

Abstract
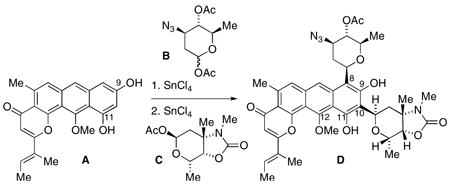
In explorations towards the total synthesis of the antitumor anthrapyran natural product kidamycin, the regioselective introduction of aminosugars angolosamine and vancosamine as C-arylglycosides has been accomplished onto hydroxylated anthrapyran aglycones. Specifically, the 9,11-dihydroxylated anthrapyran A undergoes sequential glycosylations with angolosamine synthon B and vancosamine synthon C to regio- and stereoselectively afford bis-C-glycoside D corresponding to the C-glycoside pattern of kidamycin.
The pluramycins are a relatively large family of antitumor antibiotic natural products containing the 4H-anthra[1,2-b]pyran-4,7,12-trione substructure, with variations in the side chain and substitution patterns of carbohydrates on the anthrapyran core aglycone.1 Kidamycin was isolated from Streptomyces soil bacteria in the early 1970s, as one of the earliest known members of the pluramycin antibiotics.2 Kidamycin demonstrates a wide spectrum of antimicrobial activity against anaerobic bacteria, aerobic and facultative bacteria and yeasts,3 and also has antitumor and cytotoxic properties against leukemia L-1210 as well as life-prolongation activity on mice bearing ascites tumors at single i.p. doses from just below the LD50 (18 mg/kg) to 1/16 of the LD50.4 The structure and conformation of kidamycin (1, Figure 1) was determined by NMR and X-ray crystallographic studies, exhibiting a novel meta-arrangement of two different C-arylglycosidic aminosugars, with equatorially- substituted angolosamine at C8 and axially-substituted vancosamine at C10.5 In the course of characterization studies, the acid-sensitivity of the axial C-glycoside to anomerization was discovered to favor equatorially- substituted vancosamine in isokidamycin (2), which is attributed to occur by protonation of the pyranose oxygen and elimination to a quinomethide intermediate, followed by reclosure of the pyran ring.5a Despite the interesting biological activity of kidamycin, this structurally complex compound has to date resisted total synthesis. Two syntheses of the kidamycin aglycone have been recorded,6,7 as well as the development of several methods for the regioselective construction of meta-bis-C-arylglycosides.8 In this report we will disclose the first cases in which the angolosamine and vancosamine sugars have been stereo- and regioselectively introduced onto an aglycone bearing most of the features of the natural product kidamycin. Our synthetic strategy features late-stage sequential C-glycosylation at C8 of kidamycin aglycone or related anthrapyran synthon with suitably protected angolosamine synthon, and at C10 with vancosamine synthon.
Figure 1. Structures of kidamycin and isokidamycin.

Both carbohydrate components leading to angolosamine and vancosamine were prepared utilizing tungsten-catalyzed alkynol cycloisomerization to form pyranosyl glycal intermediates. For D-angolosamine synthon 6 (Scheme 1), cycloisomerization9 of alkynyl alcohol 3 provided glycal 4.10 Removal of the silyl ether protective group allowed direct SN2 substitution11 to introduce an azide group with inversion of stereochemistry to produce 5, with non-basic nitrogen in the azide functional group.12 Treatment of this glycal 5 with hot aqueous acid13 effected one-pot hydrolysis of vinyl ether and cleavage of MOM ether to produce the diol, which was acylated to generate 6 as the angolosamine glycosyl donor. Likewise, the synthesis of L-vancosamine synthon 10 began with alkynol cycloisomerization of 7 to 8.10,14 N-Methylation of 8 was complicated by formation of bicyclic carbamate 9,15 which could be prevented by using exactly one equivalent of base. However, we recognized the advantages of the cyclic carbamate in avoiding carbonyl-group participation of the acyclic Cbz group in glycosylations as well as the convex nature of compound 9 in stereoselective glycosylation, and were able to optimize preparation of 9 by using excess base until complete conversion to the cyclic carbamate had occurred, followed by addition of iodomethane. Further derivatization of glycal 9 to glycosyl acetate 1016 broadened the choice of conditions for glycosylations.
Scheme 1. Syntheses of glycosyl donors 6 and 10 via alkynol cycloisomerization.

In 2005 we reported the synthesis of kidamycin aglycone via the advanced intermediate 11.7 Friedel-Crafts-type glycosylation17 of 11 with angolosamine synthon 5 stereo-and regioselectively provided C8-glycoside 13, albeit with low conversion (Scheme 2). Similar results were observed with the C12-thioethyl analog 12. The regioselectivity of these glycosylations was assigned by the disappearance of the C8 hydrogen resonance in comparing 1H NMR spectra for compounds 11 -14 and confirmed by the observation of a nuclear Overhauser effect between H7 and the anomeric hydrogen H1′ in compound 14.18 This glycosylation was not further optimized after we found that the O-methyl groups could not be removed from the aglycone nucleus in the presence of the fragile benzylic carbon-oxygen bond of the angolosamine carbohydrate, but we did demonstrate reduction of the azide of 13 to amine which was characterized as peracetylated 15.
Scheme 2. Glycosylations of anthrapyrans 11, 12, and 16.

Introduction of vancosamine was also explored with several substrates, with C11 phenol 16 as a representative example (Scheme 2).19,20 Initial results in SnCl4- promoted glycosylation with vancosamine synthon 10 provided a mixture of C10-glycoside anomers 17 and 18,18 with the formation of equatorially-substituted 18 attributed to Lewis acid-catalyzed anomerization in a mechanism analogous to that proposed for the isomerization of kidamycin (1) to isokidamycin (2). The positional selectivity for this glycosylation at C10 was evident by the disappearance of the C10 hydrogen resonance in the formation of compounds 17 and 18, and the anomeric stereochemistry was assigned in analogy with kidamycin (1) and isokidamycin (2), with the anomeric 1H resonance of δ 5.48 for 17 consistent with a pseudoequatorial hydrogen, relative to the shielded pseudoaxial anomeric hydrogen of 18 at δ 5.09. Lowering the reaction temperature increased stereoselectivity as well as the isolated yield for production of 17,21 but under these conditions we also observed formation of the para-C8-glycoside isomer 19, characterized by the absence of the C8 hydrogen and presence of C10 hydrogen in comparison with compounds 16-18.18
At this stage, we modified our aglycone synthesis to introduce an additional hydroxyl substituent at C9, which might be advantageous in more easily forming C-glycosides at C8 as well as C10. Ortho-lithiation22 of amide 21 and addition to the aldehyde 22, followed by desilylation provided the lactone 23. The phenol of 23 underwent addition-elimination23 with beta-chlorodienoate (24)7,24 to form compound 25, although forcing conditions were required as phenolate reactivity was diminished due to conjugation with the ester carbonyl. Hydrogenolysis of the doubly benzylic C7 lactone18 also resulted in removal of the isopropyl ethers, but global O-benzylation of both phenols and the carboxylic acid provided 26, which was easily purified. After saponification of both esters to dicarboxylic acid 27, double Friedel-Crafts cyclization25 provided the anthrapyran 28, which was converted into a variety of O-protected congeners 29 - 31 (Scheme 3).
Scheme 3. Synthesis of anthrapyrans bearing 9-hydroxyl substituent.

Glycosylation was first conducted with the bromoethyl ether-protected 30 bearing a single phenol at C9, which was anticipated to direct ortho-C-glycosylation26 at both C8 and C10. Introduction of angolosamine synthon 6 proceeded in good yield and with apparent complete stereo- and regioselectivity for C8-glycosylation17,18 in product 32 (Scheme 4). However, the attempted introduction of vancosamine via the C9-hydroxyl group instead provided the surprising result of C7-glycosylation to provide the anthrone 33, which was characterized by observation of 1H-1H coupling between the C7 hydrogen and the anomeric hydrogen of the vancosamine unit,18 as well as the white color of product 33, consistent with interruption of conjugation in the C ring at C7. Thus the aglycone substrate 31 without protective groups at either C9 or C11 phenols was subjected to glycosylation with angolosamine donor 6, providing C8-glycoside 34 in good yield, which in turn underwent regio- and stereoselective glycosylation with vancosamine synthon 10 to provide the C10-glycoside 35,18 possessing a meta-bisglycosylated anthrapyran with considerable structural similarity to kidamycin (1).
Scheme 4. Bis-glycosylations of 9-hydroxyl-substituted anthrapyrans 30 and 31.

Although we have not yet completed the synthesis of kidamycin, we have demonstrated a number of interesting and informative glycosylation transformations which have culminated in the successful preparation of the bis-C-arylglycoside 35 closely corresponding to most of the structural features of kidamycin. Continuing efforts will focus on completing the total synthesis from either the doubly glycosylated intermediate 35 or, less aggressively, by removing the C9 oxygen from intermediate 34 prior to the second glycosylation to introduce the C-vancosamine glycoside.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (R01 CA59703). We also acknowledge use of shared instrumentation provided by grants from the National Institutes of Health, National Science Foundation, and the Georgia Research Alliance (NMR spectroscopy, mass spectrometry), as well as the University Research Committee of Emory University (polarimeter).
Footnotes
Supporting Information Available: Experimental procedures and characterization data for new compounds. This material is available free of charge via the Internets at http://pubs.acs.org.
References
- 1.(a) Berdy J. Adv Appl Microbiol. 1974;18:309. [PubMed] [Google Scholar]; (b) Séquin U. Prog Chem Org Nat Prod. 1986;50:57. [Google Scholar]; (c) Rohr J, Thiericke R. Nat Prod Rep. 1992:103. doi: 10.1039/np9920900103. [DOI] [PubMed] [Google Scholar]; (d) Hansen MR, Hurley LH. Acc Chem Res. 1996;29:249. [Google Scholar]
- 2.Kanda N. J Antibiot. 1971;24:599. doi: 10.7164/antibiotics.24.599. [DOI] [PubMed] [Google Scholar]
- 3.Kanda N. J Antibiot. 1972;25:557. doi: 10.7164/antibiotics.25.557. [DOI] [PubMed] [Google Scholar]
- 4.Kanda N, Kono M, Asano K. J Antibiot. 1972;25:553. doi: 10.7164/antibiotics.25.553. [DOI] [PubMed] [Google Scholar]
- 5.(a) Furukawa M, Iitaka Y. Tetrahedron Lett. 1974;15:3287. [Google Scholar]; (b) Furukawa M, Hayakawa I, Ohta G, Iitaka Y. Tetrahedron. 1975;31:2989. [Google Scholar]
- 6.Hauser FM, Rhee RP. J Org Chem. 1980;45:3061. [Google Scholar]
- 7.Fei Z, McDonald FE. Org Lett. 2005;7:3617. doi: 10.1021/ol0509742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Dubois E, Beau JM. Carbohydr Res. 1992;228:103. doi: 10.1016/s0008-6215(00)90552-4. [DOI] [PubMed] [Google Scholar]; (b) Parker KA, Koh Yh. J Am Chem Soc. 1994;116:11149. [Google Scholar]; (c) Kaelin DE, Lopez OD, Martin SF. J Am Chem Soc. 2001;123:6937. doi: 10.1021/ja0108640. [DOI] [PubMed] [Google Scholar]; (d) Kaelin DE, Sparks SM, Plake HR, Martin SF. J Am Chem Soc. 2003;125:12994. doi: 10.1021/ja0375582. [DOI] [PubMed] [Google Scholar]; (e) Yamauchi T, Watanabe Y, Suzuki K, Matsumoto T. Synthesis. 2006:2818. [Google Scholar]
- 9.(a) McDonald FE, Reddy KS, Díaz Y. J Am Chem Soc. 2000;122:4304. [Google Scholar]; (b) Koo B, McDonald FE. Org Lett. 2007;9:1737. doi: 10.1021/ol070435s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.See Supporting Information for details on the preparation of alkynyl alcohols 3 and 7.
- 11.Thompson AS, Humphrey GR, DeMarco AM, Mathre DJ, Grabowski EJJ. J Org Chem. 1993;58:5886. [Google Scholar]
- 12.Parker KA, Ding Qj. Tetrahedron. 2000;56:10255. [Google Scholar]
- 13.Meyers AI, Durandetta JL, Munavu R. J Org Chem. 1975;40:2025. [Google Scholar]
- 14.Cutchins WW, McDonald FE. Org Lett. 2002;4:749. doi: 10.1021/ol017195f. [DOI] [PubMed] [Google Scholar]
- 15.Parker KA, Chang W. Org Lett. 2003;5:3891. doi: 10.1021/ol035479p. [DOI] [PubMed] [Google Scholar]
- 16.Lam SN, Gervay-Hague J. Org Lett. 2003;5:4219. doi: 10.1021/ol035705v. [DOI] [PubMed] [Google Scholar]
- 17.(a) Matsumoto T, Katsuki M, Suzuki K. Tetrahedron Lett. 1989;30:833. [Google Scholar]; (b) Kuribayashi T, Ohkawa N, Satoh S. Tetrahedron Letters. 1998;39:4537. doi: 10.1016/s0960-894x(98)00607-6. [DOI] [PubMed] [Google Scholar]; (c) Kuribayashi T, Ohkawa N, Satoh S. Tetrahedron Lett. 1998;39:4541. doi: 10.1016/s0960-894x(98)00607-6. [DOI] [PubMed] [Google Scholar]; (d) Shuto S, Horne G, Marwood RD, Potter BVL. Chem Eur J. 2001;7:4937. doi: 10.1002/1521-3765(20011119)7:22<4937::aid-chem4937>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 18.See Supporting Information for more details on the positional and stereochemical assignments for C-glycosylation products.
- 19.Compound 12_arose from an unsuccessful attempt to remove methyl ethers from 11 with AlCl3 / EtSH (ref. 20).
- 20.Node M, Nishide K, Fuji K, Fujita E. J Org Chem. 1980;45:4275. [Google Scholar]
- 21.The C-vancosamine glycosylation product 17 undergoes partial anomerization to a mixture of 17 and 18 upon the prolonged reaction time or higher temperature required for complete conversion of starting materials.
- 22.Falk H, Mayr E. Monatsh Chem. 1995;126:699. [Google Scholar]
- 23.(a) Hormi OEO. J Org Chem. 1988;53:880. [Google Scholar]; (b) Hormi OEO, Hirvela L. Tetrahedron Lett. 1993;34:6463. [Google Scholar]
- 24.Andrews JFP, Regan AC. Tetrahedron Lett. 1991;32:7731. [Google Scholar]
- 25.(a) Bycroft BW, Roberts JCJ. Chem Soc. 1963:4868. [Google Scholar]; (b) Devos A, Frisque-Hesbain AM, Colens A, Ghosez L. J Chem Soc, chem Commun. 1979:1180. [Google Scholar]
- 26.(a) Matsumoto T, Maeta H, Suzuki K, Tsuchihashi Gi. Tetrahedron Lett. 1988;29:3567. [Google Scholar]; (b) Matsumoto T, Katsuki M, Suzuki K. Tetrahedron Lett. 1988;29:6935. [Google Scholar]; (c) Matsumoto T, Hosoya T, Suzuki K. Tetrahedron Lett. 1990;31:4629. [Google Scholar]; (d) Ben A, Yamauchi T, Matsumoto T, Suzuki K. Synlett. 2004:225. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.