

Abstract
Mitochondrial RNA processing is an essential step for the synthesis of the components of the electron transport chain in all eukaryotic organisms, yet several aspects of mitochondrial RNA biogenesis and regulation are not sufficiently understood. RNA interactome capture identified several disease-relevant RNA-binding proteins (RBPs) with noncanonical RNA-binding architectures, including all six members of the FASTK (FAS-activated serine/threonine kinase) family of proteins. A mutation within one of these newly assigned FASTK RBPs, FASTKD2, causes a rare form of Mendelian mitochondrial encephalomyopathy. To investigate whether RNA binding of FASTKD2 contributes to the disease phenotype, we identified the RNA targets of FASTKD2 by iCLIP. FASTKD2 interacts with a defined set of mitochondrial transcripts including 16S ribosomal RNA (RNR2) and NADH dehydrogenase subunit 6 (ND6) messenger RNA. CRISPR-mediated deletion of FASTKD2 leads to aberrant processing and expression of RNR2 and ND6 mRNA that encodes a subunit of the respiratory complex I. Metabolic phenotyping of FASTKD2-deficient cells reveals impaired cellular respiration with reduced activities of all respiratory complexes. This work identifies key aspects of the molecular network of a previously uncharacterized, disease-relevant RNA-binding protein, FASTKD2, by a combination of genomic, molecular, and metabolic analyses.
Keywords: RNA-binding proteins, iCLIP, mitochondria, transcript processing, oxidative phosphorylation, Mendelian disease
INTRODUCTION
Mitochondria require a dedicated transcription and translation machinery to synthesize a subset of the membrane proteins constituting the electron transport chain (ETC). Mitochondrial DNA (mtDNA) is transcribed bidirectionally giving rise to polycistronic light-strand and heavy-strand transcripts that mature by extensive nucleolytic processing. Typically, the polycistronic precursor transcripts are processed by the excision of tRNAs flanking most protein-coding genes and the two ribosomal RNAs, a process that is referred to as the tRNA punctuation model (Ojala et al. 1981b). However, the maturation of some of the mitochondrial transcripts, such as CYB, ND5, ND6, ATP8/6, and COX3, also relies on activities other than the mitochondrial tRNA processing machinery (Brzezniak et al. 2011; Sanchez et al. 2011). As both nuclear and mitochondrial genes code for components of the ETC, intricate coordination of the output from both genomes is required to ensure proper stoichiometry of the respiratory enzyme complexes. Regulation of mitochondrial gene expression is exerted post-transcriptionally as the steady-state levels of transcripts derived from common polycistronic precursors vary extensively in their abundance (Piechota et al. 2006; Nagao et al. 2008). Approximately 1400 nuclear genes are essential for multiple aspects of mitochondrial homeostasis including, among others, assembly of the ETC and mitochondrial transcript processing (Calvo et al. 2006; Pagliarini et al. 2008; Wolf and Mootha 2014). The identification of proteins binding polyadenylated transcripts in human cells by interactome capture revealed that many proteins lacking classical RNA-binding domains associate with RNA in human and mouse cells (Baltz et al. 2012; Castello et al. 2012; Kwon et al. 2013). Potentially, these previously unknown RBPs may include factors that are relevant for mitochondrial RNA processing, as human mitochondrial RNAs bear poly(A) tails that enable their oligo(dT)-mediated capture (Chang and Tong 2012).
Mitochondria have been implicated in a multitude of cellular pathways, including oxidative phosphorylation and apoptosis, and contribute to diverse biological processes such as aging or neurodegenerative diseases (Calvo and Mootha 2010; Koopman et al. 2013). Defects of mitochondrial respiration are associated with a wide spectrum of clinical manifestations and, given the dual genetic origin of the components of the ETC, elucidating their molecular causes often is not straightforward (Fernández-Vizarra et al. 2009).
Mutations within dozens of the RNA-binding proteins detected by interactome capture, including FASTKD2 (FAS-induced serine/threonine kinase domain containing protein 2), which lacks canonical RNA-binding domains, have been associated with Mendelian diseases. Mutated FASTKD2 has been identified as the likely cause of an atypical form of infantile mitochondrial encephalomyopathy in a consanguineous family of Bedouin origin (Ghezzi et al. 2008). FASTKD2 is a member of the human FASTK protein family, formed by six architecturally related proteins named FASTK and FASTKD1-5 that localize to mitochondria (Ghezzi et al. 2008; Pagliarini et al. 2008; Simarro et al. 2010). FASTK has been proposed to act as a protein kinase (Tian et al. 1995), but critical active site residues are not conserved within the family (Simarro et al. 2010). FASTK family proteins harbor the FAST_1 and FAST_2 domains of currently unknown function as well as the RAP domain that, based on homology modeling, may act as an RNA-binding module (Lee and Hong 2004). FASTKD3 is the first FASTK family member shown to be required for mitochondrial function, but the underlying molecular mechanisms have remained unexplored (Simarro et al. 2010). Taking the RNA-binding activity of FASTKD2 (Castello et al. 2012) and early work implicating FASTK family proteins in mitochondrial physiology (Ghezzi et al. 2008; Simarro et al. 2010) as a basis, we decided to explore whether it functions in mitochondrial RNA metabolism.
RESULTS AND DISCUSSION
FASTKD2 associates with a select spectrum of mitochondrial transcripts
All members of the FASTK protein family have independently been detected as RNA-binding proteins in mRNA interactome data sets obtained from cells of murine and human origin (Baltz et al. 2012; Castello et al. 2012; Kwon et al. 2013). In particular, FASTKD2 and -4 were captured by polyadenylated transcripts in all three mRNA interactome studies published to date (Fig. 1A). With the ultimate goal of gaining insight into the molecular functions of FASTKD2, potentially helping to understand the reported hereditary form of encephalomyopathy associated with FASTKD2 mutations, we set out to identify its associated transcripts. To this end, we used the green fluorescent protein (GFP)-nanotrap system (Rothbauer et al. 2008) to trace and stringently purify FASTKD2 fused to a C-terminal GFP tag expected not to interfere with the N-terminal mitochondrial localization signal of FASTKD2 (Ghezzi et al. 2008). Although the initial validation of FASTKD2 as an RNA-binding protein confirmed the copurification of an N-terminally GFP-tagged construct with polyadenylated transcripts (Castello et al. 2012), the mitochondrial localization of this construct was not tested. C-terminally tagged FASTKD2-GFP colocalizes with mitochondria based on the overlapping staining pattern with Mitotracker dye both in transfected MCF-7 and U2OS cells (Fig. 1B).
FIGURE 1.

FASTKD2 is a mitochondrial RNA-binding protein. (A) Domain structure and detection of FASTKD proteins in published mRNA interactome data sets. (MLS) mitochondrial localization signal, (LC) low complexity region, (FAST_1/FAST_2) FAST domains. (B) Colocalization of FASTKD2-GFP with mitochondria in transiently transfected U2OS and MCF-7 cells (stained with Mitotracker dye). (C) Validation of FASTKD2-GFP as an RNA-binding protein. FASTKD2-GFP UV-cross-linked transcripts were trimmed by incubation with a serial dilution of RNase A, labeled with polynucleotide kinase and [γ-32P]ATP, complexes separated by denaturing gel electrophoresis and visualized by autoradiography after transfer to a PVDF membrane.
To assess RNA-binding of FASTKD2-GFP, we purified the protein from UV-irradiated stable cell lines, and radioactively labeled cross-linked transcripts by polynucleotide kinase treatment after gradual nucleolytic trimming. Analysis of the immunoprecipitate by gel electrophoresis and autoradiography revealed the crosslink-dependent copurification of RNA with FASTKD2-GFP (Fig. 1C). Near equal expression levels of endogenous and tagged FASTKD2 were achieved by careful titration of the tetracycline concentration used to induce expression of the tagged protein in the stably transfected cells (Fig. 2A). To maximize the purification of FASTKD2-associated transcripts and minimize the inclusion of RNAs bound to potentially interacting proteins, we applied highly stringent wash conditions and assessed the recovery of FASTKD2 by gel electrophoresis (Fig. 2B). We then isolated cross-linked transcripts from the immunoprecipitates of three individual single-cell clones using cell lines expressing the mitochondrial localization signal (MLS) of FASTKD2 (amino acids 17–68) fused to GFP as a background control.
FIGURE 2.

Identification of FASTKD2 interacting transcripts in HEK293 cells. (A) Titration of tetracycline concentrations required for equal expression of endogenous FASTKD2 and FASTKD2-GFP. The asterisk denotes a lower molecular mass species of unknown identity. (B) Analysis of FASTKD2-GFP immunoprecipitates by polyacrylamide gel electrophoresis and silver stain. (C) Crosslink site density plot for iCLIP cDNA libraries after alignment to the human genome and random barcode evaluation. Crosslink site density was calculated using the sum of annotated gene lengths for each chromosome. (D) Heat map representing significant (false discovery rate <0.05) RNA crosslink sites in MLS-GFP and FASTKD2-GFP libraries. Enrichment of light-strand transcripts is indicated in blue, enrichment of heavy-strand transcripts is indicated in red. (E) Enrichment plots for heavy- and light-strand FASTKD2-GFP crosslink sites. Enrichment and P-values were calculated using DESeq2. (F) High-confidence FASTKD2 RNA targets (log2-fold enrichment ≥4, −log10 P-value ≥ 2). Note that for a given target only the parameters for the highest scoring crosslink site are specified.
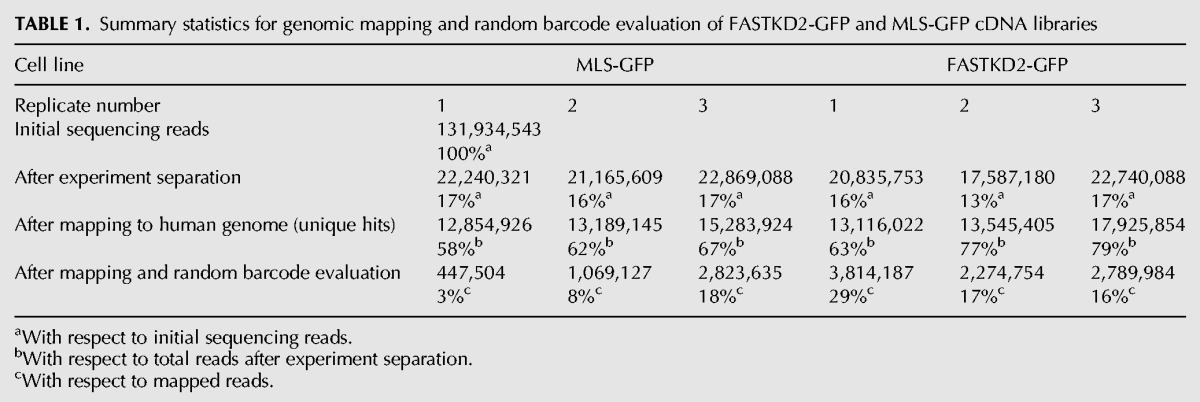
RNA was isolated from immunoprecipitates, reverse transcribed and cloned by a modified version of the iCLIP (individual nucleotide resolution UV cross-linking and immunoprecipitation) protocol (Konig et al. 2010). The circularization-based cDNA cloning procedure applied by iCLIP facilitates the detection of the crosslink site position, thus achieving a high spatial resolution of the resulting RNA–protein crosslink maps (Urlaub et al. 2002; Konig et al. 2010). We obtained cDNA libraries from three independent cell clones for both FASTKD2-GFP and the MLS-EGFP control (Table 1). These cDNA libraries were subsequently sequenced and PCR amplification artifacts removed with the help of the random barcodes that were included in the library amplification primers using the iCount pipeline (Konig et al. 2010). Alignment of the reads to the human nuclear and mitochondrial genomes revealed for all replicates of the MLS-GFP and FASTKD2-GFP libraries that the read density for mitochondrial genes was higher by several orders of magnitude than for genes encoded by the nuclear genome, a result that is consistent with the mitochondrial localization of both constructs (Fig. 2C). To gain insight into the mitochondrial target spectrum of FASTKD2, we aligned cross-link sites with a low false discovery rate (FDR) with the mitochondrial genome. The resulting peak map revealed the selective association of FASTKD2 with a subset of mitochondrial transcripts (Fig. 2D). To calculate the enrichment of a given crosslink peak in the FASTKD2-GFP versus MLS-GFP libraries, we processed the data using the DESeq2 pipeline (Anders and Huber 2010). Enrichment and P-values were then plotted for all detected crosslink peaks with a low FDR (Fig. 2E) and specific peaks defined by stringent cutoff values (log2-fold enrichment ≥4, −log10 P-value ≥2) (Fig. 2E,F). We detected several high-confidence crosslink sites within the 12S and 16S ribosomal RNAs (RNR1 and RNR2, respectively) and some of the transfer RNAs encoded on the heavy strand (Fig. 2F). In addition, high-confidence crosslink peaks are found within the mRNAs of the complex III subunit CYB, and the cytochrome c oxidase subunits COX1 and COX2 (Fig. 2F). Analysis of the high-confidence light-strand crosslink peaks suggested specific interactions of FASTKD2 with the mRNA encoding subunit 6 of respiratory complex I (ND6), 7S RNA (a short RNA of ∼200 nucleotides [nt] length also known as RNA18) and mitochondrial prolyl-tRNA (TRNP), also evident by visual inspection of the heat map of the low FDR peak data (Fig. 2D). For RNR2 the high-confidence crosslink sites cluster near the 5′ end of the transcript (pos. 1726–1883), whereas peaks within tRNA genes are preferentially located near the D-stem or the anti-codon loop. Nearly all of the highly enriched heavy-strand crosslink peaks are located in regions corresponding to mature mitochondrial transcripts. A significantly enriched peak in FASTKD2-GFP libraries corresponding to a precursor transcript maps to a region downstream from the mature 7S RNA transcript, suggesting an interaction of FASTKD2 with a precursor transcript in the region between 7S RNA and the downstream TRNP.
TABLE 1.
Summary statistics for genomic mapping and random barcode evaluation of FASTKD2-GFP and MLS-GFP cDNA libraries
Depletion of FASTKD2 affects the levels of several of its target transcripts
To analyze the consequences of FASTKD2 deficiency in a cell-based model, we established the depletion of the protein in HEK293 cells both by RNA interference (RNAi) and the Cas9 nuclease-based CRISPR system (Hsu et al. 2013). RNAi faithfully reduced the expression of FASTKD2 protein (Fig. 3A) and mRNA (Fig. 3B) to ∼15% of control levels. Complete depletion for FASTKD2 was achieved via the CRISPR/Cas9 nuclease system (Sanjana et al. 2014). To mimic the mutation causing the loss of function of FASTKD2 reported for two patients suffering from early onset encephalomyopathy (Ghezzi et al. 2008), we chose a guide RNA sequence that induces cleavage of genomic DNA in the immediate vicinity of the R432 codon of FASTKD2, which is nonsense mutated in affected patients. We isolated several independent clonal cell lines without detectable FASTKD2 expression, referred to as FASTKD2 NULL cells (Fig. 3C). Sequencing of the genomic DNA of these cell lines confirmed successful nonsense mutagenesis of all alleles encoding FASTKD2 (Fig. 3D).
FIGURE 3.

RNAi- and CRISPR-mediated depletion of FASTKD2 affect mitochondrial RNA expression and processing. (A) Western blot analysis. (B) Quantitative reverse transcriptase quantitative PCR analysis of the depletion of FASTKD2 by RNAi. (C) Analysis of expression of FASTKD2 in single-cell clones expressing Cas9 (pLenti-Cas9) or Cas9 and a guide RNA cleaving genomic DNA within the FASTKD2 gene (pLenti-Cas9 + gRNA FASTKD2). The clones marked with an asterisk are referred to as FASTKD2 NULL cell lines and were used for the subsequent analyses. (BACT) β-actin. (D) Confirmation of loss of function alleles in CRISPR-targeted FASTKD2 NULL cell lines by Sanger sequencing. Asterisks indicate premature stop codons. Fractions next to sequencing traces (starting with isoleucine 422) indicate relative frequencies of the clones corresponding to different alleles in the case of heterozygous nonsense mutations. (E) HEK293 cells were depleted of FASTKD2 by RNAi (three replicates, left panels) or CRISPR-mediated genomic deletion (three independent cell clones, right panels). RNA was extracted and subjected to Northern blot hybridization using strand-specific riboprobes. 28S rRNA was used as a loading control. (F) Band intensities obtained by quantification of the blots shown in E were normalized by 28S band intensities and are stated relative to the signals in controls (siCtrl. or pLenti-Cas9 cell clones), which were arbitrarily set to 1.
Based on the iCLIP data, we analyzed the processing and steady-state levels of FASTKD2 target RNAs in cells depleted for FASTKD2 by RNAi or CRISPR. Depletion of FASTKD2 by RNAi leaves the noncoding heavy-strand transcripts unchanged (Fig. 3E, left panels; Fig. 3F black bars). In contrast, CRISPR-mediated depletion of FASTKD2 causes a profound and selective reduction of 16S mitochondrial rRNA (RNR2) (Fig. 3E, top panel; Fig. 3F). However, the levels of 12S rRNA (RNR1), which arises from the same polycistronic heavy-strand precursor transcripts, are largely unaffected (Fig. 3E,F). Fully consistent with the results reported here, depletion of FASTKD2 by RNAi was just found in to cause misassembly of the mitochondrial ribosome and impaired mitochondrial protein synthesis in human immortalized fibroblasts (Antonicka and Shoubridge 2015). Our observations for FASTKD2 also complement a recent report implicating RAP, a protein of Arabidopsis thaliana that shares the RAP domain with FASTKD2, in precursor processing of the small ribosomal subunit RNA in chloroplasts (Kleinknecht et al. 2014). Despite this surprising similarity, our data suggest that FASTKD2 may function somewhat differently, as we did not observe the accumulation of ribosomal precursor transcripts upon depletion of FASTKD2. Moreover, the unchanged levels of RNR1 suggest that the biogenesis of the common precursor of RNR1 and RNR2 is unaffected in the absence of FASTKD2. Thus, selective depletion of RNR2 may occur due to its aberrant processing or due to ribosome misassembly. Indeed, recent studies suggest that failure to assemble RNR2 with mitochondrial ribosomal proteins leads to its selective depletion, which has been proposed to be mediated by the mitochondrial degradosome (Antonicka and Shoubridge 2015; Tu and Barrientos 2015).
The levels of isoleucyl tRNA (TRNI), one of the heavy-strand-encoded tRNAs highly enriched in FASTKD2-GFP libraries, are unaffected by depletion of FASTKD2 by RNAi and CRISPR/Cas9-mediated mutagenesis (Fig. 3E,F). We next analyzed the expression of the heavy-strand protein-encoding transcripts bound by FASTKD2. The mRNA levels of the complex IV components COX1 and COX2 are unaffected or mildly reduced by either form of depletion of FASTKD2 (Fig. 3E,F). Complex IV was found to be most severely affected in patients with mutated FASTKD2 (Ghezzi et al. 2008). We therefore also examined the expression of the mitochondrial mRNA encoding the complex IV subunit, COX3, even though this transcript did not classify as a direct FASTKD2 target (Fig. 2F). Comparable to the remaining mitochondrial complex IV subunit RNAs, the levels of COX3 mRNA appear to be mildly affected by both RNAi- and CRISPR-mediated loss of FASTKD2 (Fig. 3E,F). Similarly, both modes of FASTKD2 depletion only mildly reduce the levels of the remaining FASTKD2 protein-coding heavy-strand target transcript, the complex III subunit mRNA CYB (Fig. 3E,F).
Depletion of FASTKD2 by RNAi or CRISPR affects ND6 RNA processing and levels (Fig. 3E,F). As suggested by Jourdain et al. (2015), depletion of FASTK has a similar effect on expression of the ND6 mRNA as loss of FASTKD2. This study concludes that a mitochondrial form of FASTK regulates the exonucleolytic processing of the 3′-end of the ND6 transcript in cooperation with the mitochondrial degradosome (Jourdain et al. 2015). However, our low FDR FASTKD2 crosslink sites cluster within the coding region of the ND6 transcript, suggesting that FASTKD2 may determine the fate of the mature forms of ND6, possibly in addition to a role in precursor processing.
Expression of 7S RNA is strongly induced by both modes of depletion of FASTKD2 (Fig. 3E,F). This observation is consistent with an induction of mitochondrial transcription, a possible consequence of perturbed mitochondrial homeostasis (Metodiev et al. 2009), as 7S RNA levels have previously been proposed to correlate with de novo transcriptional activity in mitochondria (Terzioglu et al. 2013). Interfering with FASTKD2 expression by RNAi or CRISPR leaves TRNP steady-state levels unchanged (Fig. 3E,F). Taken together, both depletion of FASTKD2 by RNAi and CRISPR mutagenesis affect the levels of several mitochondrial transcripts. CRISPR-mediated loss of FASTKD2 causes transcript-specific mitochondrial RNA expression defects characterized by a drastic reduction of 16S ribosomal RNA, loss of mature ND6 mRNA and the induction of 7S RNA.
FASTKD2 is required for mitochondrial translation
To assess the physiological consequences of the RNA processing defects on mitochondrial function, we measured the metabolic parameters of control and FASTKD2-depleted cell lines. We first measured basal-, leak-, and maximal respiration by micro-respirometry (Divakaruni et al. 2014) after adaptation of the protocol to HEK293 cells. Consistent with the less severe changes in mitochondrial RNA processing (Fig. 3E,F), depletion of FASTKD2 by RNAi results in an insignificant change of the cellular oxygen consumption rate (OCR) (Fig. 4A) or the extracellular acidification rate (ECAR) (Fig. 4B), a parameter used to assess glycolytic activity (Divakaruni et al. 2014), even though FASTKD2 expression was efficiently silenced (Fig. 4C). We also measured de novo mitochondrial protein synthesis by metabolic labeling in the presence of emetine, an inhibitor of cytoplasmic but not mitochondrial translation. Even though the depletion of FASTKD2 by RNAi does not appreciably affect the steady-state levels or processing pattern of RNR2 on Northern blots (Fig. 3E, top panel on the left), it appreciably reduces global mitochondrial translation (Fig. 4D left panel). The fact that the metabolic activity of siRNA-treated cells appears largely unaltered despite a generalized reduction of mitochondrial translation suggests a remarkable capacity of the cells to buffer perturbations of mitochondrial protein synthesis for limited periods of time. Genomic deletion of FASTKD2 by CRISPR-mediated mutagenesis impairs global mitochondrial translation to an even greater extent than RNAi-mediated depletion, thus confirming the importance of the protein for mitochondrial protein synthesis (Fig. 4D right panel).
FIGURE 4.

Metabolic analysis of cells depleted of FASTKD2. (A) Oxygen consumption rate (OCR) of HEK293 cells depleted of FASTKD2 by RNAi ([siCtrl] control siRNA; [siFASTKD2] FASTKD2 targeting siRNA pool; [Basal] basal respiration; [Oligo] leak respiration in the presence of oligomycin; [FCCP1] uncoupled respiration in the presence of 200 nM FCCP (carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone); [FCCP2] uncoupled respiration in the presence of 250 nM FCCP; [A + R] residual respiration in the presence of antimycin a and rotenone). (B) Extracellular acidification rate (ECAR) of cells depleted of FASTKD2 by RNAi. Maximum ECAR measured in the presence of oligomycin (Oligo) was set to 100%. (C) Confirmation of RNAi-mediated depletion of FASTKD2 by Western blot. (D) Metabolic labeling of de novo synthesis of mitochondrial proteins in cells depleted of FASTKD2 by RNAi (left panel) or CRISPR-mediated mutagenesis (right panel) in the presence of emetine. (CBB) Coomassie brilliant blue stain. (E) OCR of HEK293 cells depleted of FASTKD2 by CRISPR. Abbreviations are as in A. (F) ECAR of cells depleted of FASTKD2 by CRISPR. (G) Relative growth of cells depleted of FASTKD2 by CRISPR in galactose media. (H) Western blot analysis of expression of FASTKD2 in crude mitochondrial preparations isolated from cells depleted of FASTKD2 by CRISPR. (I) Activity of ETC complexes in cells depleted of FASTKD2 by CRISPR. Values were normalized by the activity of citrate synthase and are stated for both cells depleted of FASTKD2 by CRISPR-mediated mutagenesis and Cas9-expressing control cells. (J) mtDNA levels of cells depleted of FASTKD2 by CRISPR. Values are stated as relative ratios of COXI (mitochondrial) to β-actin (nuclear) amplicons as determined by quantitative reverse transcriptase PCR.
Lack of FASTKD2 compromises cellular respiration
Against the background of the apparently high functional stability of mitochondrial functions to RNAi-mediated FASTKD2 depletion, we also analyzed the metabolic parameters of FASTKD2 NULL cells. Of note, CRISPR-mediated loss of FASTKD2 causes a pronounced decrease of both basal and maximal respiration (Fig. 4E, 0–20 or 40–80 min, respectively). The OCR is diminished for all three tested clonal FASTKD2 NULL cell lines, compared with the rates measured for the Cas9 control and parental cell lines. In addition to reduced oxygen consumption, each of the FASTKD2-deficient cell lines also displays an increased ECAR (Fig. 4F), suggesting that the energy deficit arising from dysfunctional oxidative phosphorylation may be compensated by an increased rate of glycolysis. As cells with defects in oxidative phosphorylation frequently exhibit diminished proliferation in media containing galactose as the main carbohydrate (Robinson et al. 1992), we compared the growth rates of FASTKD2 WT (wild type) and NULL cells in glucose- versus galactose-containing media. Whereas both FASTKD2 WT and NULL cell lines show lower proliferation rates in galactose compared to glucose media, the FASTKD2 NULL cells are far more severely affected (Fig. 4G). Therefore, FASTKD2-deficient cells display multiple signs of a marked defect in cellular respiration.
We also measured the activity of the complexes of the respiratory chain in crude mitochondria isolated from FASTKD2 NULL or the control WT cell lines (Fig. 4H). All activities were normalized to citrate synthase activity measured in the respective mitochondrial preparations. Consistent with the generalized defect in mitochondrial translation of FASTKD2-depleted cells, all mitochondrially encoded ETC complexes display diminished activities (30%–60% of WT levels, Fig. 4I). Of all ETC complexes, it appears as if NADH:ubiquinone oxidoreductase (complex I) is the most severely affected (∼30% of WT levels, Fig. 4I, first panel), consistent with one of its subunit transcripts, ND6, being strongly affected by both modes of depletion of FASTKD2. Of note, succinate dehydrogenase (complex II) activity was unaffected (Fig. 4I, second panel), consistent with its subunits and assembly factors being encoded by the nuclear genome.
Note that 7S RNA is a polyadenylated mitochondrial transcript of ∼200 nt length with poorly defined functions (Ojala et al. 1981a; Clayton 1984; Mercer et al. 2011). According to the current model of mitochondrial DNA replication, a nonpolyadenylated fragment of 7S RNA, produced by early termination, serves as an RNA primer for the replication of mitochondrial heavy-strand DNA (Agaronyan et al. 2015). To assess a possible functional consequence of altered 7S RNA expression upon depletion of FASTKD2, we determined the mitochondrial DNA content of FASTKD2 WT and NULL cell lines. mtDNA levels are slightly increased in FASTKD2 NULL cells compared to WT controls (Fig. 4J). Importantly, this excludes mtDNA depletion as a cause of the respiration defect of FASTKD2 NULL cells. Further contributions of altered 7S RNA expression to the phenotype of FASTKD2-deficient cells are difficult to assign, as this transcript still awaits further functional characterization (Ojala et al. 1981a; Clayton 1984). Taken together, CRISPR-mediated deletion of FASTKD2 has drastic effects on both mitochondrial gene expression and physiology. Both the broadly compromised cellular respiration and induced glycolysis are consistent with the drastic depletion of the large mitochondrial ribosomal subunit RNA (RNR2) associated with the complete loss of FASTKD2 expression. Nevertheless, the activity of ETC complexes remains detectable in FASTKD2 NULL cells, which suggests residual respiratory chain protein synthesis in the absence of FASTKD2.
CONCLUSIONS
The FASTK family represents an emerging family of RNA-binding proteins. Despite early evidence that FASTK family proteins localize to mitochondria and, as demonstrated for FASTKD3, affect cellular respiration (Simarro et al. 2010), mechanistic and functional information is only now beginning to emerge. A recent high-throughput screen for factors involved in mitochondrial RNA processing implicated FASTKD4 in the regulation of the turnover of a select spectrum of mitochondrial transcripts (Wolf and Mootha 2014). With the aim to identify the molecular underpinnings of a rare form of encephalomyopathy associated with mutated FASTKD2 (Ghezzi et al. 2008), we here propose a mechanistic model for its function. Our identification of the molecular targets of FASTKD2 by iCLIP yields a high-resolution map normalized to exclude PCR amplification-induced artifacts and controlled for specificity. The ensuing characterization of the expression of FASTKD2-bound transcripts upon depletion of FASTKD2 by RNAi or the CRISPR/Cas9 system provides insights into FASTKD2 function in mitochondrial biology and pathophysiology. In combination with the metabolic analyses we thus establish the physiological consequences of a genomic deletion of FASTKD2 in cultured human cells.
The effects of FASTKD2 deficiency appear to be cell type- and tissue-specific, as the originally reported complex IV deficiency was only detectable in muscle biopsies but not in fibroblasts of the affected patients (Ghezzi et al. 2008). The variable capability of different cell types to compensate for the consequences of impaired mitochondrial translation, as suggested by a recent study (Richman et al. 2015), may at least to some degree explain the apparently contrasting consequences of the loss of FASTKD2 in different tissues or cell types. Alternatively, the lack of FASTKD2 might be compensated for by further, tissue-specific factors such as other mitochondrial granule components or FASTK family proteins. It therefore remains to be determined to which degree FASTKD- and other mitochondrial RNA granule proteins cooperate or are functionally interdependent, as suggested by the comparable effects of FASTK (Jourdain et al. 2015) and FASTKD2 (this study) depletion on ND6 expression. Future work on the other members of the FASTK family of proteins will help complete the picture of their mode of function.
MATERIALS AND METHODS
Expression and detection of GFP-fusion proteins
All constructs were inserted into pcDNA5/FRT/TO-GFP, a derivative of pcDNA5/FRT/TO (Invitrogen) with a Gly/Ser-linker EGFP construct inserted between the NotI and ApaI restriction sites. The open reading frame of FASTKD2 comprising amino acids methionine 17, the conserved start codon, to glutamine 710 (Ghezzi et al. 2008) was amplified by PCR using the primers FASTKD2_F (5′-AGCTGGTACCATGAATAACAAAGCG-3′, start codon underlined) and FASTKD2_R (5′-ACGTCTCGAGTTGTGTGCTTTGCAC-3′). The mitochondrial localization signal of FASTKD2 (amino acids 16–68) was amplified using the primers FASTKD2_F and MLS_R (5′-ACGTCTCGAGCATTCTGTTATGAAAGTTATTTAAAATG-3′). The resulting amplicons were inserted into pcDNA5/FRT/TO-GFP using the XhoI and KpnI restriction sites, thus generating C-terminal GFP-fusion proteins. The constructs were introduced into HEK293 Flip-In TRex cell lines according to the manufacturer's recommendations (Life Technologies) using Effectene transfection reagents (Qiagen). For fluorescence microscopy, pcDNA5/FRT/TO-based constructs expressing FASTKD2-GFP or GFP only were transiently transfected into MCF-7 or U2OS cells. Forty-eight hours after transfection, the cells were incubated for 30 min with 250 nM Mitotracker deep red CMX ros (Life Technologies), fixed with formaldehyde and visualized using a Leica SP2 confocal microscope.
UV-cross-linking, immunoprecipitation, and labeling of FASTKD2-associated transcripts
Induced HEK293 Flp-In TRex cell lines expressing FASTKD2-GFP (10 ng/mL tetracycline, 16 h) were washed twice with PBS and irradiated with UV light at 254 nm (150 mJ/cm2) on ice. Immediately after irradiation, cells were lysed in three cell pellet volumes of lysis buffer (100 mM KCI, 5 mM MgCl2, 10 mM Tris–HCI pH 7.5, 0.5% NP-40, 4 mM DTT, 100 U/mL RNasin [Promega], 200 µM ribonucleoside vanadyl complex [NEB], EDTA-free protease inhibitor cocktail [Roche]). Lysates were incubated with 50 U/mL of DNase I (Takara) and indicated concentrations of RNaseA (Sigma) for 10 min at 37°C. Subsequently the lysates were chilled on ice and cleared by centrifugation at 16,000g for 20 min at 4°C. Fusion proteins were captured by incubation with 10 µL of pre-equilibrated magnetic particle suspension (GFP-Trap_M, Chromotek) per mL of lysate with rotation for 2 h at 4°C. Beads were recovered and washed four times with high salt buffer (500 mM NaCl, 20 mM Tris–HCl pH 7.5, 1 mM MgCl2, 0.05% NP40, 0.1% sodium dodecyl sulfate, EDTA-free protease inhibitor cocktail). Subsequently, beads were washed two times with PNK buffer (50 mM Tris–HCl pH 7.5, 50 mM NaCl, 10 mM MgCl2, 0.5% NP-40, 5 mM DTT). Beads were resuspended in the original bead suspension volume and incubated with 1 U/μL T4 polynucleotide kinase (NEB) and 0.1 μCi/μL [γ-32P]ATP (Hartmann Analytics) for 30 min at 37°C and 800 rpm to label the 5′-end of cross-linked RNAs. The magnetic beads were then washed five times with PNK buffer free of DTT. Immunoprecipitates were resolved on 4%–12% gradient gels (BioRad) and blotted on PVDF membranes. Cross-linked RNA–protein complexes were visualized by autoradiography using a phosphorimager (Fuji).
Identification of FASTKD2-associated transcripts by iCLIP
Cell lines expressing FASTKD2-GFP or MLS-GFP were subjected to UV cross-linking and lysis as described above. The cleared lysates (typically 2 mL) were then mixed 1:4 with 5× High Salt Buffer (1.25 M NaCl, 100 mM Tris–HCl pH 7.5, 0.5% sodium dodecyl sulfate). The lysates were precleared by incubation with 60 µL of equilibrated blocked magnetic particle suspension (Chromotek) per mL of lysate (1 h, 4°C, gentle rotation). GFP-fusion proteins were then captured from precleared lysates by incubation with 60 µL of GFP-Trap magnetic particle suspension (GFP-Trap_M, Chromotek) per mL of lysate (2 h, 4°C, gentle rotation). Beads were collected using a magnet and washed twice with high salt buffer. The beads were then washed twice with medium salt buffer (250 mM NaCl, 20 mM Tris–HCl pH 7.5, 1 mM MgCl2, 0.05% NP-40, 100 U/mL RNasin, 200 µM ribonucleoside vanadyl complex, EDTA-free protease inhibitor cocktail) and once with low salt buffer (150 mM NaCl, 20 mM Tris–HCl pH 7.5, 1 mM MgCl2, 0.01% NP-40, 50 U/mL RNasin, EDTA-free protease inhibitor cocktail). Beads were resuspended in 1 mL low salt buffer without RNasin and incubated (3 min, 37°C, 1 100 rpm) with 4 U of RNase I (Life Technologies) and 4 U of Turbo DNase (Life Technologies). Beads were washed once with high salt buffer, twice with low salt buffer, resuspended in 20 µL of PNK mix (70 mM Tris–HCl pH 6.5, 10 mM MgCl2, 5 mM DTT, 250 U/mL T4 polynucleotide kinase [NEB], 500 U/mL RNasin) and incubated with agitation (1 100 rpm) for 20 min at 37°C. Beads were then washed twice with high salt buffer, twice with low salt buffer and incubated for 16 h (1 100 rpm, 16°C) in 20 µL ligation mix (50 mM Tris–HCl pH 7.5, 10 mM MgCl2, 10 mM DTT, 500 U/mL T4 RNA ligase 1, 500 U/mL RNasin, 1.5 μM preadenylated linker L3 [5′-rApp-AGATCGGAAGAGCGGTTCAG-ddC-3′], 20% PEG-400 [Sigma]). The beads were washed three times with high salt buffer, twice with low salt buffer, proteins digested with proteinase K and samples processed for all subsequent steps as described previously (Konig et al. 2010). cDNA libraries obtained after PCR amplification with universal Solexa primers (25 cycles) were multiplexed and sequenced using an Illumina HiSeq2000 platform. Mapping of the retrieved reads to the hg19 assembly of the human genome (considering only single hits in the genome), random barcode evaluation, and evaluation of the significance of crosslink sites (Fig. 2D) were carried out as previously described (Konig et al. 2010). Low FDR (<0.05) crosslink site counts of the three replicates sequenced for each MLS-GFP and FASTKD2-GFP were then used to compute log2(enrichment) and log10(P-value) using DESeq2 (Anders and Huber 2010).
Cell culture, depletion of FASTKD2 by RNAi, and CRISPR-induced mutagenesis
Cells were cultured at 37°C, 95% humidity and 5% CO2 in 1× high-glucose DMEM supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin sulfate. For galactose cultivation experiments, cells were cultured in DMEM without glucose and glutamine (Life Technologies) plus 2 mM L-glutamine, 0.9 g/L D-galactose or 4.5 g/L D-glucose, 10% dialyzed fetal bovine serum, and 1 mM pyruvate. Depletion of FASTKD2 by RNAi was achieved by transfection of Dharmacon smartpool siRNAs targeting FASTKD2 using the MISSION siRNA universal negative control (Sigma) for control experiments and Lipofectamine RNAiMAX reagent (Life Technologies) as suggested by the manufacturer. Typically, cells were harvested and analyzed 72 h after transfection. To achieve the genomic deletion of FASTKD2 by CRISPR/Cas9-mediated mutagenesis, the annealed oligonucleotides FASTKD2_1269F (5′-CACCGCAAGGTTTTCAAATAAAATG-3′) and FASTKD2_1269R (5′-AAACCATTTTATTTGAAAACCTTGC-3′) were inserted into pXPR_001 (obtained from Addgene) via the BsmBI restriction sites. The resulting plasmid was introduced into HEK293 Flp-In TRex cells by lentiviral infection using standard procedures. Infected cell pools were selected with 100 µg/mL zeocin, 5 µg/mL blasticidin, and 1 µg/mL puromycin and antibiotic-resistant single-cell clones isolated by limiting dilution. Upon clonal expansion, loss of FASTKD2 expression was assessed by Western blot (Protein Tech No. 17464-1-AP). To directly identify the mutations introduced by CRISPR/Cas9-mediated mutagenesis, the genomic DNA sequence surrounding the protospacer adjacent motif was amplified using the primers FASTKD2_gSEQ_F (5′-cggtacccggggatcGGATATATTCTGTTGATCAGGACAGTTGAATTGCTGC-3′, lower case indicates homology sequences for In-Fusion cloning) and FASTKD2_gSEQ_R (5′-cgactctagaggatcAGTATGAAGAATAGATAGAATGTTTTTCATGTTTAGGGATTCAGG-3′) using Phusion DNA polymerase (NEB). PCR amplicons were either directly sequenced or inserted into linearized pUC19 by In-Fusion cloning (Clontech) following the manufacturer's recommendations. After transformation, plasmids obtained from individual colonies were subjected to Sanger sequencing.
Analysis of mitochondrial RNA processing by Northern blotting
Northern blot analysis of mitochondrial transcripts was carried out essentially as described previously (Jourdain et al. 2013). Briefly, 10 µg of total RNA were resolved by electrophoresis in 1.2% agarose–formaldehyde gels and blotted by capillary transfer on nylon membranes (Genescreen PlusR, PerkinElmer). Cross-linked membranes were probed in 50% formamide, 7% SDS, 0.2 M NaCl, 80 mM sodium phosphate (pH 7.4), 0.1 mg/mL salmon sperm DNA at 60°C or 45°C (for tRNA probes) with antisense riboprobes prepared by transcription from PCR products. Blots were briefly washed with 1× SSC, 0.1% SDS and afterwards for 30 min with 0.5× SSC, 0.1% SDS. Bands on blots were detected by phosphorimaging.
Metabolic characterization of cell lines
OCR and ECAR were determined using a Seahorse XFe96 analyzer (Seahorse Bioscience) as suggested by the manufacturer. Typically, 25,000 HEK293 cells were seeded into poly-L-lysine (PLL) coated 96-well analyzer plates (Seahorse Bioscience) and grown for 16 h to allow for attachment. OCR and ECAR were recorded in XF assay medium (base medium plus 25 mM D-glucose, 5 mM pyruvate, 4 mM L-glutamine). Leak respiration was measured after injection of 2 µM oligomycin, uncoupled respiration after injection of 200 and 250 nM FCCP and residual oxygen consumption in the presence of 0.5 µM rotenone and 0.5 µM antimycin A. To assess growth of cells in galactose-containing media, 2500 cells were seeded into PLL-coated 96-well plates and grown for 16 h in standard medium to allow for attachment. Cells were washed thrice and further grown in parallel in either galactose or glucose containing media. After 48 h, cell numbers were determined using the CyQUANT Direct cell proliferation assay (Life Technologies) as specified by the manufacturer.
Pulse labeling of mitochondrial translation products
Labeling of mitochondrial translation products was carried out as described previously (Sasarman and Shoubridge 2012). Briefly, 500,000 cells were seeded into PLL-coated wells of a six-well plate on the day before labeling. Cells were washed twice with DMEM without cysteine and methionine and incubated for 30 min (37°C, 5% CO2, 95% relative humidity) with equilibrated labeling medium (DMEM without cysteine and methionine, 2 mM glutamine, 10% dialyzed serum and 1 mM pyruvate). Cytoplasmic translation was inhibited by addition of 100 µg/mL emetine (Sigma) and plates incubated for further 5 min. Finally, translation products were labeled by addition of 200 µCi/mL of L-[35S]Methionine (Hartmann Analytics) for 1 h. Cells were then incubated for 10 min with standard culturing medium, harvested by scraping in ice-cold PBS, lysed with RIPA buffer (25 mM Tris–HCl pH 7.5, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1 mM DTT, EDTA-free protease inhibitor cocktail), cleared by centrifugation (16,000g, 10 min) and 30 µg aliquots (Bradford assay, BSA standard) analyzed by SDS gel electrophoresis (15% gels). Gels were stained with Coomassie brilliant blue, dried, and visualized by phosphorimaging.
Respiratory chain assays
Cells were resuspended in mitochondrial isolation buffer (250 mM sucrose, 50 mM KCl, 5 mM MgCl2, 20 mM Tris–HCl pH 7.5) and lysed by 12 passages through a 27 2/3 G needle. The lysate was centrifuged for 10 min at 600g at 4°C, the supernatant recovered and crude mitochondria recovered by centrifugation at 14,000g for 15 min. Mitochondrial pellets were resuspended in a minimal volume of mitochondrial isolation buffer. The protein concentration of mitochondrial preparations was determined by Bradford assay using BSA as a standard. Mitochondrial enzyme activities were recorded using a Tecan infinite M1000pro plate reader equipped with a thermostat essentially as described previously (Sauer et al. 2005). The following extinction coefficients were used to calculate relative enzyme activities from absorption values: NADH: ε340–400 nm = 6.1 mM−1 cm−1, cytochrome c: ε550–540 nm = 19.0 mM−1 cm−1, 2,6-dichlorphenol-indophenol (DCPIP): ε610–750 nm = 22.0 mM−1 cm−1, 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB): ε412 nm = 14.2 mM−1 cm−1. To assay complex I activity, mitochondria were resuspended in 25 mM potassium phosphate pH 7.5 plus 5 mM MgCl2 and disrupted by sonication in a BioRuptor device (Diagenode, 20 cycles, 30-sec duty, 30-sec pause at 4°C). Complex I activity (absorption at 340 nm against absorption at 400 nm as a background) was recorded as the rotenone-sensitive (20 µM) decylubiquinone reductase activity (50 mM potassium phosphate pH 7.2, 3 mg/mL BSA, 0.3 mM KCN, 200 µM NADH, 4 µM antimycin A, 60 µM decylubiquinone). Complex II activity (absorption at 610 nm against absorption at 750 nm as a background) was recorded in 20 mM Tris–HCl pH 7.5, 250 mM sucrose, 50 mM KCl, 5 mM MgCl2, 0.05% Triton X-100, 20 mM sodium succinate, 40 µM decylubiquinone, 60 µM DCPIP, 2 mM sodium azide. Prior to measurements, mitochondria were preincubated for 10 min at 25°C in mitochondrial isolation buffer containing 2 mM sodium azide and 20 mM succinate. Complex II–III activity was recorded as succinate:cytochrome c reductase activity (50 mM potassium phosphate pH 7.5, 20 mM sodium succinate, 1 mM KCN, 10 µM rotenone, 50 µM oxidized cytochrome c) at 550 nm using absorption at 540 nm as a background. Complex IV activity was recorded as cytochrome c oxidase activity (250 mM sucrose, 120 mM potassium phosphate pH 7.5, 5 mM MgCl2, 0.05% laurylmaltoside, 200 µM reduced cytochrome c) at 550 nm using absorption at 540 nm as a background. Complex V activity (absorption at 340 nm using absorption at 400 nm as a background) was measured by a coupled assay in assay mixtures containing 250 mM sucrose, 50 mM KCl, 5 mM MgCl2, 20 mM Tris–HCl pH 7.5, 250 µM NADH, 1 mM phosphoenolpyruvate, 2 U/mL pyruvate kinase, 2.5 U/mL lactate dehydrogenase and 2 mM ATP. Citrate synthase activity was measured at 412 nm in assay mixtures containing 200 mM Tris–HCl, 0.25% Triton X-100, 100 µM DTNB, 300 µM acetyl-coenzyme A, and 0.5 mM oxaloacetate.
Real-time PCR-based quantification of gene expression and mitochondrial DNA content
RNA was isolated using TRI reagent (Sigma) according to the manufacturer's protocol. cDNA was synthesized using Superscript II reverse transcriptase (Life Technologies) and quantitative PCR reactions were set up using a SYBR green PCR master mix (Applied Biosystems) both as specified by the manufacturer. FASTKD2 expression was detected using the primers FASTKD2_QF (5′-TCCTGAATCCCTAAACATGAAAA-3′) and FASTKD2_QR (5′-GCCATAACTTCCACGAAC TG-3′) using the β-actin transcript BACTF (5′-TCACCGGAGTCCATCACGAT-3′) and BACTR (5′-CGCGAGAAGATGACCCAGA-3′) for normalization. To determine the mitochondrial DNA content, genomic DNA was isolated as previously described (Guo et al. 2009). Mitochondrial DNA was quantified using the primers CO1F (5′-ATACTACTAACAGACCGCAACCTC-3′) and CO1R (5′-GAGATTATTCCGAAGCCTGGT-3′), whereas nuclear DNA was quantified with the primers BACTGF (5′-GAAGG ATTCCTATGTGGGCGA-3′) and BACTGR (5′-CAGGGTGAGGATGCCTCTCTT-3′).
ACKNOWLEDGMENTS
We thank E. Zielonka for help with the generation of CRISPR cell lines, B. Klaus for help with analysis of the data using DESeq2, C. Asencio, R. Horos, and J. Perez-Perri for helpful comments on the manuscript, and A. Castello, A. Jourdain, J.C. Martinou, W. Rossmanith, and all members of the Hentze laboratory for fruitful discussions. This work was supported by a European Molecular Biology Organization (EMBO) long-term fellowship (1371–2013) and a Marie Curie intra-European fellowship (MoMeFAST) to J.P., and a European Research Council advanced grant to M.W.H.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.052365.115.
Freely available online through the RNA Open Access option.
REFERENCES
- Agaronyan K, Morozov YI, Anikin M, Temiakov D. 2015. Mitochondrial biology. Replication-transcription switch in human mitochondria. Science 347: 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonicka H, Shoubridge EA. 2015. Mitochondrial RNA granules are centers for posttranscriptional RNA processing and ribosome biogenesis. Cell Rep 10: 920–932. [DOI] [PubMed] [Google Scholar]
- Baltz AG, Munschauer M, Schwanhäusser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, et al. 2012. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell 46: 674–690. [DOI] [PubMed] [Google Scholar]
- Brzezniak LK, Bijata M, Szczesny RJ, Stepien PP. 2011. Involvement of human ELAC2 gene product in 3′ end processing of mitochondrial tRNAs. RNA Biol 8: 616–626. [DOI] [PubMed] [Google Scholar]
- Calvo SE, Mootha VK. 2010. The mitochondrial proteome and human disease. Annu Rev Genomics Hum Genet 11: 25–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo S, Jain M, Xie X, Sheth SA, Chang B, Goldberger OA, Spinazzola A, Zeviani M, Carr SA, Mootha VK. 2006. Systematic identification of human mitochondrial disease genes through integrative genomics. Nat Genet 38: 576–582. [DOI] [PubMed] [Google Scholar]
- Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, et al. 2012. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149: 1393–1406. [DOI] [PubMed] [Google Scholar]
- Chang JH, Tong L. 2012. Mitochondrial poly(A) polymerase and polyadenylation. Biochim Biophys Acta 1819: 992–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton DA. 1984. Transcription of the mammalian mitochondrial genome. Annu Rev Biochem 53: 573–594. [DOI] [PubMed] [Google Scholar]
- Divakaruni AS, Paradyse A, Ferrick DA, Murphy AN, Jastroch M. 2014. Analysis and interpretation of microplate-based oxygen consumption and pH data. Methods Enzymol 547: 309–354. [DOI] [PubMed] [Google Scholar]
- Fernández-Vizarra E, Tiranti V, Zeviani M. 2009. Assembly of the oxidative phosphorylation system in humans: what we have learned by studying its defects. Biochim Biophys Acta 1793: 200–211. [DOI] [PubMed] [Google Scholar]
- Ghezzi D, Saada A, D'Adamo P, Fernandez-Vizarra E, Gasparini P, Tiranti V, Elpeleg O, Zeviani M. 2008. FASTKD2 nonsense mutation in an infantile mitochondrial encephalomyopathy associated with cytochrome c oxidase deficiency. Am J Hum Genet 83: 415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Jiang L, Bhasin S, Khan SM, Swerdlow RH. 2009. DNA extraction procedures meaningfully influence qPCR-based mtDNA copy number determination. Mitochondrion 9: 261–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. 2013. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31: 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdain AA, Koppen M, Wydro M, Rodley CD, Lightowlers RN, Chrzanowska-Lightowlers ZM, Martinou JC. 2013. GRSF1 regulates RNA processing in mitochondrial RNA granules. Cell Metab 17: 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdain AA, Koppen M, Rodley CD, Maundrell K, Gueguen N, Reynier P, Guaras AM, Enriquez JA, Anderson P, Simarro M, et al. 2015. A mitochondria-specific isoform of FASTK is present in mitochondrial RNA granules and regulates gene expression and function. Cell Rep 10: 1110–1121. [DOI] [PubMed] [Google Scholar]
- Kleinknecht L, Wang F, Stübe R, Philippar K, Nickelsen J, Bohne AV. 2014. RAP, the sole octotricopeptide repeat protein in Arabidopsis, is required for chloroplast 16S rRNA maturation. Plant Cell 26: 777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J. 2010. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol 17: 909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman WJ, Distelmaier F, Smeitink JA, Willems PH. 2013. OXPHOS mutations and neurodegeneration. EMBO J 32: 9–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SC, Yi H, Eichelbaum K, Föhr S, Fischer B, You KT, Castello A, Krijgsveld J, Hentze MW, Kim VN. 2013. The RNA-binding protein repertoire of embryonic stem cells. Nat Struct Mol Biol 20: 1122–1130. [DOI] [PubMed] [Google Scholar]
- Lee I, Hong W. 2004. RAP—a putative RNA-binding domain. Trends Biochem Sci 29: 567–570. [DOI] [PubMed] [Google Scholar]
- Mercer TR, Neph S, Dinger ME, Crawford J, Smith MA, Shearwood AM, Haugen E, Bracken CP, Rackham O, Stamatoyannopoulos JA, et al. 2011. The human mitochondrial transcriptome. Cell 146: 645–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metodiev MD, Lesko N, Park CB, Cámara Y, Shi Y, Wibom R, Hultenby K, Gustafsson CM, Larsson NG. 2009. Methylation of 12S rRNA is necessary for in vivo stability of the small subunit of the mammalian mitochondrial ribosome. Cell Metab 9: 386–397. [DOI] [PubMed] [Google Scholar]
- Nagao A, Hino-Shigi N, Suzuki T. 2008. Measuring mRNA decay in human mitochondria. Methods Enzymol 447: 489–499. [DOI] [PubMed] [Google Scholar]
- Ojala D, Crews S, Montoya J, Gelfand R, Attardi G. 1981a. A small polyadenylated RNA (7 S RNA), containing a putative ribosome attachment site, maps near the origin of human mitochondrial DNA replication. J Mol Biol 150: 303–314. [DOI] [PubMed] [Google Scholar]
- Ojala D, Montoya J, Attardi G. 1981b. tRNA punctuation model of RNA processing in human mitochondria. Nature 290: 470–474. [DOI] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, et al. 2008. A mitochondrial protein compendium elucidates complex I disease biology. Cell 134: 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piechota J, Tomecki R, Gewartowski K, Szczesny R, Dmochowska A, Kudła M, Dybczyńska L, Stepien PP, Bartnik E. 2006. Differential stability of mitochondrial mRNA in HeLa cells. Acta Biochim Pol 53: 157–168. [PubMed] [Google Scholar]
- Richman TR, Ermer JA, Davies SM, Perks KL, Viola HM, Shearwood AM, Hool LC, Rackham O, Filipovska A. 2015. Mutation in MRPS34 compromises protein synthesis and causes mitochondrial dysfunction. PLoS Genet 11: e1005089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson BH, Petrova-Benedict R, Buncic JR, Wallace DC. 1992. Nonviability of cells with oxidative defects in galactose medium: a screening test for affected patient fibroblasts. Biochem Med Metab Biol 48: 122–126. [DOI] [PubMed] [Google Scholar]
- Rothbauer U, Zolghadr K, Muyldermans S, Schepers A, Cardoso MC, Leonhardt H. 2008. A versatile nanotrap for biochemical and functional studies with fluorescent fusion proteins. Mol Cell Proteomics 7: 282–289. [DOI] [PubMed] [Google Scholar]
- Sanchez MI, Mercer TR, Davies SM, Shearwood AM, Nygård KK, Richman TR, Mattick JS, Rackham O, Filipovska A. 2011. RNA processing in human mitochondria. Cell Cycle 10: 2904–2916. [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F. 2014. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11: 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasarman F, Shoubridge EA. 2012. Radioactive labeling of mitochondrial translation products in cultured cells. Methods Mol Biol 837: 207–217. [DOI] [PubMed] [Google Scholar]
- Sauer SW, Okun JG, Schwab MA, Crnic LR, Hoffmann GF, Goodman SI, Koeller DM, Kölker S. 2005. Bioenergetics in glutaryl-coenzyme A dehydrogenase deficiency: a role for glutaryl-coenzyme A. J Biol Chem 280: 21830–21836. [DOI] [PubMed] [Google Scholar]
- Simarro M, Gimenez-Cassina A, Kedersha N, Lazaro JB, Adelmant GO, Marto JA, Rhee K, Tisdale S, Danial N, Benarafa C, et al. 2010. Fast kinase domain-containing protein 3 is a mitochondrial protein essential for cellular respiration. Biochem Biophys Res Commun 401: 440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzioglu M, Ruzzenente B, Harmel J, Mourier A, Jemt E, López MD, Kukat C, Stewart JB, Wibom R, Meharg C, et al. 2013. MTERF1 binds mtDNA to prevent transcriptional interference at the light-strand promoter but is dispensable for rRNA gene transcription regulation. Cell Metab 17: 618–626. [DOI] [PubMed] [Google Scholar]
- Tian Q, Taupin J, Elledge S, Robertson M, Anderson P. 1995. Fas-activated serine/threonine kinase (FAST) phosphorylates TIA-1 during Fas-mediated apoptosis. J Exp Med 182: 865–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu YT, Barrientos A. 2015. The human mitochondrial DEAD-Box protein DDX28 resides in RNA granules and functions in mitoribosome assembly. Cell Rep 10: 854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urlaub H, Hartmuth K, Lührmann R. 2002. A two-tracked approach to analyze RNA-protein crosslinking sites in native, nonlabeled small nuclear ribonucleoprotein particles. Methods 26: 170–181. [DOI] [PubMed] [Google Scholar]
- Wolf AR, Mootha VK. 2014. Functional genomic analysis of human mitochondrial RNA processing. Cell Rep 7: 918–931. [DOI] [PMC free article] [PubMed] [Google Scholar]