

Abstract
Exon definition is the predominant initial spliceosome assembly pathway in higher eukaryotes, but it remains much less well-characterized compared to the intron-defined assembly pathway. Addition in trans of an excess of 5′ss containing RNA to a splicing reaction converts a 37S exon-defined complex, formed on a single exon RNA substrate, into a 45S B-like spliceosomal complex with stably integrated U4/U6.U5 tri-snRNP. This 45S complex is compositonally and structurally highly similar to an intron-defined spliceosomal B complex. Stable tri-snRNP integration during B-like complex formation is accompanied by a major structural change as visualized by electron microscopy. The changes in structure and stability during transition from a 37S to 45S complex can be induced in affinity-purified cross-exon complexes by adding solely the 5′ss RNA oligonucleotide. This conformational change does not require the B-specific proteins, which are recruited during this stabilization process, or site-specific phosphorylation of hPrp31. Instead it is triggered by the interaction of U4/U6.U5 tri-snRNP components with the 5′ss sequence, most importantly between Prp8 and nucleotides at the exon–intron junction. These studies provide novel insights into the conversion of a cross-exon to cross-intron organized spliceosome and also shed light on the requirements for stable tri-snRNP integration during B complex formation.
Keywords: pre-mRNA splicing, spliceosome, exon definition, tri-snRNP, Prp8
INTRODUCTION
The spliceosome is a dynamic molecular machine formed by the interaction of five snRNPs and numerous splicing factors with the pre-mRNA (for review, see Will and Lührmann 2011). Initial assembly of the spliceosome can occur across an intron (intron definition) when the latter is relatively short, i.e., less than ∼250 nucleotides (nt) or alternatively across an exon (exon definition) (for review, see De Conti et al. 2013). Assembly of a spliceosome across an intron is initiated by the interaction of the U1 snRNP with the intron's 5′ splice site (ss), followed by the stable association of the U2 snRNP with the intron's branch site (BS) positioned 18–45 nt upstream of the 3′ss, generating the spliceosomal A complex. Stable recruitment of the U4/U6.U5 tri-snRNP to the A complex leads to formation of the precatalytic B complex. During activation (i.e., Bact complex formation), the B complex undergoes major rearrangements, including the release of the U1 and U4 snRNPs, and the recruitment of several additional proteins. Subsequent rearrangements generate the catalytically activated B* complex that carries out the first step of splicing. During the first step, the pre-mRNA is cleaved at the 5′ss and an intron-3′exon lariat intermediate is formed, generating the spliceosomal C complex. The latter is then activated for the second step of splicing, during which the lariat intron is excised and the 5′ and 3′ exons are ligated.
In contrast to pre-mRNA introns, the vast majority of exons are relatively short (on average only 170 nt in vertebrates; Sakharkar et al. 2005). When intron length exceeds ∼250 nt, as is the case for most mammalian introns (Sakharkar et al. 2005), splicing complexes initially form across the exon directly downstream from the intron to be spliced out (Robberson et al. 1990; Fox-Walsh et al. 2005). This process, called exon definition (Berget 1995), initially involves the interaction of U2 snRNP with the BS upstream of the exon, and U1 snRNP with the 5′ss downstream from it. SR proteins also directly interact with exonic splicing enhancer sequences within the exon, and bridge the U2 and U1 snRNPs via protein–protein interactions (Hoffman and Grabowski 1992). Cross-exon (CE) complexes contain, in addition to U1 and U2, loosely associated U4/U6.U5 tri-snRNP (Schneider et al. 2010b). As splicing catalysis can only occur across an intron, a rearrangement from the exon-defined to intron-organized state is required. Recent studies indicate that exon-defined splicing complexes can be directly converted to intron-organized complexes at the precatalytic B complex stage, rather than at an earlier stage as previously thought (Schneider et al. 2010b).
B complex formation during intron-defined spliceosome assembly involves the stable integration of the U4/U6.U5 tri-snRNP. However, the interactions and events leading to stable tri-snRNP association are not well understood. The DEAD-box helicase Prp28, which destabilizes the interaction between the 5′ss and U1 snRNP, and enables the interaction of the 5′ss with the ACAGAG box of U6 snRNA (Staley and Guthrie 1999), is required for stable tri-snRNP binding during B complex formation (Mathew et al. 2008). Contacts between other tri-snRNP components and the 5′ss during this stage include those between the 5′ss and loop 1 of the U5 snRNA (Newman and Norman 1992; Wyatt et al. 1992; Sontheimer and Steitz 1993), and the U5 snRNP Prp28 (Ismaili et al. 2001) and Prp8 (Reyes et al. 1996, 1999; Sha et al. 1998) proteins. In higher eukaryotes several other proteins appear to be involved in tri-snRNP recruitment/integration including the splicing factor SPF30 (Meister et al. 2001; Rappsilber et al. 2001), SR proteins (Roscigno and Garcia-Blanco 1995), the U4/U6.U5 tri-snRNP proteins 65K/hSAD1 and 110K/hSART1 (Makarova et al. 2001), and the kinase SRPK2 (Mathew et al. 2008). The tri-snRNP-associated factors hPrp31 and hPrp6 are phosphorylated by hPrp4 kinase during B complex formation and these post-translational modifications were proposed to contribute to stable tri-snRNP binding (Schneider et al. 2010a).
During B complex formation several non-snRNP proteins are recruited to the spliceosome. Foremost are a set of proteins that includes RED, MFAP1, FBP21, hSmu-1, hPrp38, and hSnu23, which are denoted B-specific, as they are recruited at this stage and released already during the subsequent activation stage (Bessonov et al. 2010; Agafonov et al. 2011). The transient association of these proteins during the B complex stage suggests that they play an essential role in B complex formation, potentially aiding in the stable integration of the tri-snRNP. While the B complex-specific proteins are conserved in higher eukaryotes, only hSnu23 and hPrp38 have homologs in the yeast S. cerevisiae. Prp38 and Snu23 are components of the tri-snRNP in S. cerevisiae (Gottschalk et al. 1999), but not in humans, suggesting that they play a different role in yeast versus human pre-mRNA splicing. Although exon definition appears to be the predominant initial spliceosome assembly pathway in higher eukaryotes, much less is known about it compared to the intron-defined assembly pathway. Indeed, the switch from a cross-exon to cross-intron organized spliceosome is currently the least well understood step of spliceosome assembly. Disruption of this crucial step can have profound effects on alternative splicing outcomes (Sharma et al. 2005; House and Lynch 2006; Bonnal et al. 2008).
Here we have dissected the cross-exon to cross-intron switch in vitro using a previously established system, in which an excess of a short 5′ss-containing RNA, which mimics the upstream 5′ss of the pre-mRNA, is added in trans after CE complexes are allowed to form (Schneider et al. 2010b). This converts the 37S CE complex (also denoted exon complex) into a faster sedimenting 45S “B-like” complex, in which the tri-snRNP is associated in a salt-stable manner, thereby mimicking the switch from a CE complex to a cross-intron spliceosomal B complex. We demonstrate that stable tri-snRNP integration during B-like complex formation is accompanied by a substantial structural change as visualized by electron microscopy. The changes in structure and stability during transition from a 37S exon to 45S B-like complex do not require the B-specific proteins nor phosphorylation of the hPrp31 protein. Mutational analyses of the 5′ss RNA oligonucleotide reveal that contacts between Prp8 and nucleotides at the exon–intron junction play a key role in the stabilization of tri-snRNP binding and concomitant structural change leading to the formation of a B-like complex. These studies provide insights into the conversion of a cross-exon to cross-intron organized spliceosome, and the requirements for stable tri-snRNP integration during spliceosomal B complex formation.
RESULTS
Displacement of U1 is not sufficient to induce stable tri-snRNP integration
37S exon splicing complexes containing U1, U2, and salt-labile bound U4/U6.U5 tri-snRNP that does not withstand native gel electrophoresis in the presence of heparin, can be generated by incubating an exon RNA (MINX) under splicing conditions in HeLa nuclear extract (Fig. 1; Schneider et al. 2010b). The MINX exon RNA contains a single exon flanked by intronic sequences containing an upstream BPS and polypyrimidine tract, which is essential for U2 snRNP binding, and a downstream, 5′ss with which the U1 snRNP interacts (Fig. 1). Additionally, three MS2-aptamers are inserted at the 3′ end to enable MS2-affinity-selection of splicing complexes. Addition of a 100-fold excess of an RNA oligonucleotide containing a 5′ss (5′ss oligo) with perfect complementarity to U1 snRNA, converts the 37S exon complex into a 45S B-like complex with stably associated tri-snRNP (Fig. 1B,C; Schneider et al. 2010b). The 5′ss oligo not only interacts with the bound tri-snRNP—apparently via base-pairing interactions with the U6 ACAGAG box (Schneider et al. 2010b)—but also displaces the U1 snRNP presumably by competing with the downstream 5′ss of the MINX exon RNA for U1 binding (Fig. 1D). Due to the large amounts of U1 snRNP in HeLa nuclear extract, these effects require the addition of a large excess of the 5′ss oligo relative to the MINX exon RNA, as the 5′ss oligo is sequestered by the free U1 in the extract. To test whether solely displacement of U1 from the downstream 5′ss is sufficient to trigger stable tri-snRNP association and the concomitant formation of a 45S complex, we allowed CE complexes to form and then added an excess of 5′ss oligo containing 2′O methylated (2′Ome) riboses. Unlike the 5′ss oligo, which binds with 1:1 stoichiometry to the 45S complex, this modified oligo does not stably bind to the CE complex, as it is not present in affinity-purified, CE complexes (Supplemental Fig. S1). However, it still displaces U1 from the 5′ss of the MINX exon RNA, as evidenced by the lack of U1 snRNA in the affinity-purified CE complexes formed in its presence (Fig. 1D). Nonetheless, the 2′Ome 5′ss oligo failed to convert the 37S exon complex into a 45S B-like complex with stably bound tri-snRNP (Fig. 1B–D). Thus, displacement of U1 from the 5′ss of the exon RNA is not sufficient to induce 45S complex formation nor stable tri-snRNP integration.
FIGURE 1.

Displacement of U1 is not sufficient to induce 45S B-like complex formation. (A) Schematic representation of the MINX exon RNA (left) and sequence of the optimized 5′ss oligonucleotide (right). Exonic nucleotides are highlighted with a box, the branch point (BP) adenosine is indicated by an A, and the polypyrimidine tract by Yn. (B,C) Cross-exon splicing complexes assembled on 32P-labeled MINX exon RNA in nuclear extract ± the 5′ss oligo or a 2′O-ribose methylated (2′Ome) version, as indicated, and analyzed on a native agarose gel in the presence of heparin (B) or on a 10%–30% glycerol gradient containing 150 mM KCl. (C) The positions of A- and B-like complexes as well as the H complex are indicated. The percentage of total radioactivity in each gradient fraction is plotted. Sedimentation values were determined using prokaryotic ribosomal subunits run in parallel. (D) Complexes in peak gradient fractions 15–18 (without, w/o oligo), 17–20 (+5′ss oligo), or 14–17 (+2′Ome 5′ss oligo) were affinity-purified, and their RNA was analyzed by denaturing PAGE and visualized by silver staining. RNA identities are indicated on the right.
Identification of abundant components of 37S exon and 45S B-like complexes
To identify factors that potentially contribute to stable U4/U6.U5 tri-snRNP binding, we subjected affinity-purified 37S exon and 45S B-like complexes to 2D gel-electrophoresis followed by mass spectrometry (MS) and identified abundant proteins present in each complex (Fig. 2). Abundant proteins (with a molecular mass above 25 kDa) in the 37S exon complex included nearly all U1, U2, and U4/U6.U5 tri-snRNP proteins plus BCLAF1, THRAP3, hnRNPA1b, hPrp4 kinase, and the SR proteins SRSF1, SRSF7, and SRSF10 (Fig. 2B). 45S B-like complexes contained a similar set of abundant proteins except hPrp4 kinase, U1 snRNP, and SR proteins were no longer abundant. In addition, the B-specific proteins were only detected (FBP21, hPrp38, and hSnu23) or first abundant (MFAP1, RED, and hSmu-1) in the 45S B-like complex (Fig. 2B), consistent with them potentially playing a role in stable tri-snRNP integration. In general, the 45S B-like complex contains the same set of abundant proteins as intron-defined B complexes assembled on the MINX pre-mRNA containing an intron flanked by two exons (Agafonov et al. 2011) with the exception that THRAP3 and BCLAF1 are not abundant in these intron-defined B complexes. The mass of abundant proteins lost and concomitantly recruited during 37S to 45S complex transition is nearly identical (∼280 kDa). Thus, the substantially higher sedimentation coefficient of the 45S B-like complex is not due to an increase in its mass, but rather suggests a structural rearrangement in the CE complex prior to or during stable integration of the tri-snRNP.
FIGURE 2.

Identification of abundant proteins in affinity-purified 37S exon and 45S B-like complexes. (A) Proteins were separated by 2D gel electrophoresis, stained with SPYRO Ruby, and the identities of protein spots were determined by mass spectrometry. Proteins smaller than 25 kDa were not analyzed. Abundant proteins and B-specific proteins only present in the 45S B-like complex are indicated in black and less abundant ones in gray. (B) Summary of abundant proteins detected by 2D gel electrophoresis in 37S exon and 45S B-like complexes. Abundant proteins in A were identified by visual inspection and are indicated by a gray box. Proteins are grouped according to their association with snRNPs or stage of recruitment. See the Supplemental Material for a detailed description as to why SRSF7, SRSF10, hPrp38, and Snu23 were also included in the group of abundant factors, despite less intensive staining, and hPrp19 and Smu-1 (in the 37S exon complex) were not considered to be abundant despite a staining intensity similar to, for example, hPrp31.
The structures of the 37S exon and 45S B-like complexes differ substantially
We next determined the structure of the 37S exon and 45S B-like complexes via negative-stain electron microscopy (EM). Well-defined, single particles were visible in the EM overview of both complexes, indicating that the increased S-value of the B-like complex is not due to aggregation (Fig. 3A,B). Classification and class averaging of these single-particle images revealed that the 37S exon complex is characterized by an elongated lower (body) domain and an upper (head) domain, which are connected via a slimmer neck domain. The size and shape of the body of the 37S exon complex are similar in most of the class averages, whereas the triangular-shaped head varies not only in size, but also in its position relative to the body and the sharpness of its appearance (Fig. 3A,E), suggesting that the head is connected via the neck in a relatively flexible manner. The class averages calculated from single particles of the 45S B-like complex appear structurally better defined with a triangular body comprised of a stump, a central mass and a foot, and a globular head domain (Fig. 3B,E). Almost identical views are seen in all class averages, indicating that the 45S complex binds in a highly preferential orientation to the carbon film. Overall it appears more compact with less conformational flexibility of the head domain compared to the 37S exon complex, which might contribute to its higher S-value. Only a small fraction (<15%) of the 37S CE complexes exhibited a morphology similar to the 45S B-like complex (data not shown). The distinct morphologies of the 37S exon and 45S B-like complexes suggest there is a structural rearrangement upon stable integration of the tri-snRNP which may contribute or be a prerequisite for its stable association.
FIGURE 3.
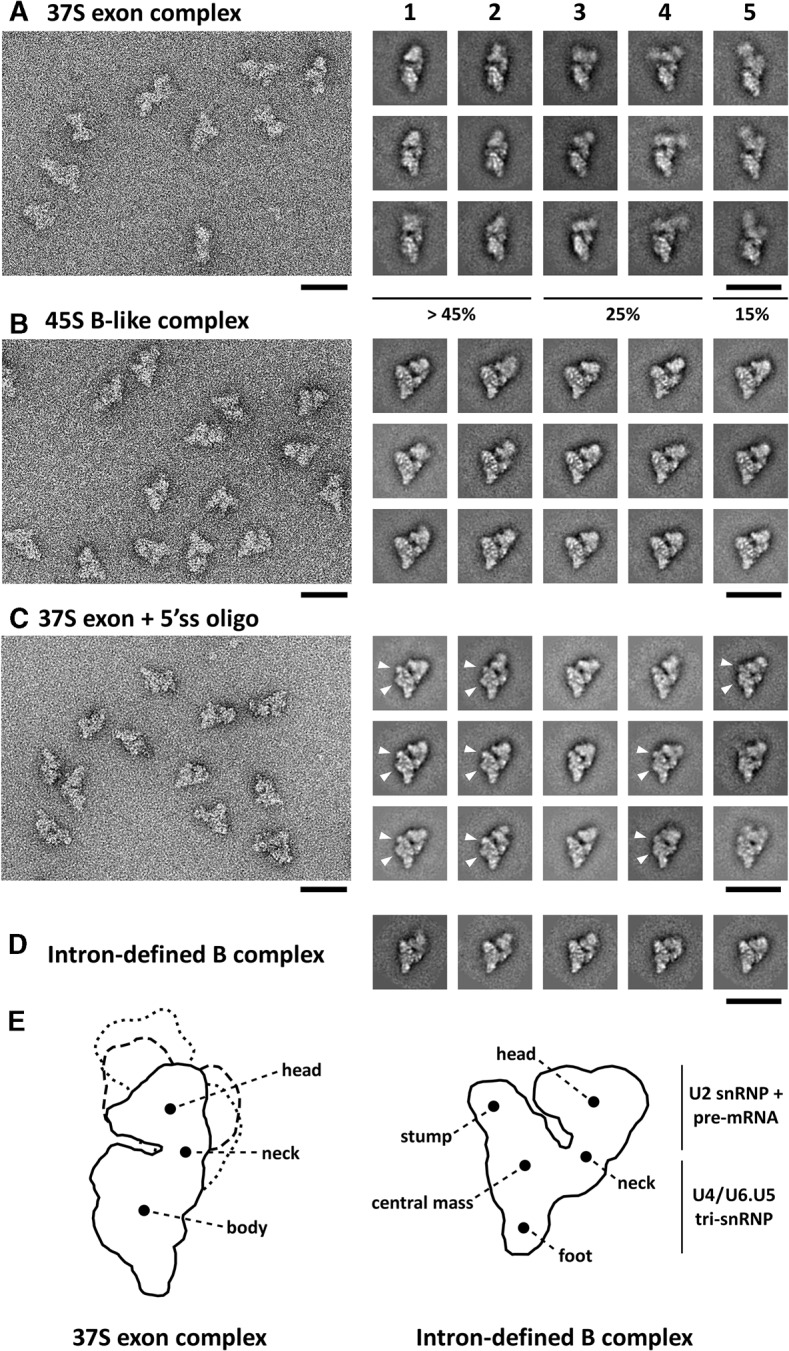
Electron microscopy of 37S exon and 45S B-like complexes. Overviews of negatively stained 37S exon (A) and 45S B-like (B) complexes or (C) affinity-purified 37S exon complexes incubated solely with the 5′ss oligo are shown on the left. Representative class averages of each complex are shown in the galleries on the right from most (1) to least (5) frequently observed classes. The relative abundance of the different classes of the 37S exon complex is indicated. Characteristic structural features of the body domain of the purified 37S exon complex + 5′ss oligo are indicated with arrows. (D) Typical class averages of an intron-defined B complex formed on the MINX pre-mRNA. Scale bars correspond to 50 nm. (E) Schematic representation of the 37S exon complex including different orientations of the head domain characteristic for galleries 1–2 (solid line), 3–4 (dashed), and 5 (dotted), and of the intron-defined B complex with main structural features labeled according to Boehringer et al. (2004).
The morphology of the 45S B-like complex is highly similar to that of an intron-defined B complex that contains stably associated tri-snRNP (Fig. 3D), except for the presence of a more pronounced cleft between the head and body domain. This suggests that the relative orientation of snRNPs within the 45S B-like and B complex are highly similar. Thus, the addition of a 5′ss oligo induces a significant change in the structure of the 37S exon complex such that it adopts an organization characteristic for a B complex with stably integrated tri-snRNP. This is consistent with the idea that the interaction of the 37S exon complex with a 5′ss in trans mimics the situation in a pre-mRNA, where the upstream 5′ss is present in cis.
The 5′ss oligo alone induces a structural change in purified 37S exon complexes
We next tested whether the recruitment of proteins to the 45S B-like complex—in particular the B-specific proteins—is a prerequisite for stable tri-snRNP binding and the accompanying change in its morphology. Thus, 37S exon complexes were affinity purified from a splicing reaction and subsequently incubated solely with either the unmodified or the 2′Ome modified 5′ss oligo. Addition of the unmodified 5′ss oligo but not the modified oligo, shifted the majority of the exon complex into a faster migrating (∼45S) complex that sediments on a glycerol gradient (containing 75 mM salt) in the same fractions as affinity-purified B-like complexes assembled in nuclear extract (Fig. 4A). Affinity purification of complexes peaking in gradient fractions 15–17, revealed that they contain the U4/U6.U5 tri-snRNP in addition to U1 and U2 snRNP and the MINX exon RNA, based on their RNA composition (Fig. 4B). The presence of U1, even after addition of a 100-fold excess of the 5′ss oligo, is likely due to the fact that the affinity-purified complexes plus 5′ss oligo were incubated on ice (due to the labile nature of the purified 37S exon complexes) in contrast to the reaction performed in the presence of nuclear extract, which was incubated at 30°C. This also demonstrates that the presence of U1 snRNP does not hinder the shift to a 45S complex. When analyzed on a glycerol gradient containing 150 mM salt, tri-snRNP remained bound to affinity-purified 37S exon complexes incubated with the 5′ss oligo, but not to 37S exon complexes alone (Supplemental Fig. S2). That is, under these conditions the purified 37S exon complex migrated as a single peak in fractions 10–12 with an S-value <30S, and contained solely U2 and exon RNA. In contrast, the complexes generated by incubating affinity-purified 37S exon complexes with the 5′ss oligo (henceforth called purified exon + 5′ss oligo complexes) contained stoichiometric amounts of U2 and the tri-snRNP, but no longer U1, confirming that the tri-snRNP is associated in a more salt-stable manner (Supplemental Fig. S2). Taken together, these data demonstrate that all of the factors required for salt-stable tri-snRNP integration and the shift from a 37S to 45S complex are present in the affinity-purified 37S exon complex and only require its interaction with the 5'ss oligo. Thus, B complex-specific proteins do not appear to play a major role in this process.
FIGURE 4.
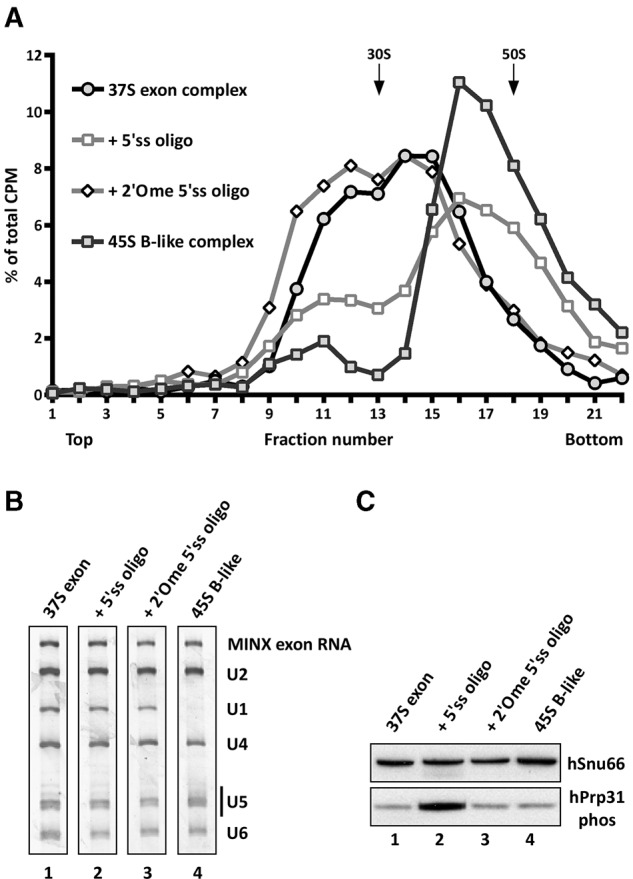
Addition of the 5′ss oligo to the affinity-purified 37S exon complex induces a shift to a 45S complex. (A) Glycerol-gradient centrifugation (75 mM KCl) of affinity-purified 45S B-like complexes or 37S exon complexes incubated alone or solely with the indicated 5′ss oligonucleotide. The percentage of total radioactivity is plotted for each gradient fraction. (B) RNA composition of complexes from peak gradient fractions 14–16 (37S exon or + 2′Ome 5′ss oligo) or fractions 16–18 (+5′ss oligo or 45S B-like). RNA was separated by denaturing PAGE and visualized by silver staining. Positions of the snRNAs and MINX exon RNA are indicated. (C) Western blot of the indicated complexes ± the 5′ss oligo immunostained with antibodies against the phosphorylated form of hPrp31 (hPrp31 phos). Antibodies against hSnu66 were used to ensure equal loading.
As the incubation of purified 37S exon complexes with the 5′ss oligo was performed on ice in the absence of additional ATP, ATP hydrolysis and thus the activity of helicases or kinases also do not appear to be required for the shift in S-value and stable tri-snRNP integration. Consistent with previous results (Schneider et al. 2010b), phosphorylation of hPrp31 was strongly enhanced in 45S B-like complexes formed in extract as compared to 37S exon complexes (Fig. 4C). However, no increase in hPrp31 phosphorylation was observed with the purified exon + 5′ss oligo complexes. Thus the shift to a 45S complex with tri-snRNP bound in a salt-stable manner is not dependent on hPrp31 phosphorylation.
Negative-stain EM of purified exon + 5′ss oligo complexes, gradient purified at 150 mM salt, revealed single, well-defined particles (Fig. 3C), indicating that the increased S-value is not due to particle aggregation. In the vast majority of the 2D class averages (which did not vary substantially from one another), the overall dimensions and appearance of this complex were very similar to that of the 45S B-like complex assembled in splicing extract (Fig. 3), including a similar relative orientation of the head and body. However, small differences can be seen. The body of the purified exon + 5′ss oligo complex contained an additional protrusion and a less pronounced stump (Fig. 3C, indicated with arrows), potentially due to the absence of B-specific proteins. Nonetheless, these results indicate that the recruitment of B-specific proteins is not responsible for the major change in exon complex morphology observed by EM. Thus, the major structural rearrangement accompanying stable tri-snRNP interaction is not dependent on protein factors that are not already present in the affinity-purified 37S exon complex, including the B-specific proteins, but depends on the interaction of the 5′ss oligo.
Effect of 5′ss oligo mutations on stable tri-snRNP binding during B-like complex formation
We next introduced a variety of mutations into the 5′ss oligo and analyzed their effects on 45S B-like complex formation to determine which RNA–RNA and/or RNA–protein interactions play a role in inducing the structural rearrangement and accompanying stable integration of the tri-snRNP. Previous studies indicated that intron nucleotides of the 5′ss oligo base pair with the U6 snRNA ACAGAG box in the 45S B-like complex (Schneider et al. 2010b), while exon nucleotides potentially interact with U5 snRNA loop I. Disruption of the base-pairing potential between loop I of U5 snRNA and the 5′ss oligo by mutating exonic nucleotides −2 to −3 to uridines (U5:5′ss), had little effect on B-like complex formation in nuclear extract as assayed by native gel electrophoresis in the presence of heparin (Fig. 5B, lane 3), or by the less stringent glycerol-gradient centrifugation (Supplemental Fig. S3). Mutation of intron nucleotides +5 to +8 (U6:5′ss), which disrupts base-pairing interactions with the U6 ACAGAG box, impaired, but did not abolish formation of B-like complexes on native gels (Fig. 5B, lane 4), but had no effect on 45S complex formation when assayed by gradient centrifugation (Fig. 5C). However, when the 5′ss oligo contained both mutations (U5:5′ss + U6:5′ss), 45S B-like complex formation was completely abolished. In addition, the 5′ss oligo no longer copurified with exon complexes, indicating that it no longer was stably bound by the tri-snRNP (Supplemental Table S1; Supplemental Fig. S5). Taken together these results indicate that neither the U5/5′ss nor U6/5′ss interaction alone is absolutely necessary for the conformational change leading to a 45S complex, but that the U6/5′ss interaction potentially contributes to the stability of tri-snRNP association in the presence of heparin.
FIGURE 5.
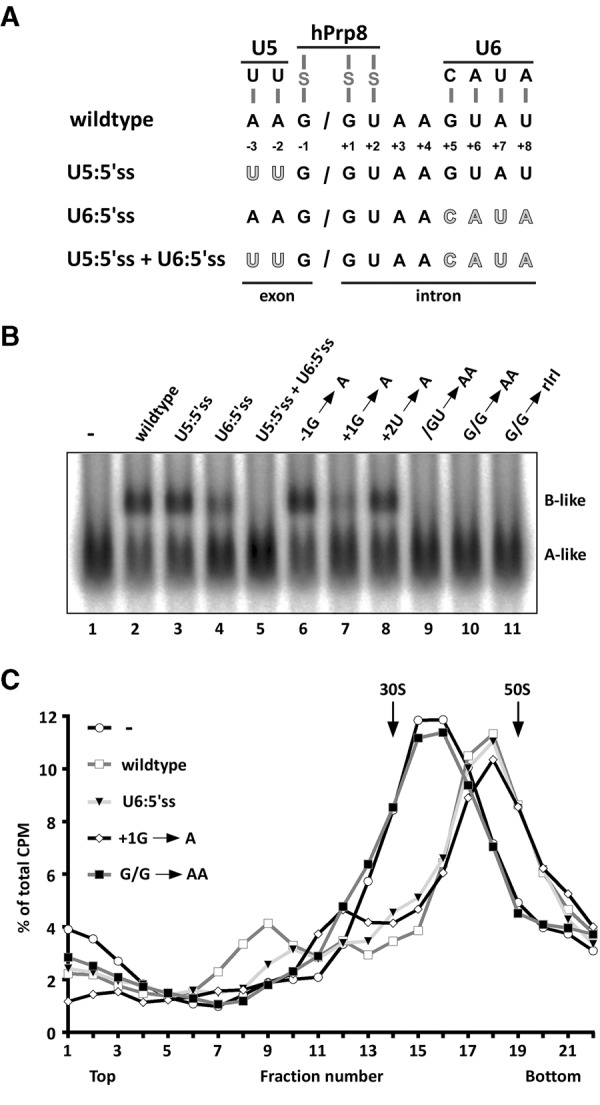
Mutation of nucleotides at the exon–intron junction of the 5′ss oligo abolishes 45S B-like complex formation. (A) Schematic of interactions between the 5′ss oligonucleotide and U5 snRNA, U6 snRNA, and hPrp8, and overview of mutations introduced within the 5′ss oligonucleotide sequence to abolish base-pairing with U5 and/or U6 snRNA. Mutated nucleotides are highlighted in gray. (B) Native gel analysis of B-like complex assembly upon addition of the indicated 5′ss oligonucleotides to the splicing reaction after allowing cross-exon complex formation. Samples were analyzed on a 2% (w/v) LMP agarose gel in the presence of heparin and visualized by autoradiography. The identities of the spliceosomal complexes are indicated. (C) Glycerol-gradient centrifugation (150 mM KCl) of spliceosomal complexes formed on 32P-MINX exon RNA in splicing reactions upon addition of the indicated 5′ss oligonucleotides. The percentage of total radioactivity is plotted for each gradient fraction.
Next, we monitored the impact of mutations within the conserved G/GU nucleotides at the exon–intron junction that are not involved in RNA–RNA base-pairing interactions, but interact with the U5 hPrp8 protein in intron-defined B complexes formed during trans- and cis-splicing (Reyes et al. 1996, 1999). To confirm that hPrp8 interacts with the 5′ss oligo exon–intron junction in our 45S B-like complexes, we first generated 45S complexes using a 32P end-labeled 5′ss oligo containing a 6-thio-G substitution at position +1 of the intron, which did not impair B-like complex formation (Fig. 6A). UV crosslinking of these complexes followed by RNase A digestion, revealed a major crosslinked protein migrating at ∼250 kDa and only two other very weak crosslinks at ∼100 and ∼35 kDa (Fig. 6B). Immunoprecipitation with hPrp8-specific antibodies confirmed that the crosslinked protein at ∼250 kDa is hPrp8 (Fig. 6C). Similar results, albeit with different hPrp8 crosslinking efficiencies, were obtained using 5′ss oligos with 6-thio-G at position −1 or 4-thio-U at position +2 (Supplemental Fig. S4), demonstrating that predominantly hPrp8 contacts the exon–intron boundary nucleotides of the 5′ss oligo.
FIGURE 6.

Exon–intron junction nucleotides of the 5′ss oligo interact with the hPrp8 protein in 45S B-like complexes. (A) The (+1) 6-thio-G modification does not hinder B-like complex formation as assayed by native gel electrophoresis performed as previously described. (B) 45S B-like complexes assembled in the presence of a 5′-32P-labeled (+1) 6-thio-G modified 5′ss oligo were subjected to glycerol-gradient centrifugation, and affinity-purified complexes were irradiated at 365 nm (lanes 2,3,5,6) to induce protein–RNA crosslinks and subsequently treated with RNase A (lanes 3,6). Proteins were separated on a 10%–13% polyacrylamide gel and visualized by silver staining (lanes 1–3) or by a phosphoimager (lanes 4–6). (*) Position of MINX exon–RNA. (C) Immunoprecipitation confirms that the major crosslinked protein is hPrp8. 45S B-like complexes ± UV irradiation were denatured and immunoprecipitation was performed with PAS-bound antibodies against hPrp8, as indicated. Immunoprecipitated proteins were analyzed by Western blotting with hPrp8-specific antibodies (upper panel) and proteins crosslinked to the 32P-labeled 5′ss oligo were detected by autoradiography (lower panel).
5′ss oligos containing the single point mutations −1G → A or +2U → A induced B-like formation as efficiently as the wildtype (Fig. 5B), while +1G → A strongly impaired B-like complex formation in the presence of heparin, but had no effect on 45S complex formation as assayed by glycerol-gradient centrifugation (Fig. 5C; Supplemental Fig. S3). Thus, like the U6:5′ss mutant, this point mutation reduces the stability of tri-snRNP association in the presence of heparin, but does not affect 45S complex formation nor the salt-stable binding of the tri-snRNP as assayed by gradient centrifugation. Mutation of two neighboring exon–intron junction nucleotides (i.e., G/G → AA or /GU → AA) completely inhibited 45S complex formation (Fig. 5B,C). Although the base-pairing potential with U5 or U6 is not altered, oligos containing the double mutations were no longer bound by CE complexes, whereas the wild-type and −1G → A oligos were retained (Supplemental Fig. S5; Supplemental Table S1). This indicates that protein–RNA interactions, which may also be needed for establishing or maintaining the short U5/5′ss and U6/5′ss base-pairing interactions, are required for stable binding of the 5′ss oligo. Mutation of the two G's at the exon–intron junction to riboinosine (rIrI) also inhibited stable tri-snRNP integration (Fig. 5) and the transition to a 45S complex (Supplemental Fig. S3), suggesting a very specific and functionally important recognition of these guanosine bases by hPrp8. As the 2′Ome 5′ss oligo also did not induce 45S B-like complex formation (Fig. 1), nor was bound by the 37S exon complex (Supplemental Fig. S1), the physical association of the 5′ss oligo also appears to involve recognition of its ribose backbone. This is in line with previous results showing that modifications of the ribose backbone at position −2 to +3 of a 5′ss oligo inhibited the formation of intron-defined spliceosomes during trans-splicing (Sha et al. 1998). Taken together, these results support the idea that hPrp8 plays a key role in binding the 5′ss oligo and further in mediating the structural change that leads to/accompanies stable integration of the tri-snRNP.
DISCUSSION
Here we have investigated the switch from a cross-exon to cross-intron organized spliceosome, one of the most poorly understood steps of spliceosome assembly. Our data provide valuable new insights into the requirements for stable U4/U6.U5 tri-snRNP integration during this process, which likely also apply to precatalytic B complex formation during the intron-defined assembly pathway. They reveal that interaction of a cross-exon spliceosomal complex with a 5′ss oligo added in trans triggers a major conformational change that is visible by EM and accompanies the stable integration of the tri-snRNP. This conformational change does not require the B-specific proteins, which are recruited during this stabilization process. Instead it is triggered by the interaction of tri-snRNP components with the 5′ss sequence, most importantly hPrp8.
A simplified system that mimics the switch from a CE complex to a precatalytic B complex
Investigation of the switch from a cross-exon defined to a cross-intron organized spliceosome, using multiple intron and exon-containing pre-mRNAs, has proven difficult due to the inability to first form solely a CE complex and then chase it into a cross-intron organized complex. However, by adding in trans an excess of a 5′ss containing RNA, we can convert a CE complex, formed on a single exon RNA substrate, into a B-like spliceosomal complex that is compositionally and structurally highly similar to an intron-defined B complex. That is, both B-like and B complexes not only have the same snRNA composition, but also an almost identical set of abundant proteins (Fig. 2; Agafonov et al. 2011), exhibit identical sedimentation coefficients of ∼45S (data not shown), and are morphologically nearly indistinguishable under the electron microscope (Fig. 3). This indicates a very similar spatial organization of their RNA and protein components, including a similar if not identical arrangement of U2 and the tri-snRNP. The tri-snRNP is associated with both complexes in a heparin-resistant manner, consistent with similar molecular interactions tethering the tri-snRNP to each complex. Previous studies also showed that the B-like complex is a functional intermediate that is catalytically active in trans splicing of the 5′ss oligo (Schneider et al. 2010b). Thus, data presented here underscore the similarities between the B-like and intron-defined B complex and thus provide additional evidence that the simplified in vitro system that we employ here indeed mimics the switch from a CE complex to a cross-intron B complex. Thus the factors contributing to stable tri-snRNP integration during B-like complex formation, which we identify in this study, also likely play key roles in this process during intron-defined B complex formation.
Exchange of abundant spliceosomal proteins during the transition from a 37S exon to 45S B-like complex
Abundant components of the 37S exon and 45S B-like complexes potentially play key structural roles in these RNP complexes. A comparison of the protein composition of these complexes by 2D gel electrophoresis not only identified those proteins that are stoichiometrically or near stoichiometrically present, but also revealed which proteins are recruited and released during the stabilization of tri-snRNP binding and formation of a B-like complex. Identical to a cross-intron B complex, formation of a 45S B-like complex was accompanied by the recruitment of the B-specific proteins RED, MFAP1, FBP21, hSmu-1, hPrp38, and hSnu23 (Agafonov et al. 2011). In contrast, the U1 snRNP, SR proteins, and hPrp4 kinase (hPrp4K), which is required for stable B complex formation in vitro (Schneider et al. 2010a), were lost. hPrp4K phosphorylates the U4/U6.U5 tri-snRNP-associated factors hPrp31 and hPrp6 during intron-defined B and 45S B-like complex formation (Schneider et al. 2010a,b). Although hPrp4K is present in the 37S exon complex, it does not appear to be active, as seen by the very low phosphorylation levels of hPrp31 in the 37S exon complex (Fig. 4C), suggesting that its activity is repressed and first triggered upon interaction of the 37S exon complex with the 5′ss oligo. The structural change accompanying 45S complex formation might increase the accessibility of the phosphorylation sites on hPrp31 and hPrp6 or lead to the establishment of new protein–protein interactions that trigger hPrp4K activity. However, this conformational rearrangement at the same time appears to induce changes that also result in the release of hPrp4K.
Phosphorylation of hPrp31 and/or hPrp6 was previously proposed to play a decisive role in stable tri-snRNP integration during B-like and B complex formation (Schneider et al. 2010a,b). Here we provide evidence that this is not the case, at least not in vitro; addition of the 5′ss oligo to purified 37S exon complexes induces salt-stable binding of the tri-snRNP, but phosphorylation of hPrp31 does not increase, indicating that the transition to a 45S complex is not dependent on its phosphorylation and presumably also not that of hPrp6. However, the presence of the MS2-MBP protein used for affinity-selection of 37S exon complexes precludes the analysis of the purified exon + 5′ss oligo complexes by native gel electrophoresis in the presence of heparin. Thus, we cannot rule out that these phosphorylation events, while not required for the structural change, may still contribute to highly stable, heparin-resistant tri-snRNP binding, and also that they may potentially play an important role in vivo. However, mutations in the 5′ss oligo that severely reduced B-like complex formation on native gels in the presence of heparin (such as +1G → A) did not correlate with a reduction in hPrp31 phosphorylation (Supplemental Fig. S3).
B-specific proteins are not required for salt-stable tri-snRNP integration
We show that addition of the 5′ss oligo alone to affinity-purified 37S exon complexes (i.e., in the absence of nuclear extract) induced the formation of a 45S complex with a stably integrated tri-snRNP. Thus, all factors required for salt-stable tri-snRNP binding are already present in the purified 37S exon complex. As the B-specific proteins are for the most part absent or present in low amounts in the 37S exon complex, these results show that the B-specific proteins are not essential for salt-stable tri-snRNP binding. However, we cannot exclude that they contribute to stable tri-snRNP integration at higher salt concentrations or in the presence of heparin, as it was not possible to analyze affinity-purified CE complexes on native gels when heparin was added. However, when we checked by Western blotting the amount of B-specific proteins and also site-specifically phosphorylated hPrp31 present in various affinity-purified B-like complexes, including those with reduced heparin-resistance (i.e., formed in the presence of the U6:5′ss or +1G → A 5′ss oligos), there was no detectable difference (Supplemental Fig. S3). These results show that this apparent difference in complex stability does not correlate with the amount of B-specific proteins present. These results are consistent with the idea that the B-specific proteins, as well as phosphorylation of hPrp31, are not major determinants for the heparin-resistant (i.e., highly stable) binding of the tri-snRNP. As the B-specific proteins do not appear to contribute substantially to B complex formation, and are already displaced upon formation of the Bact complex, they likely are involved in one or more of the RNP or compositional rearrangements that occur during the B to Bact transition.
Stable binding of the tri-snRNP likely depends on interactions with U2 proteins
Characterization of abundant factors in the 45S B-like complex showed that it is mainly composed of U2 snRNP and tri-snRNP proteins in addition to the B-specific proteins. As we showed that the B-specific proteins are not essential to stabilize the interaction of the tri-snRNP, tri-snRNP interactions with U2 proteins likely play a key role in this process. Indeed, the tri-snRNP is initially recruited to the CE complex via interactions with the U2 snRNP, including formation of U2/U6 helix II (Schneider et al. 2010b). Yeast-2-hybrid experiments identified interactions between the U2-associated SF3b proteins and U5-specific proteins, especially hPrp8, hBrr2, and hSnu114 (Hegele et al. 2012). Thus, interactions between these proteins potentially play a role in the salt-stable integration of the tri-snRNP during B-like complex formation. Interaction of the 5′ss oligo with the tri-snRNP, in particular hPrp8, may trigger the formation of new protein–protein interactions involving U2 and U5 proteins, or stabilize existing ones (see below). Our studies did not reveal a role for several factors previously implicated in stable tri-snRNP integration in the cross-intron B complex, including SPF30 (Meister et al. 2001; Rappsilber et al. 2001) or SR proteins (Roscigno and Garcia-Blanco 1995). That is, neither the 37S exon nor the 45S B-like complexes contained abundant amounts of SPF30, and SR proteins abundant in the 37S exon complex are displaced upon formation of the 45S B-like complex.
37S exon complexes undergo a major structural change upon 5′ss oligo binding
Comparison of the EM class averages generated for the 37S exon and 45S B-like complexes demonstrated that stable tri-snRNP integration during 45S B-like formation is accompanied by a major rearrangement in the structure of the complex. Previous immuno-EM studies mapped the U2 snRNP to the head domain of intron-defined, human B complexes (Wolf et al. 2009). Immuno-EM studies with S. cerevisiae B complexes, indicated that U5-associated proteins are located in the body domain, while the neck domain is mainly composed of U4/U6 di-snRNP-specific proteins (N Rigo, B Kastner, R Lührmann, in prep.). A comparison of the EM structures of the 37S exon and 45S B-like complex suggests that the position of the head domain relative to the body is the main structural difference between these complexes. Thus, the conformational change triggered by the interaction with the 5′ss oligo, probably involves a rearrangement of the tri-snRNP (body) with respect to the U2 snRNP (head), leading then to the stable tri-snRNP integration.
RNA structure probing of affinity-purified 37S exon and 45S B-like complexes showed little change in U6 snRNA accessibility, aside from reduced accessibility of the U6 ACAGAG box in the 45S B-like complex (Schneider et al. 2010b), which apparently is due to interaction of the 5′ss oligo with this region of U6. This supports the idea that the observed structural change is not due to major changes in RNA–protein or RNA–RNA interactions. In the 37S exon complex, the U4/U6.U5 tri-snRNP establishes base-pairing interactions solely with the U2 snRNA, via U2/U6 helix II, and does not appear to interact directly with the MINX exon RNA, at least not via RNA–RNA interactions (Schneider et al. 2010b). Thus, the interaction of the 5′ss oligo probably leads to a more stable orientation between U2 snRNP and the tri-snRNP by inducing changes in protein–protein interactions. Consistent with this idea, the addition of a 5′ss oligo to HeLa nuclear extract results in formation of a U2/U4/U6/U5 tetra-snRNP (Konforti and Konarska 1994), suggesting that the interaction of the 5′ss oligo with the U4/U6.U5 tri-snRNP induces or stabilizes interactions between U2 snRNP and the tri-snRNP even in the absence of pre-mRNA.
Sequence requirements for the 5′ss oligo to induce a structural change
By introducing mutations within the 5′ss oligo, we provided novel insights into RNA–RNA and RNA–protein interactions that play a role in the stabilization of U4/U6.U5 tri-snRNP binding and the structural change that generates a 45S B-like complex. The changes in composition, structure, and stability, which occur during formation of a 45S B-like complex, were induced by addition of solely the 5′ss oligo to the splicing reaction. The 5′ss is recognized by multiple factors during intron-defined cis-splicing and also trans-splicing, including the U5 and U6 snRNAs, and the U5 Prp8 protein. In previous studies, a highly specific crosslink between intron position +2 (relative to the exon–intron junction) and the RNase H domain of hPrp8 was identified, which coincides with B complex formation (Reyes et al. 1996, 1999). hPrp8 was also shown via crosslinking to contact 5′ss nucleotides spanning the 5′ss exon–intron junction in intron-defined B complexes (Sha et al. 1998). Consistent with this result, we demonstrated via site-specific protein–RNA crosslinking that hPrp8 contacts the G/GU nucleotides of the 5′ss oligo at the exon–intron junction in stably assembled 45S B-like complexes, and that Prp8 is the major crosslinked protein (Fig. 6).
Stable binding of the 5′ss oligo to the exon complex appears to be a prerequisite for the transition from a 37S to 45S complex, as we did not observe 45S complex formation when the 5′ss oligo did not remain bound during MS2-affinity purification (Supplemental Table S1). Binding of the 5′ss oligo thus correlates with the structural change transforming the 37S exon complex into a 45S B-like complex. Multiple contacts involving not only Prp8, but also U6/5′ss base-pairing interactions appear to contribute to stable 5′ss oligo binding, and the resulting conformational change from a 37S to 45S complex. Disruption of U5/5′ss or U6/5′ss base-pairing potential individually had no effect on the formation of a 45S complex as assayed by glycerol-gradient centrifugation. Thus, these interactions are not required to induce the structural change in the complex. While the U5:5′ss or U6:5′ss oligos copurified with the complexes formed on the exon RNA, the double mutant did not. This suggests that both U5 and U6 contacts contribute to 5′ss oligo binding, assuming that these mutations do not additionally alter protein–RNA interactions; for example, hPrp8 interactions with the 5′ss oligo may be dependent on U6/5′ss interactions and vice versa. In the U5:5′ss and U6:5′ss double mutant, the highly conserved GU at positions +1 to +2 is not altered and thus a major determinant for hPrp8 binding is still present. Nonetheless, the 5′ss oligo no longer copurifies with the CE complexes, indicating that contacts between exon–intron junction nucleotides and Prp8 are not sufficient. Likewise, mutation of /GU to /AA, G/G to A/A, or G/G to rI/rI also leads to loss of 5′ss oligo binding indicating that hPrp8 contacts with these nucleotides are necessary, but not sufficient for 5′ss oligo binding and the accompanying conformational change. Our results are consistent with previous studies showing that the interaction of a 5′ss RNA oligonucleotide with the U2/U4/U6/U5 tetra-snRNP in HeLa nuclear extract is also dependent on U6/5′ss and most importantly Prp8/5′ss interactions, with mutations at intron positions +1 and +2 exhibiting the most pronounced negative effects on 5′ss binding (Konforti and Konarska 1994).
Heparin-resistant (i.e., highly stable) tri-snRNP binding during B-like complex formation was only observed if the 5′ss oligo was stably bound and the structural rearrangement transforming the 37S exon complex into a 45S B-like complex had occurred. However, the latter conformational change did not always translate into heparin-resistant tri-snRNP binding. For example, the G → A transition at position +1, as well as the U6:5′ss mutation, reduced substantially stable tri-snRNP binding in the presence of heparin, but had little or no effect on 45S B-like complex formation as assayed by gradient centrifugation. This suggests there are multiple protein–protein, protein–RNA, and/or RNA–RNA interactions that contribute incrementally to stable tri-snRNP integration, and that these mutations weaken or abolish only a subset of them. In previous studies, addition of a 2′Ome oligo complementary to the U6 ACAGAG box completely blocked B-like formation on native gels in the presence of heparin (Schneider et al. 2010b), further indicating that the U6/5′ss base-pairing interaction is an important determinant for highly stable tri-snRNP association. The more dramatic effect observed in this case suggests that other molecular interactions were also disturbed by the addition of this anti-U6 oligonucleotide.
The most severe impairment of B-like formation was observed upon mutation of nucleotides at the exon–intron junction which are contacted primarily by hPrp8. This provides evidence that not only RNA–RNA interactions between the 5′ss oligo and the snRNAs, but also additional protein–RNA interactions, in particular hPrp8, are required for stable binding of the 5′ss oligo and the accompanying structural changes leading to B-like complex formation. Recognition of the 5′ss by hPrp8 also involves the exocyclic part of the GG dinucleotide at the exon–intron junction, as mutation to riboinosine nucleotides inhibit B-like complex formation (Fig. 5). It also appears to involve recognition of the ribose backbone by Prp8, based on the loss of B-like formation when a 2′Ome 5′ss oligo was used. In summary, base-pairing with the U6 ACAGAG box is not required to trigger the observed structural rearrangement during transition to a 45S B-like complex, but instead the latter likely requires a highly specific interaction of hPrp8 with nucleotides at the exon–intron junction. Given the compositional, structural and functional similarities between the B-like and the intron-defined B complex, these findings likely also hold true for the formation of an intron-defined B complex where the 5′ss is present in cis.
Molecular mechanism whereby the Prp8/5′ss interaction could lead to stable tri-snRNP binding
The hPrp8-RNase H domain crosslinks with the +2 position of the 5′ss in B complexes formed via cis- and trans-splicing (Reyes et al. 1996, 1999). We detected crosslinks between nucleotides −1 to +2 of the 5′ss oligo and hPrp8 in stably assembled, 45S B-like complex and it is likely that these crosslinks are also located in Prp8′s RNase H domain. The latter was recently shown to adopt both an open and closed conformation (Schellenberg et al. 2013), and structural studies of the C-terminal part of hPrp8 revealed considerable movements of the RNase H domain with respect to hPrp8′s reverse transcriptase and endonuclease domains (Galej et al. 2013). These alternative positions of the RNase H domain were proposed to transmit conformational changes of the RNA network during splicing. Binding of the 5′ss oligo by hPrp8 during B-like complex formation might induce a conformational change in the RNase H domain of hPrp8 that is transmitted to other domains of the protein. These rearrangements might in turn induce changes in interactions between proteins of the U4/U6.U5 tri-snRNP and the U2 snRNP, and as a consequence, result in the structural transition to a complex with stably bound tri-snRNP.
MATERIALS AND METHODS
Exon and B-like complex formation
Splicing complexes were assembled on 32P-labeled, uncapped MINX exon RNA in HeLa nuclear extract essentially as described previously (Schneider at al. 2010b) except 37S exon complexes were assembled by incubating at 30°C for 6 min, and 45S B-like complexes were formed by incubating for 3 min and then adding a 100-fold excess of a 5′ss containing RNA oligonucleotide (AAGGUAAGUAU) (Eurofins MWG Operon) and incubating for an additional 3 min. Spliceosomal complexes were analyzed by agarose gel electrophoresis in the presence of 0.65 µg/µL heparin (Das and Reed 1999) and bands were visualized with a Typhoon phosphoimager (GE Healthcare).
MS2-affinity-selection of splicing complexes
Spliceosomal complexes were isolated by MS2-affinity-selection essentially as previously described (Bessonov et al. 2010). Briefly, MINX exon RNA was incubated with a 20-fold molar excess of MS2-MBP fusion protein and then added to a standard, 1.0 mL splicing reaction. The latter was loaded onto a 14 mL linear 10%–30% (v/v) glycerol gradient containing G-150 buffer (20 mM HEPES-KOH pH 7.9, 1.5 mM MgCl2, 150 mM KCl), centrifuged at 25,000 rpm for 15 h at 4°C in a Sorvall TST 41.14 rotor and fractions were harvested from the top. The distribution of 32P-labeled MINX exon RNA across the gradient was determined by Cherenkov counting. Peak fractions containing the spliceosomal complexes were pooled and loaded onto amylose beads (NEB). The matrix was washed with G-75 buffer (20 mM HEPES-KOH pH 7.9, 1.5 mM MgCl2, 75 mM KCl) and complexes were eluted with G-75 buffer containing 20 mM maltose.
Stabilization of affinity-purified exon complexes
MS2-affinity-purified 37S exon complexes in G-75 buffer containing 20 mM maltose were incubated with a 100-fold excess of 5′ss oligonucleotide for 15 min on ice. The reaction was loaded onto a linear 10%–30% (v/v) glycerol gradient containing G-75 or G-150 buffer as indicated, and centrifuged at 60,000 rpm for 135 min at 4°C in a Sorvall TH660 rotor.
2D gel electrophoresis and mass spectrometry
Two-dimensional gel-electrophoresis of affinity-purified spliceosomal complexes was performed as described in Agafonov et al. (2011), except the second dimension was performed with an 8% acrylamide gel, and proteins were stained with RuBPS (RubiLAB). For mass spectrometry, Coomassie-stained protein spots were cut out of the 2D gel and proteins were digested in-gel with trypsin and extracted. The extracted peptides were analyzed in a liquid chromatography coupled electrospray ionization quadruple time-of-flight mass spectrometer (LTQ Orbitrap XL) under standard conditions. Proteins were identified by searching fragment spectra against the NCBI nonredundant (nr) database using Mascot as a search engine.
Electron microscopy
For EM, affinity-purified complexes were subjected to a second, linear 10%–30% (v/v) glycerol gradient containing G-75 (37S exon complex) or G-150 (45S B-like and purified exon + 5′ss oligo complex) buffer, and 0%–0.1% (v/v) glutaraldehyde (Kastner et al. 2008) and centrifuged at 60,000 rpm for 2 h in a TH660 rotor at 4°C. Gradients were harvested manually in 175 µL fractions from the top. Particles in peak fractions were negatively stained by the single-carbon film method adopted from Golas et al. (2003). Images were recorded at 160 kV and a magnification of 88,000 with a CM200 FEG electron microscope (Philips) at RT and twofold binning on a 4 k × 4 k CCD camera (TVIPS). For each data set, 10,000–13,000 individual single-particle images were collected and subjected to single-particle image-processing using the software package IMAGIC-5 (van Heel et al. 1996). The image-processing encompassed a reference-free alignment, after which the images were subjected to multivariate statistical analysis and classification (van Heel and Frank 1981; Dube et al. 1993; van Heel 1984). The resulting class averages were used as reference images in subsequent rounds of alignment until the class averages were stable.
UV crosslinking and Western blotting
5′ss RNA oligonucleotides containing either 6-thioguanosine (position −1 or +1 relative to the 5′ss) or 4-thiouridine (position +2) were radioactively labeled at their 5′-end using [γ-32P]-ATP and T4 polynucleotide kinase. Spliceosomal complexes were allowed to form after addition of radiolabeled 5′ss oligo and then affinity-purified. Samples were irradiated at 365 nm for 10 min and recovered protein (±RNase A digestion) was analyzed by SDS-PAGE and visualized by silver staining or by autoradiography. For immunoprecipitation of hPrp8-RNA crosslinks, UV-crosslinked complexes were disrupted by RNase digestion and incubated with 0.05% SDS and 0.5% Triton X-100 for 15 min at 37°C. The mixture was added to anti-hPrp8-bound PAS beads (preblocked with PBS containing 0.5 mg/mL BSA and 0.05 mg/mL tRNA) and incubated head-over-tail at RT for 2 h. After washing with G-150 buffer containing 0.05 % SDS and 0.5 % Triton X-100, the bound material was eluted by addition of SDS loading dye and incubated at 96°C for 10 min, separated by SDS-PAGE and analyzed by Western blot or autoradiography. For Western blotting, proteins separated by SDS-PAGE were transferred to a nitrocellulose membrane and incubated with antibodies against hPrp8 (Lauber et al. 1996), hSnu66 (Makarova et al. 2001), or phosphorylated hPrp31 (Schneider et al. 2010a). Proteins were detected using an ECL detection kit (GE Healthcare).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to T. Conrad for cultivation of HeLa cells and P. Dube for help in electron microscopy studies. We thank U. Plessman, M. Raabe, H. Kohansal, and G. Heyne for excellent technical assistance. This work was supported by a grant (LU 294/15-1) from the Deutsche Forschungsgemeinschaft (DFG) to R.L.
Author contributions: C.B., N.R., D.E.A., and B.K. performed the experiments. C.B., C.L.W., and R.L. wrote the paper. All authors were involved in the planning of experiments and the interpretation of their results.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.053991.115.
REFERENCES
- Agafonov DE, Deckert J, Wolf E, Odenwälder P, Bessonov S, Will CL, Urlaub H, Lührmann R. 2011. Semiquantitative proteomic analysis of the human spliceosome via a novel two-dimensional gel electrophoresis method. Mol Cell Biol 31: 2667–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berget SM. 1995. Exon recognition in vertebrate splicing. J Biol Chem 270: 2411–2414. [DOI] [PubMed] [Google Scholar]
- Bessonov S, Anokhina M, Krasauskas A, Golas MM, Sander B, Will CL, Urlaub H, Stark H, Lührmann R. 2010. Characterization of purified human Bact spliceosomal complexes reveals compositional and morphological changes during spliceosome activation and first step catalysis. RNA 16: 2384–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehringer D, Makarov EM, Sander B, Makarova OV, Kastner B, Lührmann R, Stark H. 2004. Three-dimensional structure of a pre-catalytic human spliceosomal complex B. Nat Struct Mol Biol 11: 463–468. [DOI] [PubMed] [Google Scholar]
- Bonnal S, Martinez C, Forch P, Bachi A, Wilm M, Valcárcel J. 2008. RBM5/Luca-15/H37 regulates Fas alternative splice site pairing after exon definition. Mol Cell 32: 81–95. [DOI] [PubMed] [Google Scholar]
- Das R, Reed R. 1999. Resolution of the mammalian E complex and the ATP-dependent spliceosomal complexes on native agarose mini-gels. RNA 5: 1504–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Conti L, Baralle M, Buratti E. 2013. Exon and intron definition in pre-mRNA splicing. Wiley Interdiscip Rev RNA 4: 49–60. [DOI] [PubMed] [Google Scholar]
- Dube P, Tavares P, Lurz R, van Heel M. 1993. The portal protein of bacteriophage SPP1: a DNA pump with 13-fold symmetry. EMBO J 12: 1303–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox-Walsh KL, Dou Y, Lam BJ, Hung SP, Baldi PF, Hertel KJ. 2005. The architecture of pre-mRNAs affects mechanisms of splice-site pairing. Proc Natl Acad Sci 102: 16176–16181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galej WP, Oubridge C, Newman AJ, Nagai K. 2013. Crystal structure of Prp8 reveals active site cavity of the spliceosome. Nature 493: 638–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golas MM, Sander B, Will CL, Lührmann R, Stark H. 2003. Molecular architecture of the multiprotein splicing factor SF3b. Science 300: 980–984. [DOI] [PubMed] [Google Scholar]
- Gottschalk A, Neubauer G, Banroques J, Mann M, Lührmann R, Fabrizio P. 1999. Identification by mass spectrometry and functional analysis of novel proteins of the yeast [U4/U6.U5] tri-snRNP. EMBO J 18: 4535–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegele A, Kamburov A, Grossmann A, Sourlis C, Wowro S, Weimann M, Will CL, Pena V, Lührmann R, Stelzl U. 2012. Dynamic protein-protein interaction wiring of the human spliceosome. Mol Cell 45: 567–580. [DOI] [PubMed] [Google Scholar]
- Hoffman BE, Grabowski PJ. 1992. U1 snRNP targets an essential splicing factor, U2AF65, to the 3′ splice site by a network of interactions spanning the exon. Genes Dev 6: 2554–2568. [DOI] [PubMed] [Google Scholar]
- House AE, Lynch KW. 2006. An exonic splicing silencer represses spliceosome assembly after ATP-dependent exon recognition. Nat Struct Mol Biol 13: 937–944. [DOI] [PubMed] [Google Scholar]
- Ismaili N, Sha M, Gustafson EH, Konarska MM. 2001. The 100-kDa U5 snRNP protein (hPrp28p) contacts the 5′ splice site through its ATPase site. RNA 7: 182–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastner B, Fischer N, Golas MM, Sander B, Dube P, Boehringer D, Hartmuth K, Deckert J, Hauer F, Wolf E, et al. 2008. GraFix: sample preparation for single-particle electron cryomicroscopy. Nat Methods 5: 53–55. [DOI] [PubMed] [Google Scholar]
- Konforti BB, Konarska MM. 1994. U4/U5/U6 snRNP recognizes the 5′ splice site in the absence of U2 snRNP. Genes Dev 8: 1962–1973. [DOI] [PubMed] [Google Scholar]
- Lauber J, Fabrizio P, Teigelkamp S, Lane WS, Hartmann E, Lührmann R. 1996. The HeLa 200 kDa U5 snRNP-specific protein and its homologue in Saccharomyces cerevisiae are members of the DEXH-box protein family of putative RNA helicases. EMBO J 15: 4001–4015. [PMC free article] [PubMed] [Google Scholar]
- Makarova OV, Makarov EM, Lührmann R. 2001. The 65 and 110 kDa SR-related proteins of the U4/U6.U5 tri-snRNP are essential for the assembly of mature spliceosomes. EMBO J 20: 2553–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Hartmuth K, Mohlmann S, Urlaub H, Ficner R, Lührmann R. 2008. Phosphorylation of human PRP28 by SRPK2 is required for integration of the U4/U6-U5 tri-snRNP into the spliceosome. Nat Struct Mol Biol 15: 435–443. [DOI] [PubMed] [Google Scholar]
- Meister G, Hannus S, Plöttner O, Baars T, Hartmann E, Fakan S, Laggerbauer B, Fischer U. 2001. SMNrp is an essential pre-mRNA splicing factor required for the formation of the mature spliceosome. EMBO J 20: 2304–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AJ, Norman C. 1992. U5 snRNA interacts with exon sequences at 5′ and 3′ splice sites. Cell 68: 743–754. [DOI] [PubMed] [Google Scholar]
- Rappsilber J, Ajuh P, Lamond AI, Mann M. 2001. SPF30 is an essential human splicing factor required for assembly of the U4/U5/U6 tri-small nuclear ribonucleoprotein into the spliceosome. J Biol Chem 276: 31142–31150. [DOI] [PubMed] [Google Scholar]
- Reyes JL, Kois P, Konforti BB, Konarska MM. 1996. The canonical GU dinucleotide at the 5′ splice site is recognized by p220 of the U5 snRNP within the spliceosome. RNA 2: 213–225. [PMC free article] [PubMed] [Google Scholar]
- Reyes JL, Gustafson EH, Luo HR, Moore MJ, Konarska MM. 1999. The C-terminal region of hPrp8 interacts with the conserved GU dinucleotide at the 5′ splice site. RNA 5: 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robberson BL, Cote GJ, Berget SM. 1990. Exon definition may facilitate splice site selection in RNAs with multiple exons. Mol Cell Biol 10: 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscigno RF, Garcia-Blanco MA. 1995. SR proteins escort the U4/U6.U5 tri-snRNP to the spliceosome. RNA 1: 692–706. [PMC free article] [PubMed] [Google Scholar]
- Sakharkar MK, Perumal BS, Sakharkar KR, Kangueane P. 2005. An analysis on gene architecture in human and mouse genomes. In Silico Biol 5: 347–365. [PubMed] [Google Scholar]
- Schellenberg MJ, Wu T, Ritchie DB, Fica S, Staley JP, Atta KA, LaPointe P, MacMillan AM. 2013. A conformational switch in PRP8 mediates metal ion coordination that promotes pre-mRNA exon ligation. Nat Struct Mol Biol 20: 728–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M, Hsiao HH, Will CL, Giet R, Urlaub H, Lührmann R. 2010a. Human PRP4 kinase is required for stable tri-snRNP association during spliceosomal B complex formation. Nat Struct Mol Biol 17: 216–221. [DOI] [PubMed] [Google Scholar]
- Schneider M, Will CL, Anokhina M, Tazi J, Urlaub H, Lührmann R. 2010b. Exon definition complexes contain the tri-snRNP and can be directly converted into B-like precatalytic splicing complexes. Mol Cell 38: 223–235. [DOI] [PubMed] [Google Scholar]
- Sha M, Levy T, Kois P, Konarska MM. 1998. Probing of the spliceosome with site-specifically derivatized 5′ splice site RNA oligonucleotides. RNA 4: 1069–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Falick AM, Black DL. 2005. Polypyrimidine tract binding protein blocks the 5′ splice site-dependent assembly of U2AF and the prespliceosomal E complex. Mol Cell 19: 485–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontheimer EJ, Steitz JA. 1993. The U5 and U6 small nuclear RNAs as active site components of the spliceosome. Science 262: 1989–1996. [DOI] [PubMed] [Google Scholar]
- Staley JP, Guthrie C. 1999. An RNA switch at the 5′ splice site requires ATP and the DEAD box protein Prp28p. Mol Cell 3: 55–64. [DOI] [PubMed] [Google Scholar]
- van Heel M. 1984. Multivariate statistical classification of noisy images (randomly oriented biological macromolecules). Ultramicroscopy 13: 165–183. [DOI] [PubMed] [Google Scholar]
- van Heel M, Frank J. 1981. Use of multivariate statistics in analysing the images of biological macromolecules. Ultramicroscopy 6: 187–194. [DOI] [PubMed] [Google Scholar]
- van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M. 1996. A new generation of the IMAGIC image processing system. J Struct Biol 116: 17–24. [DOI] [PubMed] [Google Scholar]
- Will CL, Lührmann R. 2011. Spliceosome structure and function. Cold Spring Harb Perspect Biol 3: a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf E, Kastner B, Deckert J, Merz C, Stark H, Lührmann R. 2009. Exon, intron and splice site locations in the spliceosomal B complex. EMBO J 28: 2283–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt JR, Sontheimer EJ, Steitz JA. 1992. Site-specific cross-linking of mammalian U5 snRNP to the 5′ splice site before the first step of pre-mRNA splicing. Genes Dev 6: 2542–2553. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.