE 4.
Effective inhibitory concentrations of elbasvir against variant repliconsa
| Replicon | EC50 |
EC90 |
||
|---|---|---|---|---|
| Mean ± SD (nM) | Fold shiftb | Mean ± SD (nM) | Fold shiftb | |
| 1a_H77 (WT) | 0.007 ± 0.004 | 0.017 ± 0.009 | ||
| 1a_M28T | 0.108 ± 0.035 | 15 | 0.378 ± 0.180 | 22 |
| 1a_M28V | 0.009 ± 0.006 | 1 | 0.016 ± 0.012 | 1 |
| 1a_M28A | 0.427 ± 0.220 | 61 | 1.527 ± 0.688 | 91 |
| 1a_Q30Dc | 3.7 ± 1.7 | 925 | 8.6 ± 3.5 | 1,433 |
| 1a_Q30H | 0.045 ± 0.033 | 6 | 0.108 ± 0.067 | 6 |
| 1a_Q30R | 0.114 ± 0.087 | 16 | 0.407 ± 0.212 | 24 |
| 1a_L31V | 0.431 ± 0.130 | 61 | 2.143 ± 0.737 | 127 |
| 1a_L31M | 0.070 ± 0.031 | 10 | 0.245 ± 0.162 | 15 |
| 1a_L31F | 0.673 ± 0.384 | 96 | 2.203 ± 1.605 | 131 |
| 1a_H58D | 0.041 ± 0.017 | 6 | 0.168 ± 0.066 | 10 |
| 1a_A92P | 0.007 ± 0.003 | 1 | 0.017 ± 0.005 | 1 |
| 1a_Y93H | 1.543 ± 0.782 | 220 | 5.930 ± 2.900 | 351 |
| 1a_Y93N | 6.552 ± 1.253 | 929 | 22.50 ± 8.544 | 1,333 |
| 1a_Y93C | 0.078 ± 0.004 | 11 | 0.208 ± 0.038 | 12 |
| 1a_Q30D_Y93Nc | 86.4 ± 60.1 | 21,600 | 257.7 ± 105.7 | 42,950 |
| 1b_con1 (WT) | 0.003 ± 0.002 | 0.006 ± 0.004 | ||
| 1b_L28M | 0.006 ± 0.003 | 2 | 0.020 ± 0.006 | 3 |
| 1b_L31M | 0.003 ± 0.001 | 1 | 0.010 ± 0.004 | 2 |
| 1b_L31F | 0.046 ± 0.015 | 15 | 0.265 ± 0.206 | 44 |
| 1b_L31V | 0.013 ± 0.008 | 4 | 0.057 ± 0.029 | 10 |
| 1b_Y93H | 0.050 ± 0.030 | 17 | 0.400 ± 0.200 | 67 |
| 1b_V121I | 0.0005 ± 0.0004 | 0.2 | 0.0010 ± 0.0007 | 0.2 |
| GT3_S52 (WT) | 0.14 ± 0.09 | 0.49 ± 0.19 | ||
| 3a_A30K | 7.0 ± 1.7 | 50 | 20 ± 6.5 | 41 |
| 3a_L31F | 20 ± 2 | 143 | 45 ± 8 | 92 |
| 3a_E92Kd | ||||
| 3a_Y93H | 68 ± 40 | 485 | 159 ± 56 | 324 |
RAV, resistance-associated variant; WT, wild type; EC50, effective concentration to inhibit growth by 50% compared to WT growth; EC90, effective concentration to inhibit growth by 90% compared to WT growth. The genotype 1a results were generated with a transient replicon system (except where indicated), whereas the genotype 1b and genotype 3a data were generated with a stable replicon system.
Relative to WT referent.
Results were generated with a stable replicon system where genotype 1a_H77 (WT referent) yielded an EC50 of 0.004 nM and an EC90 of 0.006 nM.
The replicon was unable to be generated due to poor fitness.
(v) Fitness of RAVs in a replicon assay.
The fitness of resistant variants was assessed in a replicon formation assay, in which the replicon RNA from resistant variation were transfected into Huh7 cells and replicon colonies were selected and counted. In this assay, substitutions at amino acid positions 30 and 31 in genotype 1a replicons were well tolerated (replicative capacity ranging from 41% to 100% of that of the wild type), whereas variations at position 93 reduced replicon fitness by ≥85% (Table 5). In particular, Y93H dramatically reduced replication to <1% compared to that of the wild-type genotype 1a replicon. Genotype 1b replication was sensitive to changes at amino acids 31 and 93. L31V/F and Y93H/C caused a dramatic decrease in genotype 1b fitness, reducing the colony number to <2% of that of the wild type. In contrast, Y93H/C in genotype 3 replicons modestly reduced replicative capacity to 50% of that of the wild type.
TABLE 5.
Replicative capacity (fitness) of variants in genotype 1a_H77 or genotype 1b_con1 replicons
| Genotype | Replicon variation | Fitness (% of wild-type control replication) |
|---|---|---|
| 1a | H77 (WT) | 100 |
| Q30D | 70 | |
| Q30E | 57 | |
| Q30K | 100 | |
| L31V | 41 | |
| Y93N | 6 | |
| Y93H | 1 | |
| Q30D_Y93N | 15 | |
| 1b | con1 (WT) | 100 |
| L31F | 0.1 | |
| L31V | 1.7 | |
| Y93C | 0.5 | |
| Y93H | 0.5 | |
| 3a | S52 (WT) | 100 |
| Y93H | 50 |
Phase 1b clinical trial results. (i) Subject accounting.
A total of 48 patients received elbasvir or placebo in an elbasvir dose-ranging study (3), including 17 patients with genotype 1a, 13 patients with genotype 1b, and 18 with genotype 3. All patients completed the 5-day course of therapy. Mislabeling of 7 samples from 3 patients (1 each infected with genotype 1a, 1b, or 3) occurred at the sequencing facility; these samples were therefore excluded from the analysis. Baseline virus from 1 additional genotype 3 patient could not be amplified. Consequently, only sequence data from the other 44 patients (including 16 with genotype 1a, 12 with genotype 1b, and 16 with genotype 3) were used for the resistance analyses.
Variants with NS5A substitutions at positions 28, 30, 58, and 93 (which had been previously identified as potential RAVs for NS5A inhibitors) were detected at baseline in 7 of 44 patients (15.9%) (Table 6). Except for possibly 1 patient infected with genotype 3 in the 10-mg elbasvir dose group, baseline NS5A variants did not appear to impact the magnitude of viral load reduction during treatment. In particular, M28V or Q30R in genotype 1a and Y93H in genotype 1b at baseline had little impact on the magnitude of viral load reduction during treatment.
TABLE 6.
Patients harboring baseline NS5A variants at positions 28, 30, 58, and 93, detected by population sequencing in the phase 1b triala
| Identification no. of patients with key baseline NS5A variant(s) | No. of evaluable patients in the corresponding full cohortb | Cohort specification |
Baseline NS5A polymorphism(s) by population sequencing | Variation(s) and % prevalence by clonal sequencing | Baseline HCV RNA level (viral load) (IU/ml) | Max viral load reduction (log10 IU/ml) in: |
||
|---|---|---|---|---|---|---|---|---|
| HCV subgenotype | Elbasvir dose (mg) | Patient | Cohortc | |||||
| 1 | 4 | 1a | 5 | M28V | M28V, 94 | 2.77 × 105 | −4.04 | −4.09 |
| 2 | 5 | 1a | 50 | Q30R | Not tested | 1.29 × 107 | −3.92 | −4.17 |
| 3 | 1 | 1b | 5 | P58S | Not tested | 6.71 × 106 | −4.11 | Not applicable |
| 4 | 5 | 1b | 50 | Y93H/Y | Y93H, 64 | 1.19 × 107 | −4.46 | −4.82 |
| 5 | 3 | 3 | 10 | A30A/E/K/T | A30K, 44; Y93H, 15 | 4.29 × 106 | <1 | −1.43 |
| 6 | 5 | 3 | 50 | P58S | Not tested | 8.17 × 106 | −2.62 | −3.24 |
| 7 | 2 | 3 | 0 (placebo) | A30A/L/S/V Y93H | Not tested | 3.99 × 106 | <1 | −0.28 |
Sequence data from 44 patients were used for the resistance analysis. Variants with NS5A substitutions at positions 28, 30, 58, and 93 were detected at baseline in 7 of 44 patients (15.9%) by population sequencing. Only data for patients in dosing cohorts where at least 1 patient had a NS5A polymorphism detected at baseline are shown. Seven cohorts enrolled 1 patient each with baseline variants and overall included a total of 25 patients. The last 2 columns allow comparison of the maximum viral load reduction in the patient with baseline variants to the other patients in the same cohort. All patients completed the 5-day course of monotherapy, including elbasvir in patients 1 through 6 and placebo in patient 7.
All evaluable patients with the same subgenotype infection treated with the same dose of elbasvir.
The geometric mean reduction in HCV RNA levels for the entire cohort of patients with the same subgenotype treated with the same dose of elbasvir exclusive of the 1 patient with the specified NS5A variant was computed for comparison.
Postbaseline sequencing was performed for 35/36 elbasvir recipients (97.2%) during follow-up visits. The sole exception was a genotype 1b patient who had HCV RNA levels of <1,000 IU/ml at all follow-up visits. These postbaseline sequences were compared to pretreatment results to identify treatment-emergent variants selected by elbasvir. The most common postbaseline variants (present in >10% of patients) encoded M28T, Q30R, L31V, or Y93H in genotype 1a, L31V or Y93H in genotype 1b, and A30K, L31F, and Y93H in genotype 3 (Fig. 1). Of substitutions at the 4 amino acid positions indicated in the graph, Y93H/C/N and L31V/M/I/F were the most prevalent, occurring in 83% (29/35) and 54% (19/35) of the postbaseline sequences obtained from the 35 patients. Postbaseline variations at position 30 were observed in 37% (12/35) of the patients, including 11 genotype 1a sequences with Q30R and 1 genotype 3a sequence with A/E/K/T30K. M28T, L28M, or V28A was noted in 20% (7/35) of the patients.
FIG 1.
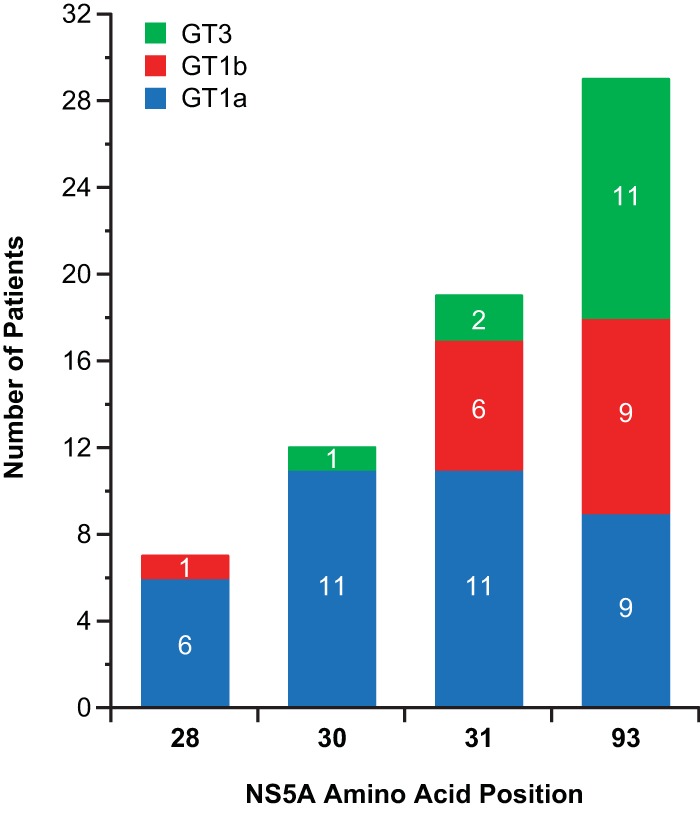
Prevalence of specific amino acid substitutions at established NS5A resistance loci detected by population sequencing after monotherapy with elbasvir in the phase 1b study. The column graph displays the most common postbaseline variants identified in patients infected with genotype (GT) 1a, 1b, or 3 in the phase 1b study. Among the polymorphisms examined, substitutions at amino acids 28, 30, 31, and 93 were each observed in more than 10% of evaluable patients.
For genotype 1a patients, 10/11 and 8/11, respectively, with variants at positions 30 and 31 still had changes at these loci at the last follow-up. For genotype 1b patients, 8/9 with variants at position 93 had persistent changes at this locus at the last follow-up. For genotype 3 patients, 11/11 with variants at position 93 had persistent changes at this locus at the last follow-up.
(ii) Genotype 1 infections.
All doses of elbasvir led to rapid HCV RNA reductions of 3.7 to 5.1 log10 IU/ml in genotype 1 infections (3). At the same dose, larger viral load reductions were achieved in genotype 1b infections than in infections with genotype 1a. The viral load decline after discontinuation of 5-day elbasvir monotherapy was more sustained in genotype 1b patients than in genotype 1a patients at the same elbasvir dose. In general, the types and prevalence of postbaseline RAVs selected within each subgenotype were similar across dosing levels.
In the 2 genotype 1a patients with pretreatment M28V or Q30R polymorphisms, >3-log viral load reductions were achieved with 5 mg and 50 mg dosing of elbasvir, respectively. In the patient with baseline M28V (which did not confer elbasvir resistance in vitro) treated with the 5-mg dose of elbasvir, Q30H/Q and L31L/V were additionally detected during the posttreatment follow-up period along with M128V/A. Clonal analysis identified linkages between M28Vand L31V as well as between M28A and Q30H, but no linkage was found between L31V and Q30H. Only M28V was detectable at the last follow-up on day 61 by population sequencing. In the genotype 1a patient with baseline Q30R (associated with a 24-fold increase in elbasvir EC90 in vitro) treated with the 50-mg dose of elbasvir, Q30R was detected with L31V from the end of treatment until the last follow-up on day 56. Clonal sequencing was not performed for this patient. One genotype 1b patient with Y93H/Y and A92T/A mixtures at baseline treated with the 50-mg dose of elbasvir achieved a >4-log viral load reduction (even though Y93H was associated with a 67-fold increase in elbasvir EC90 in vitro). After treatment cessation, clonal sequencing identified L28M linked to Y93H and L31V linked to A92K, without linkage between Y93H and either L31V or A92K. At the last visit on day 59, only the Y93H/Y and L31V/L mixtures were detected by population sequencing.
(iii) Genotype 3 infections.
Antiviral responses were less robust for genotype 3 than for genotype 1 infections in the 10-mg elbasvir dosing group, but mean HCV RNA reductions of ∼3 log were achieved at the 50- and 100-mg doses (3). Posttreatment Y93H was found in all 10 patients in the 50- and 100-mg elbasvir dose groups and persisted through the last follow-up in each case. L31F was also detected in 2 of these patients.
One of the 3 evaluated patients with genotype 3 infection in the 10-mg dose group harbored a baseline A30A/E/K/T mixture and had a <1-log10 IU/ml reduction in the level of HCV RNA at nadir compared to a mean 1.43-log10 drop in viremia in the other 2 patients in the 10-mg dosing group without detectable RAVs at baseline. Population sequencing showed that A30A/E/K/T converted to A30K (which conferred a 41-fold increase in elbasvir EC90 in vitro) from treatment discontinuation through the last follow-up on day 61.
DISCUSSION
In preclinical experiments, elbasvir exhibited more potent antiviral activity against genotype 1a and 1b (EC90, 0.006 nM) than against genotype 3 replicons (EC90, 0.12 nM). In de novo resistance selection assays, elbasvir suppressed the emergence of resistant genotype 1 and genotype 3 colonies in a dose-dependent manner. Under the same selection pressure (expressed as multiples of the elbasvir EC90), resistant colonies emerged less frequently in genotype 1b than genotype 1a at 10× or 100× EC90. At 1,000× EC90, the frequencies of resistant colonies were similar in genotype 1a and 1b replicons; most of these RAVs involved more than one resistance locus, suggesting that an elbasvir dose of 1,000× EC90 can suppress RAVs involving only a single amino acid change. In genotype 3, increasing the selection pressure from 10× EC90 to 100× EC90 significantly reduced the number of resistant colonies, although a further increase to 1,000× EC90 did not lead to an incremental reduction. Increased doses selected cells that were highly resistant and contained single and double amino acid substitutions. Although elbasvir retained meaningful activity against some clinically relevant NS5A variants, susceptibility in vitro was significantly reduced against genotype 1a and 3 variants harboring particular substitutions at certain loci, especially at position 93 (Y93N and Y93H, respectively). In contrast, substitutions in genotype 1b (including Y93H) caused much less loss of potency. The sequence context of NS5A might impact the level of resistance beyond what can be attributed to individual RAVs (17). Cross-resistance to NS3 protease inhibitors would not be expected (18).
In a subsequent small dose-escalating study of elbasvir given as 5-day monotherapy, viral load reductions were greater for genotype 1 than genotype 3 infections, especially at lower elbasvir doses (3). The decreases in HCV RNA levels were generally more pronounced and longer lasting in infections caused by genotype 1b than by genotype 1a. Robust antiviral responses were observed in the presence of baseline M28V or Q30R variants in genotype 1a infection or the baseline Y93H variant in genotype 1b infections. Variations at resistance loci common to other NS5A inhibitors emerged following exposure to elbasvir monotherapy. Population sequencing detected NS5A RAVs less frequently in genotype 1b than in genotype 1a infections. Viral rebound following cessation of therapy was generally slower with genotype 1b than with genotype 1a. With clonal sequencing, polymorphisms could be found in a large proportion of the viral population irrespective of genotype. The evolution of variants was dynamic, with the linkage of substitutions changing over time.
The antiviral activity and resistance profile of elbasvir observed in patients were generally predictable from the preclinical findings. Similar RAVs were selected in vitro and in vivo. Population sequencing of viruses from patients enrolled in the phase 1b trial confirmed M28T, Q30R, L31V, and Y93H in genotype 1a, L31V and Y93H in genotype 1b, and A30K, L31F, and Y93H in genotype 3 as the predominant RAVs selected by elbasvir monotherapy. Variants containing these substitutions had reduced susceptibility to elbasvir in HCV replicons with variable replicative capacity. The phenotypic impact of minority variants has not yet been established in most cases.
The preclinical genotypic and phenotypic analyses presaged the clinical findings in the phase 1b study (3). Fewer resistant colonies were selected by elbasvir among genotype 1b than genotype 1a or 3 quasispecies across the dosing range tested in the laboratory. These in vitro observations were consistent with the greater antiviral effect seen in patients with genotype 1b infections relative to genotype 1a or 3 infections during the clinical trial. In contrast, in vitro replicative capacity (“viral fitness”) did not reliably explain viral-load kinetics in vivo. Variants that impaired fitness in the colony formation assay (illustrated by Y93 substitutions in genotype 1a) often rebounded as major species, persisting after cessation of elbasvir monotherapy.
The intriguing observation that some RAVs with impaired fitness in the colony formation assay rebounded as major species in patients after cessation of elbasvir monotherapy may reflect assay or biological factors. Since in vitro and in vivo measurements have different targets, discordance might be expected in some cases. While the replicon assay yields the number of cells that support HCV replication in the laboratory, viral load measurements give the actual number of circulating viruses in the patient. The impact of a substitution within NS5A may to some degree depend on the surrounding context, including the degree of phosphorylation (19). Adaptive amino acid changes elsewhere in and/or outside NS5A in the virus not incorporated into the replicon could enhance replication of certain RAVs in vivo. Most known drug-associated substitutions have been located in domain I, and much less is known about the impact of variants in domains II and III (20). Substitutions in domains II and III typically do not directly cause a shift in replicon EC50; however, the effect of these substitutions on viral replication in the setting of polymorphisms in domain I has not been systematically investigated.
The differential impact of RAVs on the outcomes of genotype 1a and 1b infections in the clinical trials was consistent with the lower degree of resistance observed in vitro with substitutions in genotype 1b compared to genotype 1a replicons. In the pivotal C-EDGE study of treatment-naive patients (21), SVR12 was attained in 2 of 9 (22%) evaluable patients infected with genotype 1a with baseline NS5A RAVs conferring >5-fold-decreased susceptibility to elbasvir, as opposed to 9 of 10 (90%) patients with baseline NS5A RAVs conferring ≤5-fold-decreased susceptibility and 133 of 135 (99%) patients without baseline NS5A RAVs. In contrast, 16 of 17 (94%) evaluable patients with genotype 1b infections harboring baseline NS5A RAVs conferring >5-fold-decreased susceptibility achieved SVR12.
Results with other NS5A inhibitors suggest the generalizability of this observation (22). For example, in patients with baseline NS5A RAVs treated with the NS5A inhibitor daclatasvir combined with asunaprevir (a NS3 protease inhibitor) and beclabuvir (a nonnucleoside NS5B polymerase inhibitor), SVR12 was achieved in 25 (74%) of the 34 patients with genotype 1a infection versus all 17 (100%) patients with genotype 1b infection (23). There was no evident association between baseline NS3 or NS5B variants and SVR12. As found in the C-EDGE trials (21, 24), the UNITY-1 findings again indicated a clinically meaningful impact of baseline NS5A RAVs on the outcome of interferon-sparing treatment exclusively in genotype 1a as opposed to genotype 1b infections.
Elbasvir plus grazoprevir (an investigational NS3/4A protease inhibitor) as a once-daily, oral, single fixed-dose combination tablet is currently being developed for treatment of chronic HCV infection (1, 2, 21, 24–26). Further analyses of the phase 3 trials of grazoprevir-elbasvir will soon provide more critical data concerning the safety and efficacy of this novel double direct-acting antiviral combination.
ACKNOWLEDGMENTS
We are indebted to all the patients, health care providers, and investigators participating in the parent study. We also thank Robert Nachbar, Donald Graham, David Nickle, Robert Chase, Nicolas Morin, and Ernest Asante-Appiah of Merck for their contributions to the analysis. We are likewise indebted to Karyn Davis from Merck for technical assistance in the preparation of the manuscript.
Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., is developing elbasvir (MK-8742) as a component of combination therapy for chronic HCV infections. The company sponsored and funded the clinical study and the derivative analysis reported here. A penultimate version of the paper was reviewed by the sponsor.
As present or former employees of Merck, authors may own stock and/or stock options in the company. All authors had full access to any pertinent data upon request. Each coauthor approved an essentially final version of the manuscript. The opinions expressed in this report represent the consensus of the authors and do not necessarily reflect the formal position of Merck.
REFERENCES
- 1.Lawitz E, Gane E, Pearlman B, Tam E, Ghesquiere W, Guyader D, Alric L, Bronowicki JP, Lester L, Sievert W, Ghalib R, Balart L, Sund F, Lagging M, Dutko F, Shaughnessy M, Hwang P, Howe AY, Wahl J, Robertson M, Barr E, Haber B. 2015. Efficacy and safety of 12 weeks versus 18 weeks of treatment with grazoprevir (MK-5172) and elbasvir (MK-8742) with or without ribavirin for hepatitis C virus genotype 1 infection in previously untreated patients with cirrhosis and patients with previous null response with or without cirrhosis (C-WORTHY): a randomised, open-label phase 2 trial. Lancet 385:1075–1086. doi: 10.1016/S0140-6736(14)61795-5. [DOI] [PubMed] [Google Scholar]
- 2.Sulkowski M, Hezode C, Gerstoft J, Vierling JM, Mallolas J, Pol S, Kugelmas M, Murillo A, Weis N, Nahass R, Shibolet O, Serfaty L, Bourliere M, DeJesus E, Zuckerman E, Dutko F, Shaughnessy M, Hwang P, Howe AY, Wahl J, Robertson M, Barr E, Haber B. 2015. Efficacy and safety of 8 weeks versus 12 weeks of treatment with grazoprevir (MK-5172) and elbasvir (MK-8742) with or without ribavirin in patients with hepatitis C virus genotype 1 mono-infection and HIV/hepatitis C virus co-infection (C-WORTHY): a randomised, open-label phase 2 trial. Lancet 385:1087–1097. doi: 10.1016/S0140-6736(14)61793-1. [DOI] [PubMed] [Google Scholar]
- 3.Yeh W, Lipardi C, Jumes P, De Lepeleire I, Van Den Bulk N, Caro L, Huang X, Mangin E, Nachbar R, Gane E, Popa S, Ghicavii N, Wagner F, Butterton JR. 2013. MK-8742, a HCV NS5A inhibitor with a broad spectrum of HCV genotypic activity, demonstrates potent antiviral activity in genotype-1 and -3 HCV-infected patients. Hepatology 58:438A. [Google Scholar]
- 4.Coburn CA, Meinke PT, Chang W, Fandozzi CM, Graham DJ, Hu B, Huang Q, Kargman S, Kozlowski J, Liu R, McCauley JA, Nomeir AA, Soll RM, Vacca JP, Wang D, Wu H, Zhong B, Olsen DB, Ludmerer SW. 2013. Discovery of MK-8742: an HCV NS5A inhibitor with broad genotype activity. ChemMedChem 8:1930–1940. doi: 10.1002/cmdc.201300343. [DOI] [PubMed] [Google Scholar]
- 5.Welsch C, Zeuzem S. 2012. Clinical relevance of HCV antiviral drug resistance. Curr Opin Virol 2:651–655. doi: 10.1016/j.coviro.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 6.American Association for the Study of Liver Diseases and Infectious Diseases Society of North America. 2015. HCV guidance: recommendations for testing, managing, and treating hepatitis C. http://www.hcvguidelines.org Accessed 29 January 2015. [Google Scholar]
- 7.HCV Phenotype Working Group, HCV Drug Development Advisory Group. 2015. Clinically relevant HCV drug resistance mutations figure and tables. Ann Forum Collab HIV Res 14:1–610. http://www.idsociety.org/uploadedFiles/IDSA/Hepatitis_C/For_IDSA_Members/ForumforCollaborativeHIV-ClinicallyRelevantHCVDrug.pdf Accessed 29 January 2015. [Google Scholar]
- 8.Barnard RJ, Howe JA, Ogert RA, Zeuzem S, Poordad F, Gordon SC, Ralston R, Tong X, Sniukiene V, Strizki J, Ryan D, Long J, Qiu P, Brass CA, Albrecht J, Burroughs M, Vuocolo S, Hazuda DJ. 2013. Analysis of boceprevir resistance associated amino acid variants (RAVs) in two phase 3 boceprevir clinical studies. Virology 444:329–336. doi: 10.1016/j.virol.2013.06.029. [DOI] [PubMed] [Google Scholar]
- 9.Sullivan JC, De Meyer S, Bartels DJ, Dierynck I, Zhang EZ, Spanks J, Tigges AM, Ghys A, Dorrian J, Adda N, Martin EC, Beumont M, Jacobson IM, Sherman KE, Zeuzem S, Picchio G, Kieffer TL. 2013. Evolution of treatment-emergent resistant variants in telaprevir phase 3 clinical trials. Clin Infect Dis 57:221–229. doi: 10.1093/cid/cit226. [DOI] [PubMed] [Google Scholar]
- 10.Ribeiro RM, Li H, Wang S, Stoddard MB, Learn GH, Korber BT, Bhattacharya T, Guedj J, Parrish EH, Hahn BH, Shaw GM, Perelson A. 2012. Quantifying the diversification of hepatitis C virus (HCV) during primary infection: estimates of the in vivo mutation rate. PLoS Pathog 8:e1002881. doi: 10.1371/journal.ppat.1002881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cento V, Mirabelli C, Salpini R, Dimonte S, Artese A, Costa G, Mercurio F, Svicher V, Parrotta L, Bertoli A, Ciotti M, Di Paolo D, Sarrecchia C, Andreoni M, Alcaro S, Angelico M, Perno CF, Ceccherini-Silberstein F. 2012. HCV genotypes are differently prone to the development of resistance to linear and macrocyclic protease inhibitors. PLoS One 7:e39652. doi: 10.1371/journal.pone.0039652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hara K, Rivera MM, Koh C, Demino M, Page S, Nagabhyru PR, Rehermann B, Liang TJ, Hoofnagle JH, Heller T. 2014. Sequence analysis of hepatitis C virus from patients with relapse after a sustained virological response: relapse or reinfection? J Infect Dis 209:38–45. doi: 10.1093/infdis/jit541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou S, Liu R, Baroudy BM, Malcolm BA, Reyes GR. 2003. The effect of ribavirin and IMPDH inhibitors on hepatitis C virus subgenomic replicon RNA. Virology 310:333–342. doi: 10.1016/S0042-6822(03)00152-1. [DOI] [PubMed] [Google Scholar]
- 14.Liu R, Abid K, Pichardo J, Pazienza V, Ingravallo P, Kong R, Agrawal S, Bogen S, Saksena A, Cheng KC, Prongay A, Njoroge FG, Baroudy BM, Negro F. 2007. In vitro antiviral activity of SCH446211 (SCH6), a novel inhibitor of the hepatitis C virus NS3 serine protease. J Antimicrob Chemother 59:51–58. [DOI] [PubMed] [Google Scholar]
- 15.Koletzki D, Pattery T, Fevery B, Vanhooren L, Stuyver LJ. 2013. Amplification and sequencing of the hepatitis C virus NS3/4a protease and the NS5b polymerase regions for genotypic resistance detection of clinical isolates of subtypes 1a and 1b. Methods Mol Biol 1030:137–149. doi: 10.1007/978-1-62703-484-5_12. [DOI] [PubMed] [Google Scholar]
- 16.Leitner T, Halapi E, Scarlatti G, Rossi P, Albert J, Fenyö EM, Uhlen M. 1993. Analysis of heterogeneous viral populations by direct DNA sequencing. Biotechniques 15:120–127. [PubMed] [Google Scholar]
- 17.Hernandez D, Zhou N, Ueland J, Monikowski A, McPhee F. 2013. Natural prevalence of NS5A polymorphisms in subjects infected with hepatitis C virus genotype 3 and their effects on the antiviral activity of NS5A inhibitors. J Clin Virol 57:13–18. doi: 10.1016/j.jcv.2012.12.020. [DOI] [PubMed] [Google Scholar]
- 18.Howe AY, Black S, Curry S, Ludmerer SW, Liu R, Barnard RJ, Newhard W, Hwang PM, Nickle D, Gilbert C, Caro L, DiNubile MJ, Mobashery N. 2014. Virologic resistance analysis from a phase 2 study of MK-5172 combined with pegylated interferon/ribavirin in treatment-naive patients with hepatitis C virus genotype 1 infection. Clin Infect Dis 59:1657–1665. doi: 10.1093/cid/ciu696. [DOI] [PubMed] [Google Scholar]
- 19.Xiong W, Yang J, Wang M, Wang H, Rao Z, Zhong C, Xin X, Mo L, Yu S, Shen C, Zheng C. 2015. Vinexin β interacts with hepatitis C virus NS5A, modulating its hyperphosphorylation to regulate viral propagation. J Virol 89:7385–7400. doi: 10.1128/JVI.00567-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim S, Welsch C, Yi MK, Lemon SM. 2011. Regulation of the production of infectious genotype 1a hepatitis C virus by NS5A domain III. J Virol 85:6645–6656. doi: 10.1128/JVI.02156-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeuzem S, Ghalib R, Reddy KR, Pockros PJ, Ben Ari Z, Zhao Y, Brown DD, Wan S, DiNubile MJ, Nguyen BY, Robertson MN, Wahl J, Barr E, Butterton JR. 2015. Grazoprevir-elbasvir combination therapy for treatment-naive cirrhotic and noncirrhotic patients with chronic HCV genotype 1, 4, or 6 infection: a randomized trial. Ann Intern Med 163:1–13. doi: 10.7326/M15-0785. [DOI] [PubMed] [Google Scholar]
- 22.Gao M. 2013. Antiviral activity and resistance of HCV NS5A replication complex inhibitors. Curr Opin Virol 3:514–520. doi: 10.1016/j.coviro.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 23.Poordad F, Sievert W, Mollison L, Bennett M, Tse E, Bräu N, Levin J, Sepe T, Lee SS, Angus P, Conway B, Pol S, Boyer N, Bronowicki JP, Jacobson I, Muir AJ, Reddy KR, Tam E, Ortiz-Lasanta G, de Lédinghen V, Sulkowski M, Boparai N, McPhee F, Hughes E, Swenson ES, Yin PD. 2015. Fixed-dose combination therapy with daclatasvir, asunaprevir, and beclabuvir for noncirrhotic patients with HCV genotype 1 infection. JAMA 313:1728–1735. doi: 10.1001/jama.2015.3860. [DOI] [PubMed] [Google Scholar]
- 24.Kwo P, Gane E, Peng C-Y, Pearlman B, Vierling J, Serfaty L, Buti Ferret M, Sharfran S, Stryszak P, Lin L, Gress J, Wahl J, Barr E, Robertson M, Haber B. 2015. Efficacy and safety of grazoprevir/elbasvir +/− RBV for 12 weeks in patients with HCV G1 or G4 infection who previously failed peginterferon/RBV: C-EDGE treatment-experienced trial, abstr P0886. In 2015 Int Liver Congress: 50th Annu Meet Eur Assoc Study Liver (EASL), Vienna, April 22 to 26, 2015. [Google Scholar]
- 25.Rockstroh JK, Nelson M, Katlama C, Lalezari J, Mallolas J, Bloch M, Matthews G, Saag MS, Zamor P, Orkin C, Gress J, Klopfer S, Shaughnessy M, Wahl J, Nguyen BY, Barr E, Platt H, Robertson M, Sulkowski S. 2015. Efficacy and safety of grazoprevir (MK-5172) and elbasvir (MK-8742) in patients with hepatitis C virus and HIV co-infection (C-EDGE CO-INFECTION): a non-randomised, open-label trial. Lancet HIV 2:e319–e327. doi: 10.1016/S2352-3018(15)00114-9. [DOI] [PubMed] [Google Scholar]
- 26.Forns X, Gordon SC, Zuckerman E, Lawitz E, Calleja JL, Hofer H, Gilbert C, Palcza J, Howe AY, DiNubile MJ, Robertson MN, Wahl J, Barr E, Buti M. 2015. Grazoprevir and elbasvir plus ribavirin for chronic HCV genotype-1 infection after failure of combination therapy containing a direct-acting antiviral agent. J Hepatol 63:564–572. doi: 10.1016/j.jhep.2015.04.009. [DOI] [PubMed] [Google Scholar]