

Abstract
Ribosomal protein S4 nucleates assembly of the 30S ribosome 5′ and central domains, which is crucial for the survival of cells. Protein S4 changes the structure of its 16S rRNA binding site, passing through a non-native intermediate complex before forming native S4-rRNA contacts. Ensemble FRET was used to measure the thermodynamic stability of non-native and native S4 complexes in the presence of Mg2+ ions and other 5′-domain proteins. Equilibrium titrations of Cy3-labeled 5′-domain RNA with Cy5-labeled protein S4 showed that Mg2+ ions preferentially stabilize the native S4-rRNA complex. In contrast, ribosomal proteins S20 and S16 act by destabilizing the non-native S4-rRNA complex. The full cooperative switch to the native complex requires S4, S16, and S20 and is achieved to a lesser degree by S4 and S16. The resulting thermodynamic model for assembly of the 30S body illustrates how ribosomal proteins selectively bias the equilibrium between alternative rRNA conformations, increasing the cooperativity of rRNA folding beyond what can be achieved by Mg2+ ions alone.
Keywords: RNA–protein interactions, ribosomal protein S4, FRET, ribosome assembly
INTRODUCTION
The biosynthesis of new ribosomes is necessary for cell growth and is regulated in response to starvation or other stress conditions. To ensure each ribosomal subunit assembles completely, the ribosomal proteins bind to the 16S rRNA in a hierarchy that arises from protein-dependent conformational changes in the 16S rRNA (Held et al. 1974; Held and Nomura 1975; Nowotny and Nierhaus 1988). Biochemical and biophysical studies have confirmed the cooperative binding of proteins to the central and 3′ domains of the 16S rRNA (Stern et al. 1989; Jagannathan and Culver 2003; Grondek and Culver 2004; Recht and Williamson 2004). Nevertheless, the molecular mechanisms underlying such cooperative behavior and its influence on the assembly landscape are yet to be fully understood, particularly when ribosomal proteins influence regions of the rRNA remote from their immediate binding sites.
The 5′ domain of the E. coli 16S rRNA forms the body of the 30S subunit (Fig. 1A), and folds rapidly and independently of other domains during the 30S assembly (Powers et al. 1993; Weitzmann et al. 1993; Adilakshmi et al. 2008; Mulder et al. 2010). Hydroxyl radical footprinting experiments showed that the 16S 5′-domain RNA alone folds to a native-like structure in the presence of 10 mM MgCl2 (Adilakshmi et al. 2005). Some native interactions in helices 12 and 18 were not observed in the absence of proteins, however, and RNA tertiary interactions between helices 6, 10, 15, and 17 formed 10–60 sec later than other interactions owing to alternative base-pairing of these helix junctions (Adilakshmi et al. 2005). Thus, Mg2+ alone is insufficient for complete assembly of the 30S body.
FIGURE 1.
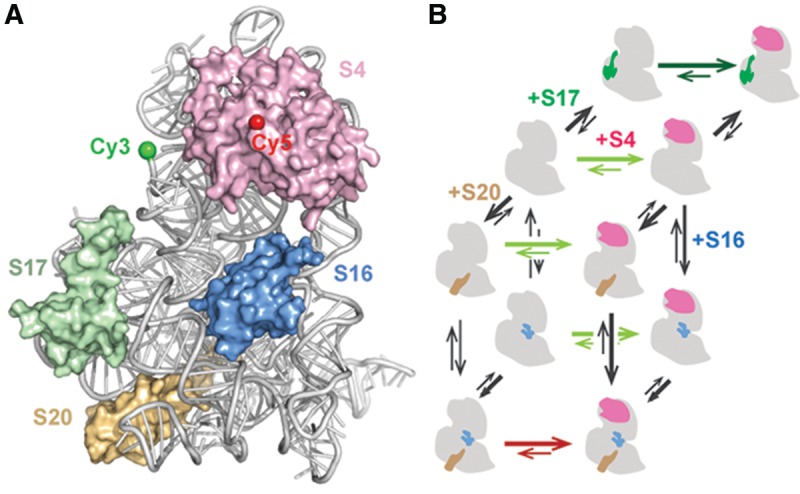
Assembly of the 16S 5′ domain. (A) Proteins S4 (pink), S17 (green), S16 (blue), and S20 (wheat) bound to nucleotides 21–556 of the Escherichia coli 16S rRNA 5′-domain RNA (gray) in the 70S ribosome (2i2p). Green and red spheres indicate positions of Cy3 and Cy5, respectively. (B) Thermodynamic cycles for assembly of the 16S 5′ domain. Thermodynamically favorable steps predicted by the Nomura map are shown with thick arrows. Green and red arrows show a preference for the native and intermediate S4-5′-domain RNA complexes, respectively.
Three primary assembly proteins (S4, S17, and S20) each bind a different helix junction in the 16S 5′ domain (Powers and Noller 1995a; Brodersen et al. 2002) and enable recruitment of the secondary assembly protein S16 (Fig. 1). Protein S4 recognizes a five-way junction between 16S helices 3, 4, 16, 17, and 18 (Fig. 1A; Stern et al. 1986; Gerstner et al. 2001; Bellur and Woodson 2009) and nucleates in vitro assembly of the 5′ and central domains (Nowotny and Nierhaus 1988). Time-resolved footprinting and smFRET experiments showed that S4 binding induces a series of conformational changes in the 5WJ, leading to the native S4-rRNA complex (Mayerle et al. 2011; Kim et al. 2014). Remarkably, S4 binding also stabilizes tertiary interactions throughout the 5′-domain RNA, which includes interactions as much as 60 Å away from the S4 binding site. Protein S17 interacts with 16S helix 11 and also indirectly stabilizes tertiary interactions in the lower half of the 5′ domain (Ramaswamy and Woodson 2009a). Binding of protein S20 to helices 6, 8, 9, and 10 stabilizes those helix junctions and mediates interactions with helix 44 later in 30S assembly (Dutca and Culver 2008; Ramaswamy and Woodson 2009a).
As each primary ribosomal protein favors specific conformations of the 16S 5′-domain, allosteric interactions between protein binding sites likely govern the thermodynamics of assembly. We established a FRET assay to measure the populations of native and non-native (intermediate) S4-rRNA complexes (Kim et al. 2014) at varying Mg2+ concentrations and in the presence of different 5′-domain proteins. In the mature 30S ribosome, 16S helix 3 docks under a pseudoknot in helix 18 (530 loop), resulting in efficient energy transfer from a Cy3 donor placed near the end of helix 3 and a Cy5 acceptor on protein S4 (Fig. 1A). During S4 recognition, however, the system passes through an intermediate in which helix 3 is flipped away from helix 18 and S4, resulting in a low FRET state. Hydroxyl radical footprinting showed that proteins S4, S17, and S20 favored the flipped intermediate state, but addition of protein S16 switched the complexes to the native conformation (Ramaswamy and Woodson 2009b).
In this study, we used a FRET assay to measure the influence of Mg2+ ions and 5′-domain proteins on the thermodynamics of S4 binding (Fig. 1B). The population distribution between intermediate and native S4-rRNA complexes enabled us to understand the different roles played by magnesium ions and ribosomal proteins. Changes in the equilibrium between the native and intermediate complexes arising from thermodynamic cooperativity between 5′-domain proteins will also be discussed.
RESULTS AND DISCUSSION
Design of dye-labeled 5′-domain RNA
The RNA was labeled by hybridizing a Cy3-DNA oligomer to a 3′-extension of the E. coli 16S 5′-domain RNA. To design the extension, 30–40-nt RNA sequences with high G–C content were screened for predicted alterations to the 5WJ secondary structure using Mfold. Ten extended 5′-domain RNA sequences (Supplemental Table S1) with the largest preference for the native secondary structure (>5 kcal/mol) were tested for their ability to base pair with a complementary oligodeoxynucleotide using electrophoretic mobility gel shift assays. Subnanomolar binding affinities were observed for several RNA–oligomer pairs (Fig. 2A). Some 3′ extensions such as E9 produced multiple RNA–primer complex bands and were not considered further (Fig. 2A).
FIGURE 2.
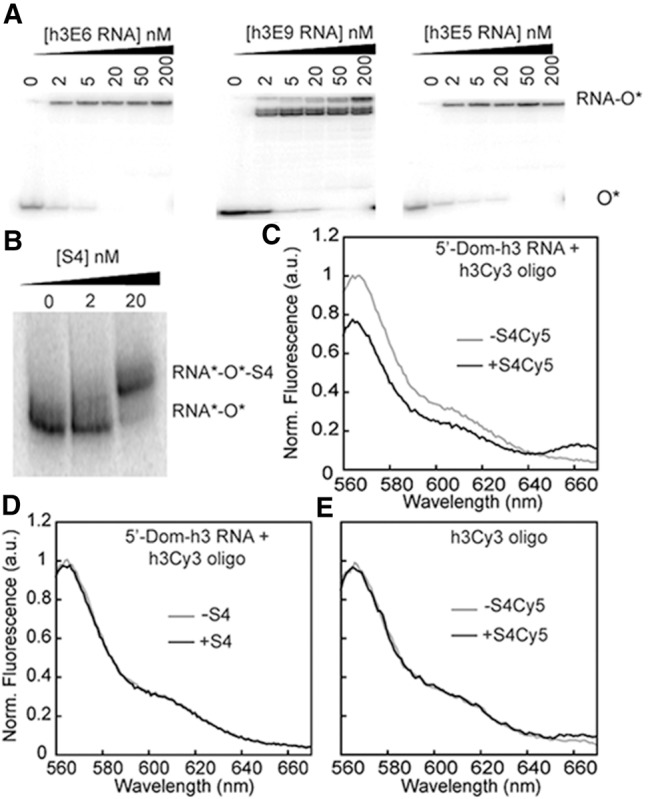
FRET assay for S4-rRNA interactions. (A) DNA oligonucleotides complementary to 3′-extensions of 5′ domains (Supplemental Table S1) were tested for annealing by EMSA. Sample gels for Oligo 6 (left panel), Oligo 9 (center), and Oligo 5 (right). The 5′-Dom-h3E9 RNA adopts multiple conformations. 5′-Dom-h3E5 RNA and Oligo5 was selected for fluorescence experiments. (B) EMSA shows that protein S4 binds the extended RNA–DNA complex. (C) FRET upon addition of 10 nM S4-Cy5 to 5′-Dom-h3E5 RNA-h3-oligo-Cy3 complex (1.5 nM each in 20 mM MgCl2) at 37°C. (D) Addition of unlabeled S4 (10 nM) at similar conditions shows no significant change in donor fluorescence. (E) Addition of acceptor-labeled protein S4 to h3Cy3 oligo shows no FRET signal.
Native gel mobility shift assays showed that E. coli S4 stably binds the extended 5′-domain RNA–oligomer complex, as expected (Fig. 2B). The 5′-domain RNA-Cy3–oligomer complex was next titrated with Cy5-labeled S4 protein (Materials and Methods), resulting in a maximum average FRET efficiency EFRET = 0.18 for the 5′-domain-S4 complex (Fig. 2C). The decrease in Cy3 donor fluorescence and increase in Cy5 acceptor fluorescence was not observed in control experiments performed with unlabeled protein S4, unlabeled RNA–oligomer complex or labeled oligomer plus labeled-S4 protein (no RNA) (Fig. 2D,E).
Protein S4 and 5′-domain RNA complexes are in equilibrium
To obtain equilibrium constants for S4 binding to the flipped and docked conformations of the 5′-domain RNA, the observed increase in EFRET with S4-Cy5 concentration was fit to a four-state thermodynamic cycle (Fig. 3A). Our model assumes that the free RNA can exist in two states (helix 3 flipped or docked) and that both states can bind S4. Dissociation equilibrium constants for both native (docked) and flipped complexes, KN and KF, were determined by nonlinear least-squares fits of S4 titration curves to a quadratic form of the binding polynomial for two complexes (Equation 1). We used a quadratic equation because S4 binds the rRNA very tightly, with KN = 0.08 nM in 20 mM MgCl2 (Supplemental Table S2). The fits yielded K2, which describes the relative population distribution between the docked (high FRET) and flipped (low FRET) complexes, and (1 + K1) KN, which describes the overall probability of forming the native complex (see Supplemental Material).
FIGURE 3.
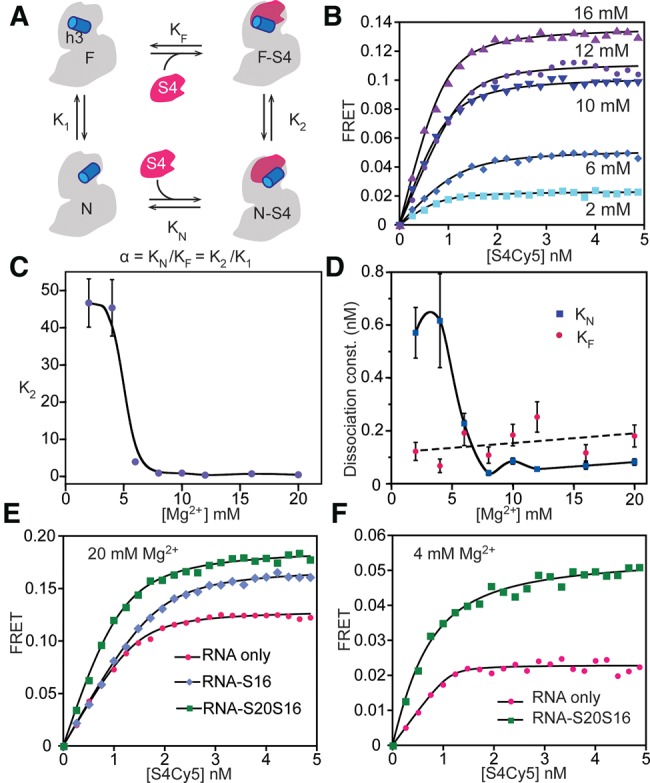
Thermodynamic cycle for S4 binding. (A) Four-state model for S4 titrations contains high-FRET native (N-S4) and low-FRET intermediate (F-S4) S4-bound complexes and corresponding unbound states (N and F). (B) Titrations with S4-Cy5 at different [Mg2+]. Lines represent the best least-squares fit to Equation 1. The initial slope is sensitive to β = KN(1 + K1). The endpoints are proportional to (EN + EF K2)/(1 + K2). See Supplemental Material for further details. (C) Ratio of N-S4 and F-S4 complexes, K2, as a function of [Mg2+]. The smooth curve is only intended to guide the eye. (D) Equilibrium dissociation constant for the native complex (KN, blue diamonds), using K1 from hydroxyl radical footprinting of free 5′-domain RNA. The calculated dissociation constant for the flipped complex (KF, red circles) does not increase with Mg2+. (E) S4-5′-domain complexes give higher FRET signal in 20 mM MgCl2 in the presence of both proteins S16 and S20 (green squares) compared to only S16 (blue diamonds) or without any additional protein (pink circles). (F) Titration curves in 4 mM [Mg2+]. S4-RNA complexes formed in the presence of proteins S20 and S16 (green) show high FRET compared to S4 alone (pink).
To evaluate K1 and KN, we used information from earlier hydroxyl radical footprinting experiments on the 16S 5′-domain RNA that measured the fraction of free RNA in the native conformation (helix 3 docked) at different Mg2+ concentrations and in the presence of different primary assembly proteins (Ramaswamy and Woodson 2009a). Estimates of K1 from footprinting data allowed us to calculate binding constants KF, KN, and the linkage α = KN/KF = K2/K1, which describes how strongly S4 binding is coupled to docking of helix 3 compared to the free RNA. Therefore, these experiments quantify previously observed changes in the equilibrium between the intermediate and native states (K2) in the presence of magnesium ions and primary and secondary assembly proteins (S17, S20, and S16) that bind the 5′-domain RNA.
Mg2+ ions stabilize the native S4 complex
Previous smFRET experiments in which the 5′-domain RNA was tethered to a microscope slide showed that higher Mg2+ concentrations favor the docked (native) complex (Kim et al. 2014). These experiments only counted long-lived complexes that could be captured on the slide. To measure the effect of Mg2+ on S4 binding in solution, Cy3-5′-domain RNA was titrated with S4-Cy5 in 0–20 mM MgCl2 (Fig. 3B). A weak FRET signal in the absence of Mg2+ (data not shown) indicated that Mg2+ is essential for stable S4 binding, as expected (Gerstner et al. 2001). A small but measurable FRET signal in 2 mM MgCl2 increased with Mg2+ concentration to a maximum FRET efficiency at 12 mM MgCl2 (Fig. 3B). This observation corresponded to a shift from the flipped intermediate complex (K2 = 47 ± 6) in 2 mM MgCl2 toward the native docked conformation in 12 mM MgCl2 (K2 = 0.37 ± 0.01) (Fig. 3C; Supplemental Table S2).
Our thermodynamic model showed that this shift arises from a substantial stabilization of the native S4 complex by Mg2+ ions. The equilibrium constant K1 for docking helix 3 taken from footprinting experiments decreased 10-fold over our Mg2+ titration, from 10 in 2 mM MgCl2 to 1 in 20 mM MgCl2 (Supplemental Table S2). Taking these values for K1, the dissociation constant for the native complex also decreased from KN = 0.6 ± 0.1 nM in 2 mM MgCl2 to KN = 0.04 ± 0.01 nM in 8 mM MgCl2 (Fig. 3D) while the dissociation constant for the flipped complex, KF, remained roughly constant, with α = 4.7 to 0.45. Alternatively, the data can be described almost as well by a simpler model that assumes constant linkage α = 0.5 between S4 binding and rRNA folding, and an average S4 affinity KN = 0.074 nM. For this model to be true, the folding equilibrium of the free RNA must vary ∼10 times more with Mg2+ than predicted by the hydroxyl radical footprinting experiments, from K1 = 100 in 2 mM MgCl2 to 1 in 20 mM MgCl2.
In both models, Mg2+ ions stabilize the native rRNA even in the absence of S4, as seen in footprinting experiments (Powers and Noller 1995b; Bellur and Woodson 2009; Ramaswamy and Woodson 2009a). The high-resolution crystal structure of the E. coli ribosome (Berk et al. 2006; PDB ID 2I2P) reveals ordered, partially dehydrated magnesium ions coordinated with the right-angle junction between 16S helices 3 and 4 and the pseudoknot in helix 18. The need to fill these specific Mg2+ binding sites may explain why high Mg2+ concentrations favor the native structure of the S4-RNA complex.
Interestingly, we observed a slight increase in the amount of flipped complex at very high Mg2+ concentrations (Fig. 3C). Gerstner et al. (2001) observed a similar trend for the binding of Bacillus stearothermophilus S4 to 16S rRNA. That K2 reaches a minimum ∼12 mM MgCl2 suggests that Mg2+ ions alone are not sufficient to fully populate the native complex, particularly at low physiological Mg2+ concentrations.
Effects of primary assembly proteins
To quantitatively measure how other 5′-domain proteins affect S4-rRNA interactions, S4 titrations were carried out in 20 mM Mg2+ plus a large excess (15 nM) of proteins S16, S17, and S20 in different combinations (Fig. 3E; Supplemental Table S3). Since protein S20 promotes proper folding of the “lower” junction between 16S helices 7-8-9-10, one might expect that S20 increases sampling of native contacts in the upper 5WJ where S4 binds. However, S20 did not substantially alter the equilibrium between flipped and docked 5′-domain-S4 complexes (K2 = 0.75 ± 0.02) or S4 binding affinity (KN = 0.16 ± 0.02).
Protein S17 stabilizes the tertiary structure of the 5′-domain to a larger extent than protein S20 (Ramaswamy and Woodson 2009a). S17 lowered dissociation constants for both S4 complexes (KN = 0.03 ± 0.03 and KF = 0.014 ± 0.014; Supplemental Table S3), indicating a thermodynamic advantage for S4 recruitment when S17 is also bound to the 16S rRNA (ΔΔGF○=−1.6 ± 0.6 kcal/mol; (ΔΔGN○=−2.0 ± 0.6 kcal/mol, Fig. 4). Protein S17 may also create a kinetic advantage for S4 recruitment. Single-molecule FRET studies showed that nearly all S4 binding trajectories pass through the flipped intermediate, which offers a low free energy route to the native complex (Kim et al. 2014). It will be interesting to determine whether S17, which binds the 16S rRNA upstream of S4, helps improve the efficiency of S4 addition.
FIGURE 4.
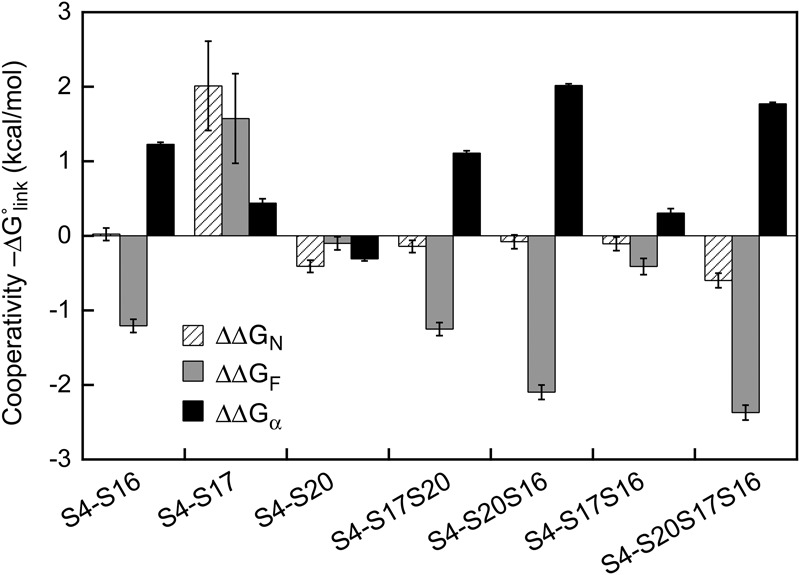
Thermodynamic linkage between S4 and other 5′-domain proteins. The linking free energy, ΔGlink, for the native complex (striped bars), flipped complex (gray bars), and α (black bars) was calculated as ΔGlink○=ΔGS4+j○−ΔGS4○ in which j indicates a combination of ribosomal proteins S16, S17, and S20. Free energies for S4 binding, ΔGS4○=RT ln KdS4, were evaluated at 37°C (Supplemental Table S3). ΔΔGα○=ΔΔGF○−ΔΔGN○ represents how other 5′-domain proteins alter the linkage between S4 binding and Mg2+-dependent folding of 16S helix 3.
Proteins S16 and S20 destabilize the intermediate S4-rRNA complex
In the Nomura assembly map, addition of secondary assembly protein S16 to the 16S rRNA depends on protein S4, and to a lesser degree protein S20. S16 binds to a pocket created by 16S helices 7, 15, and 17, that lies on the interface between the 5WJ recognized by S4 and the lower junction recognized by S20 (Schluenzen et al. 2000; Wimberly et al. 2000). Protein S16 did not stabilize the native S4 complex (KN = 0.08 ± 0.01 nM), either on its own or with S20 (hatched bars, Fig. 4). When proteins S16 and S20 were added together, however, the population skewed toward the native S4 complex (K2 = 0.02 ± 0.01; Fig. 3E), reflecting a much stronger cooperativity between S4 binding and the native 16S conformation (ΔG°α = −2.5 kcal/mol with S4–S20–S16 versus 0.5 kcal/mol for S4 alone; Fig. 4). This preference was not achieved by stabilizing the native complex (ΔΔGN○≈0; Fig. 4). Instead, S16 and S20 substantially weakened the affinity of S4 for the flipped conformation (ΔΔGF○=2.1 kcal/mol Fig. 4). Therefore, ribosomal proteins can select against certain assembly intermediates, unlike Mg2+ ions.
This preference for the native conformation of the S4-16S complex and destabilization of the flipped complex was maintained when S17 was included in the S4 titration (ΔΔGF○=2.4 kcal/mol, ΔΔGα○=−1.8 kcal/mol; Fig. 4). S20 alone had almost no effect on the stability of the flipped complex (Fig. 4). In contrast, protein S17 alone or combined with S16 but not S20 showed little preference for the native (K2 = 0.53 ± 0.02; ΔΔG°α = −0.31 kcal/mol). Therefore, two different protein combinations (S4, S16, and S20 versus S4, S16, and S17) form stable complexes with the 16S 5′ domain, but favor different conformations and exhibit differing degrees of cooperativity with S4 (Fig. 4).
Finally, we tested whether the ribosomal proteins will have similar effects on S4 binding at more physiological Mg2+ concentrations (Fig. 3F). In 4 mM MgCl2, most of the S4 complexes occupy the flipped intermediate state (K2 = 45 ± 8; α = 9 ± 2). The combination of S16 and S20 substantially reverses this preference (α = 0.74 ± 0.0408), however, stabilizing the native complex about twofold (KN = 0.27 ± 0.04 versus 0.6 ± 0.2) while destabilizing the intermediate complex about fivefold (KF = 0.36 ± 0.08 nM). Nevertheless, S4 binds the rRNA more tightly in 20 mM Mg2+ without S20 and S16 (KN = 0.08 ± 0.01 nM) than in 4 mM Mg2+ with S20 and S16. This observation further shows that ribosomal proteins and Mg2+ play distinct roles in ribosomal assembly.
Thermodynamic cooperativity is important for the assembly of other multiprotein RNA complexes such as signal recognition particle or splicing complexes (Menichelli et al. 2007; Wysoczanski et al. 2014). The thermodynamic cooperativity between protein S4 and other ribosomal proteins measured in this study was comparable to that obtained in the previous studies on central domain proteins (Recht and Williamson 2001). By using a FRET assay sensitive to the conformation of 16S helix 3, we were able to assign the cooperative free energy to two different S4 complexes formed during 5′-domain assembly. While Mg2+ ions preferentially lower the free energy of the native structure, the ribosomal proteins mainly act by stabilizing both complexes (S17) or destabilizing the flipped intermediate complex (S16). First, this analysis indicates that the hierarchical relationship between S4, S20, and S16 in the Nomura assembly map is achieved by elevating the free energy of an alternative non-native conformation, rather than lowering the free energy of the native complex. Second, S17 and S20 have almost opposite effects on the conformation of helix 3, especially when S16 is also present. This reversal in the preference for the flipped conformation could prevent helix 3 from docking prematurely, ensuring that all 5′-domain proteins are present and the pre-rRNA is properly folded before adding later assembly proteins such as S12 and S5. Although the rpsQ and rpsT genes that encode S17 and S20 are dispensable in E. coli, deletion strains grow poorly (Bubunenko et al. 2007). Our results suggest why these primary binding proteins are important for streamlining the ribosomal assembly process.
MATERIALS AND METHODS
Modified 5′-domain RNA for FRET measurements
The 5′ domain of E. coli 16S rRNA (nt 21–556, E. coli numbering) was extended at the 3′-end to base pair with a fluorophore-labeled complementary DNA oligomer (Dorywalska et al. 2005) as previously described (Kim et al. 2014). The best sequence extension, h3E5 (Supplemental Table S1) did not interfere with S4 binding or alter the 5′-domain secondary structure and provided the highest affinity (Kd ≤ 0.5 nM) for the DNA oligomer of 10 sequence extensions tested. Extended 5′-domain RNAs were transcribed in vitro with T7 RNA polymerase and purified by 4% PAGE using standard protocols. The absorbance at 260 nm (ε = 5.38 µM−1 cm−1) was used to determine the RNA concentration.
DNA oligomer binding assays
32P-labeled anti-h3ext oligomers (0.5 nM, 10 µL) were incubated with 0–100 nM extended 5′-domain RNAs in HK buffer (80 mM K-HEPES pH 7.6, 330 mM KCl, 6 mM 2-mercaptoethanol) at 70°C for 5 min, then cooled to 25°C for 5 min. An 8 µL aliquot of each sample was mixed with 2 µL loading buffer (30% glycerol, 2% xylene cyanol, 2% bromophenol blue) and 2 µL of this mixture was loaded on a native 8%–20% bi-layer polyacrylamide gel containing 10 mM MgCl2. The fraction of RNA-bound oligomer was measured using a Molecular Dynamics Phosphorimager. The primer-extended 5′-domain RNA complex was stable to purification on size exclusion spin columns and immobilization on microscope slides over the range of 2–20 mM MgCl2 concentrations used here (Kim et al. 2014).
Ribosomal protein purification and labeling
The double mutant S4 protein (S4:C32S,S189C) was prepared by site-directed mutagenesis (QuikChange, Stratagene) of pET24b/rpS4, and overexpressed and purified by cation exchange chromatography (UNO S6, BioRad) as previously described (Culver and Noller 1999). Isolated proteins were dialyzed overnight into 80 mM K-HEPES pH 7.6, 1 M KCl, 1 mM TCEP with three buffer changes and stored at −80°C in 500 µL aliquots.
For labeling, protein S4:C32S,S189C (15 µM, 500 μL) was incubated 30 min in 80 mM K-HEPES at pH 7.6, 1 M KCl, 1 mM TCEP, 3 M urea at 20°C. A sixfold molar excess of maleimide-linked Cy5 (GE Healthcare) was added to protein S4 and incubated at 20°C for another 2 h. The reaction was quenched by adding 24.5 mL 20 mM Tris–HCl pH 7.0, 6 M urea, and 6 mM 2-mercaptoethanol. The final KCl concentration was adjusted to 20 mM before loading on a BioRad Uno-S6 column to remove excess unreacted dye, followed by overnight dialysis against 80 mM K-HEPES pH 7.6, 1 M KCl, 6 mM 2-mercaptoethanol. The labeling efficiency was ∼100%, as measured by SDS-PAGE as previously described (Hickerson et al. 2005). The concentration of the labeled protein was determined from A650 (ε650 = 2.5 × 105 M−1 cm−1). The concentration of unlabeled S4 protein was determined from A280 (ε280 = 17,843 M−1 cm−1). Both labeled and unlabeled proteins were stored at −80°C in a light-tight box.
Ribosomal proteins S16, S17, and S20 were overexpressed and purified as previously described (Culver and Noller 1999).
S4-RNA binding
Extended 5′-domain RNA was base paired with Cy3-labeled h3E oligomer (1.6 nM each) as described above, then refolded at 37°C for 15 min in 0–20 mM MgCl2. Proteins S16, S17, and S20 (15 nM) were added as stated in the text, and incubated another 15 min at 37°C before titration of the RNA complex (500 µL) with small volumes of S4-Cy5 in the same folding buffer in a quartz fluorescence cuvette at 37°C. The sample was incubated 1 min after each S4 addition before recording the fluorescence emission from 550 to 700 nm with 540 nm excitation (Horiba Fluorolog-3). Excitation and emission slits were 2 and 5 nm, respectively. Fluorescence intensities at 566 nm (donor) and 663 nm (acceptor) were used to calculate the FRET efficiency E = I663/(I663 + I566) at each S4 concentration. Reported titration curves were the average of two or three independent titrations.
The change in FRET efficiency was fit to a binding equation that assumes two bound states and two unbound states (Fig. 2A),
| (1) |
In Equation 1, ST and RT are the total RNA and protein concentrations, respectively, β = KN (1 + K1) KN is the overall dissociation constant for the high-FRET native complex, and K2 is the equilibrium constant between the flipped and native complexes. The average FRET efficiencies for the native and flipped complexes, EN = 0.18 and EF = 0.02, were assumed to equal the maximum and minimum observed FRET values and were held constant, while K2, β, and RT were allowed to vary. Because EFRET ≈ (EN + EF K2)/(1 + K2) when ST≫β, the endpoint of each titration depends on the FRET efficiencies of the native and flipped complexes and the equilibrium constant K2 between these complexes. A derivation of Equation 1 is given in the Supplemental Material.
The conformation of the free RNA, K1, was obtained from ƒN = 1/(1 + K1) measured by protection of 16S nt 501–504 from hydroxyl radical footprinting of the 5′-domain RNA (Ramaswamy and Woodson 2009a) in the same buffer and at the same temperature as used for the binding experiments reported here. Footprinting experiments on the RNA with S20 or S17 were used to calculate K1 for titrations that included those proteins; S16 does not appreciably bind the 5′-domain RNA in the absence of S4. These values of K1 were used to calculate KN, KF, and α, as described in the Supplemental Material.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Mayerle for helpful advice. This work was supported by a grant from the National Institutes of Health (R01 GM60819) to S.A.W.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.051292.115.
REFERENCES
- Adilakshmi T, Ramaswamy P, Woodson SA. 2005. Protein-independent folding pathway of the 16 S rRNA 5′ domain. J Mol Biol 351: 508–519. [DOI] [PubMed] [Google Scholar]
- Adilakshmi T, Bellur DL, Woodson SA. 2008. Concurrent nucleation of 16S folding and induced fit in 30S ribosome assembly. Nature 455: 1268–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellur DL, Woodson SA. 2009. A minimized rRNA-binding site for ribosomal protein S4 and its implications for 30S assembly. Nucleic Acids Res 37: 1886–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berk V, Zhang W, Pai RD, Cate JH. 2006. Structural basis for mRNA and tRNA positioning on the ribosome. Proc Natl Acad Sci 103: 15830–15834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodersen DE, Clemons WM Jr, Carter AP, Wimberly BT, Ramakrishnan V. 2002. Crystal structure of the 30 S ribosomal subunit from Thermus thermophilus: structure of the proteins and their interactions with 16 S RNA. J Mol Biol 316: 725–768. [DOI] [PubMed] [Google Scholar]
- Bubunenko M, Baker T, Court DL. 2007. Essentiality of ribosomal and transcription antitermination proteins analyzed by systematic gene replacement in Escherichia coli. J Bacteriol 189: 2844–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culver GM, Noller HF. 1999. Efficient reconstitution of functional Escherichia coli 30S ribosomal subunits from a complete set of recombinant small subunit ribosomal proteins. RNA 5: 832–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorywalska M, Blanchard SC, Gonzalez RL Jr, Kim HD, Chu S, Puglisi JD. 2005. Site-specific labeling of the ribosome for single-molecule spectroscopy. Nucleic Acids Res 33: 182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutca LM, Culver GM. 2008. Assembly of the 5′ and 3′ minor domains of 16S ribosomal RNA as monitored by tethered probing from ribosomal protein S20. J Mol Biol 376: 92–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstner RB, Pak Y, Draper DE. 2001. Recognition of 16S rRNA by ribosomal protein S4 from Bacillus stearothermophilus. Biochemistry 40: 7165–7173. [DOI] [PubMed] [Google Scholar]
- Grondek JF, Culver GM. 2004. Assembly of the 30S ribosomal subunit: positioning ribosomal protein S13 in the S7 assembly branch. RNA 10: 1861–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held WA, Nomura M. 1975. Escherichia coli 30 S ribosomal proteins uniquely required for assembly. J Biol Chem 250: 3179–3184. [PubMed] [Google Scholar]
- Held WA, Ballou B, Mizushima S, Nomura M. 1974. Assembly mapping of 30 S ribosomal proteins from Escherichia coli. Further studies. J Biol Chem 249: 3103–3111. [PubMed] [Google Scholar]
- Hickerson R, Majumdar ZK, Baucom A, Clegg RM, Noller HF. 2005. Measurement of internal movements within the 30 S ribosomal subunit using Förster resonance energy transfer. J Mol Biol 354: 459–472. [DOI] [PubMed] [Google Scholar]
- Jagannathan I, Culver GM. 2003. Assembly of the central domain of the 30S ribosomal subunit: roles for the primary binding ribosomal proteins S15 and S8. J Mol Biol 330: 373–383. [DOI] [PubMed] [Google Scholar]
- Kim H, Abeysirigunawarden SC, Chen K, Mayerle M, Ragunathan K, Luthey-Schulten Z, Ha T, Woodson SA. 2014. Protein-guided RNA dynamics during early ribosome assembly. Nature 506: 334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayerle M, Bellur DL, Woodson SA. 2011. Slow formation of stable complexes during coincubation of minimal rRNA and ribosomal protein S4. J Mol Biol 412: 453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menichelli E, Isel C, Oubridge C, Nagai K. 2007. Protein-induced conformational changes of RNA during the assembly of human signal recognition particle. J Mol Biol 367: 187–203. [DOI] [PubMed] [Google Scholar]
- Mulder AM, Yoshioka C, Beck AH, Bunner AE, Milligan RA, Potter CS, Carragher B, Williamson JR. 2010. Visualizing ribosome biogenesis: parallel assembly pathways for the 30S subunit. Science 330: 673–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotny V, Nierhaus KH. 1988. Assembly of the 30S subunit from Escherichia coli ribosomes occurs via two assembly domains which are initiated by S4 and S7. Biochemistry 27: 7051–7055. [DOI] [PubMed] [Google Scholar]
- Powers T, Noller HF. 1995a. Hydroxyl radical footprinting of ribosomal proteins on 16S rRNA. RNA 1: 194–209. [PMC free article] [PubMed] [Google Scholar]
- Powers T, Noller HF. 1995b. A temperature-dependent conformational rearrangement in the ribosomal protein S4.16 S rRNA complex. J Biol Chem 270: 1238–1242. [DOI] [PubMed] [Google Scholar]
- Powers T, Daubresse G, Noller HF. 1993. Dynamics of in vitro assembly of 16 S rRNA into 30 S ribosomal subunits. J Mol Biol 232: 362–374. [DOI] [PubMed] [Google Scholar]
- Ramaswamy P, Woodson SA. 2009a. Global stabilization of rRNA structure by ribosomal proteins S4, S17, and S20. J Mol Biol 392: 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy P, Woodson SA. 2009b. S16 throws a conformational switch during assembly of 30S 5′ domain. Nat Struct Mol Biol 16: 438–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recht MI, Williamson JR. 2001. Central domain assembly: thermodynamics and kinetics of S6 and S18 binding to an S15-RNA complex. J Mol Biol 313: 35–48. [DOI] [PubMed] [Google Scholar]
- Recht MI, Williamson JR. 2004. RNA tertiary structure and cooperative assembly of a large ribonucleoprotein complex. J Mol Biol 344: 395–407. [DOI] [PubMed] [Google Scholar]
- Schluenzen F, Tocilj A, Zarivach R, Harms J, Gluehmann M, Janell D, Bashan A, Bartels H, Agmon I, Franceschi F, et al. 2000. Structure of functionally activated small ribosomal subunit at 3.3 angstroms resolution. Cell 102: 615–623. [DOI] [PubMed] [Google Scholar]
- Stern S, Wilson RC, Noller HF. 1986. Localization of the binding site for protein S4 on 16 S ribosomal RNA by chemical and enzymatic probing and primer extension. J Mol Biol 192: 101–110. [DOI] [PubMed] [Google Scholar]
- Stern S, Powers T, Changchien LM, Noller HF. 1989. RNA-protein interactions in 30S ribosomal subunits: folding and function of 16S rRNA. Science 244: 783–790. [DOI] [PubMed] [Google Scholar]
- Weitzmann CJ, Cunningham PR, Nurse K, Ofengand J. 1993. Chemical evidence for domain assembly of the Escherichia coli 30S ribosome. FASEB J 7: 177–180. [DOI] [PubMed] [Google Scholar]
- Wimberly BT, Brodersen DE, Clemons WM Jr, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. 2000. Structure of the 30S ribosomal subunit. Nature 407: 327–339. [DOI] [PubMed] [Google Scholar]
- Wysoczanski P, Schneider C, Xiang S, Munari F, Trowitzsch S, Wahl MC, Lührmann R, Becker S, Zweckstetter M. 2014. Cooperative structure of the heterotrimeric pre-mRNA retention and splicing complex. Nat Struct Mol Biol 21: 911–918. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.