

Abstract
Background:
Neurofibromatosis type 2 (NF2) is an autosomal dominant syndrome with a frequency of 1 in 25,000 live births and a penetrance of almost 100% by the sixth decade of life. The main tumors occurring in NF2 patients are bilateral vestibular schwannomas, other peripheral, cranial and spinal nerve schwannomas, intracranial and intraspinal meningiomas, ependymomas, and gliomas.
Case Description:
We report the case of a 6-year-old boy who presented with a 1-month history of nausea and recurrent vomiting. Physical examination was positive for ataxic gait and left-sided facial nerve palsy. Family history was positive for NF2 in the patient's father and paternal uncle. Magnetic resonance imaging brain revealed a solid enhancing lesion arising from the right cerebellar cortex, which was effacing the fourth ventricles and causing hydrocephalus. Craniotomy and excision of the lesion were performed. Histopathology report confirmed the diagnosis to be desmoplastic medulloblastoma. Based on the patients’ subsequent history and family history, he was diagnosed to be a case of NF2.
Conclusion:
This is the first case of medulloblastoma occurring in a patient with NF2 and raises the possibility of an association between medulloblastoma and NF2.
Keywords: Association, brain neoplasm, medulloblastoma, neurofibromatosis type 2
BACKGROUND
Neurofibromatosis type 2 (NF2) is an autosomal dominant syndrome with a frequency of 1 in 25,000 live births and a penetrance of almost 100% by the sixth decade of life.[2] It has a variable presentation, resulting from the mutation in the tumor suppressor gene merlin, which is located on chromosome 22q, and is a predominantly intracranial condition with its characteristic bilateral vestibular schwannomas.[15] The other main tumors occurring in NF2 patients are other peripheral, cranial and spinal nerve schwannomas, intracranial and intraspinal meningiomas, ependymomas and gliomas.[6] Four large clinical studies have been conducted which confirm the aforementioned clinical picture.[4,10,11,12] These tumors are mostly benign and grow slowly, but their location within the central nervous system can cause great morbidity and mortality.[9]
Clinical presentation of most patients with NF2 includes hearing loss which is usually unilateral, with or without tinnitus.[6] NF2 is diagnosed clinically after a patient fulfills a predefined criteria.[5]
Herein, we report a case NF2 presenting with recurrent vomiting and headache that was diagnosed with medulloblastoma. This, to the best of our knowledge, is the first case of medulloblastoma occurring in an NF2 patient.
CASE REPORT
A 6-year-old boy of average height and weight presented to the ER in 2003 with complaints of repeated vomiting and headache for 1-month. He was a student of a local public school, his past medical history being unremarkable; his immunization status was complete, and there were no known allergies. In family history, the patient's father and uncle (father's brother) were known to have NF2. On physical examination, he was vitally stable. Positive findings included left-sided facial nerve palsy and ataxic gait.
After a series of initial investigations, magnetic resonance imaging (MRI) brain was done. The report described a solid mass arising from the right cerebellar cortex, which was isointense to the gray matter on T1-weighted images and hypo to isointense to the gray matter on T2-weighted images. Postcontrast images showed an intense enhancement, which was almost homogenous. The mass was causing effacement of the fourth ventricles with dilatation of the third and lateral ventricles. The vertical height of the lesion was 4.2 cm; AP dimension was 4 cm, and the transverse diameter was 4.8 cm. There was no evidence of intracranial hemorrhage. Gray and white matter signals of supratentorial brain were within normal limits. No midline structural defect was seen. The differential diagnoses of medulloblastoma or astrocytoma were made.
The patient underwent craniotomy and excision of the lesion with the insertion of a ventriculoperitoneal shunt. The histopathology report described a malignant infiltrating tumor present in sheets [Figure 1]. The tumor was composed of small oval to indented basophilic cells, exhibiting nuclear hyperchromasia [Figures 1 and 2]. Extensive necrosis [Figure 3] and karyorrhexis were identified with areas of hemorrhage. Brisk mitotic figures were also seen [Figure 4]. The tumor cells showed positivity for immunohistochemical stain CD 56 [Figure 5]. Focal positivity for immunohistochemical stain glial fibrillary acidic protein was also seen [Figure 6]. Based on these findings a final diagnosis of desmoplastic medulloblastoma was made.
Figure 1.
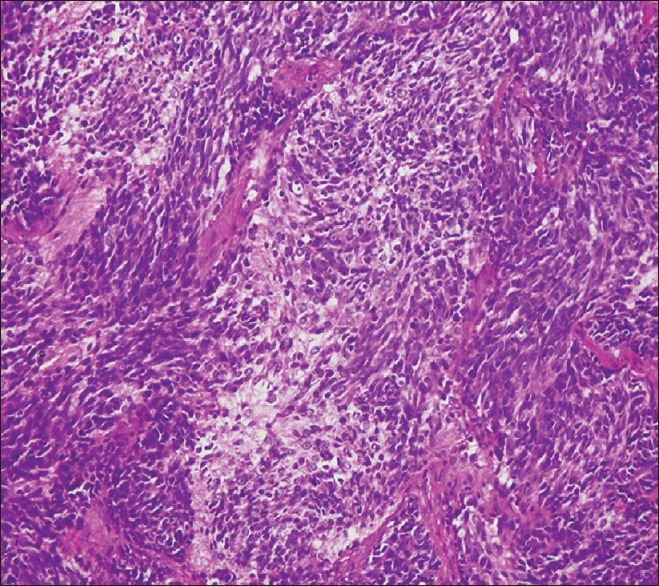
Patternless sheets of primitive appearing neoplastic cells with hyperchromatic nuclei with neuropil
Figure 2.
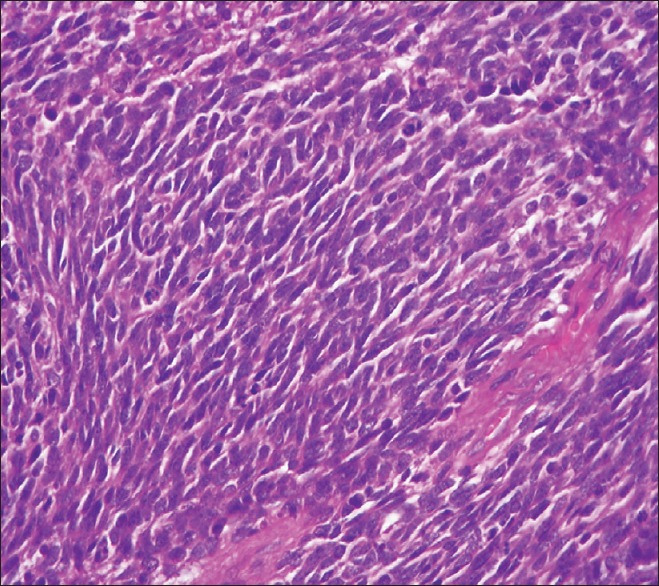
Primitive appearing neoplastic cells with hyperchromatic nuclei, scant cytoplasm, and indistinct cell borders
Figure 3.
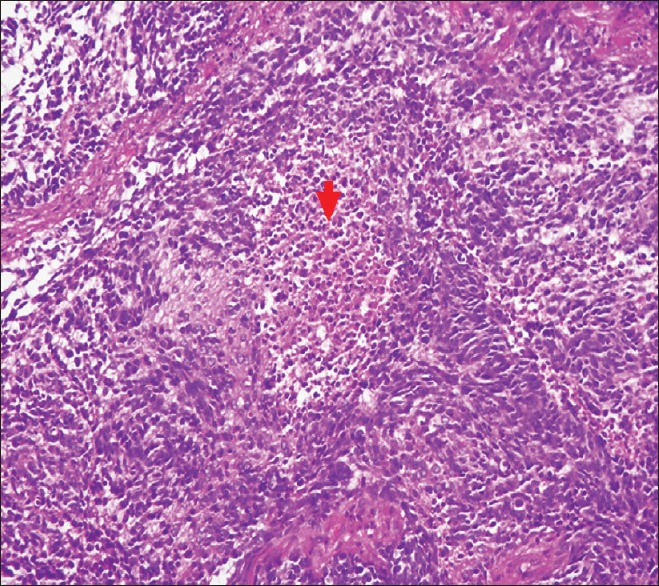
Tumor with areas of necrosis as indicated by the arrow (H and E, ×20)
Figure 4.
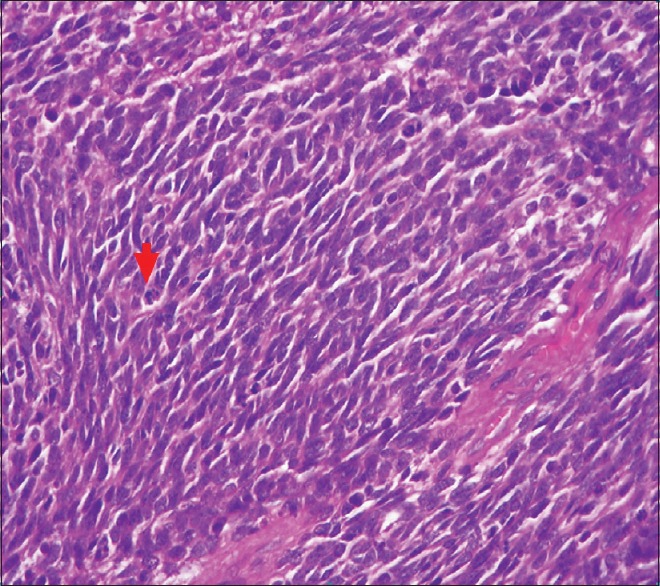
Neoplastic cells showing prominent mitotic figures as pointed out by the arrow (H and E, ×40)
Figure 5.
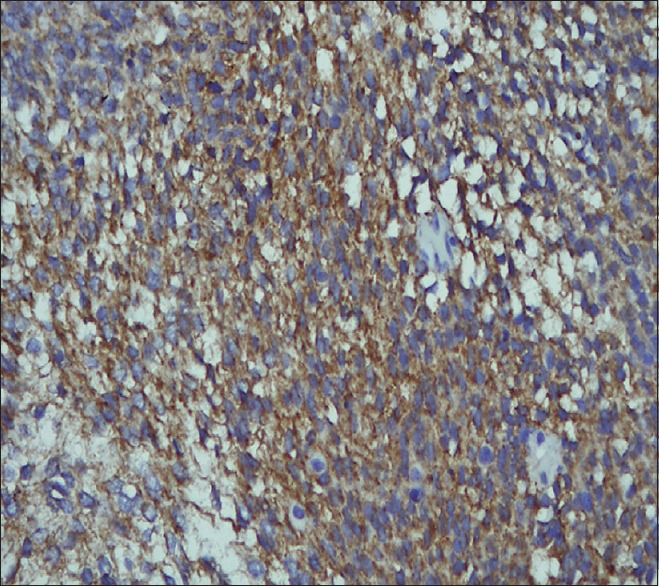
Tumor cells showing positivity for immunohistochemical stain CD56
Figure 6.
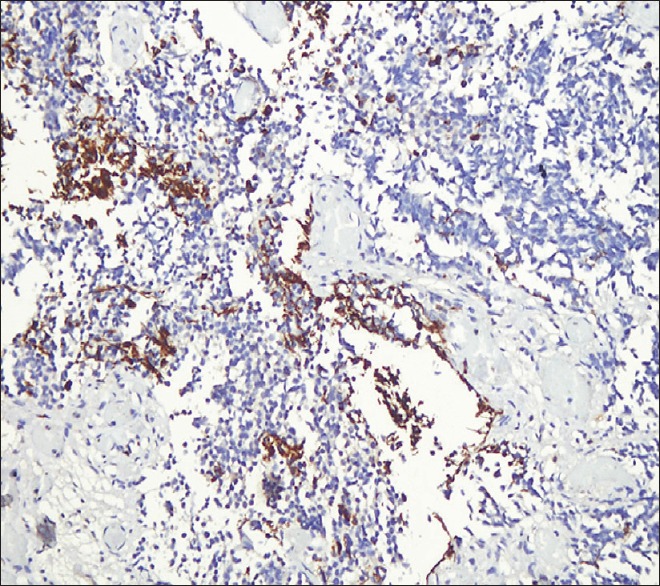
Tumor cells showing focal positivity for immunohistochemical stain glial fibrillary acidic protein
The patient underwent metastatic workup which was negative. After discharge, he was kept under close follow-up. Pediatric oncology team was taken on board and after discussing the case at the tumor board meeting, a multidisciplinary approach was taken and both chemotherapy and radiotherapy were administered.
In 2003, he was found to have a 1.0 cm × 1.5 cm skin colored nodule on this left forearm, which was excised with histopathology of the lesion revealing benign peripheral nerve sheath tumor.
In 2004, during a routine MRI scan the patient was found to have a cerebellar lesion for which he underwent craniotomy and excision of the lesion. Biopsy of the excised specimen revealed postchemotherapy and radiotherapy gliosis. He was then again kept under close follow-up. He complained of vision disturbances in 2006 and after an ophthalmological exam, was diagnosed to have right eye cataract for which he underwent phacoemulsification and insertion of the intraocular lens.
Till 2010, his routine follow-up MRI scans showed no significant pathological changes. However, in 2010, he again started complaining of vision disturbances and ophthalmological examination revealed right-sided posterior capsular opacification and left-sided cataract. He underwent left capsulectomy.
Over the span of next 3 years the patient remained well however, in 2013, he was again brought to Aga Khan University Hospital with complaints of headache, vomiting, and gait disturbances. MRI brain showed right superior frontal, inferior frontal, parasagittal and left posterior parietal meningiomas; excision of the lesions was performed.
Considering his case history and his family history, a diagnosis of NF2 was considered for him. A neurologist was taken on board; the NIH criterion was applied, and the patient was diagnosed to have NF2.
After the surgery in 2013, the patient has not been reported any symptoms. The follow-up MRI scans have not revealed any positive findings.
DISCUSSION
NF is a genetic disorder of the nervous system. It became widely recognized in the 19th century,[1] but it has a wide pictorial history that traces back to the 13th century.[3] The neuromas of NF were first described, in 1849.[3] Von Recklinghausen's is credited with the discovery of NF and coined the name of this disorder, in 1882.[3] Research on NF increased between 1909 and 1990 after Joseph Merick, the famous Elephant Man, was erroneously diagnosed with NF1.[3]
NF is considered to have two distinct types, NF1 and NF2.[14] NF1 has more peripheral manifestations and NF2 carries predominantly central manifestations. Over the past two decades, our knowledge of the genetics and management of NF has dramatically increased. The two forms of NF have been shown to be two distinct entities, both at clinical and molecular levels.[14] NF also has other rarer forms with outlying phenotypes and atypical presentations. These unconventional and atypical forms have been delineated and include hereditary spinal NF, schwannomatosis, familial intestinal NF, autosomal dominant “café au lait” spots alone, Watson syndrome, autosomal dominant “neurofibromas” alone, Noonan syndrome and the syndrome of multiple naevi, multiple schannomas, and multiple vaginal leiomyomas.[14]
NF2 is often a devastating autosomal dominant disorder which until recently was confused with its more common namesake NF1.[6] NF2 is a multiple neoplasia syndrome that results from a mutation in NF2 tumor suppressor gene on chromosome 22.[2] Affected patients carry a dominant loss of function mutation of the merlin gene on chromosome 22. Merlin is a cytoskeletal protein that functions as a tumor suppressor by facilitating E-cadherin-mediated contact inhibition.
NF2 inevitably develops schwannomas, typically affecting both the vestibular nerves, resulting in hearing loss and deafness. Most of the patients present with hearing loss which is unilateral at onset and may be accompanied or preceded by tinnitus.[7] Vestibular schannomas may also cause dizziness or imbalance as first symptoms. Nausea, vomiting or true vertigo are rare symptoms, occurring more commonly in the later stages.[7] The other tumors seen in NF2 are schwannomas of other cranial, spinal, and peripheral nerves. Furthermore, NF2 is associated with intracranial and intraspinal meningiomas and low-grade central nervous system malignancies like ependymomas.[7] As mentioned previously, both meningiomas and schwannomas were seen in our patient. Ophthalmic manifestations are also present in NF2 and include reduced juvenile cataracts, as seen in our patient, and retinal hamartomas.[8] About 70% of NF2 cases have skin tumors with intracutaneous plaque like lesions or more deep-seated tumors.[7]
Medulloblastoma is an aggressive posterior fossa brain tumor. Although medulloblastoma has been reported in patients with NF1, one study found the prevalence of posterior fossa tumors in NF1 to be 0.83%, there is no reported case of medulloblastoma occurring in a patient with NF2.[13]
To the best of our knowledge, this is the first case of medulloblastoma in a patient with NF2. The literature review conducted for this report could not find any case report or research study relating medulloblastoma and NF2. This raises the possibility of an association between these two disease entities and shows that even NF2 can also have this posterior fossa brain stem tumor.
Footnotes
Contributor Information
Jan Kalimullah, Email: kohatian3659@yahoo.com.
Abdul Malik Amir Humza Sohail, Email: ameer.hamzasohail@gmail.com.
Rai Dilawar Shahjehan, Email: dilawarmunir@gmail.com.
Sabeehuddin Siddique, Email: sabeeh.siddique@aku.edu.
Muhammad Ehsan Bari, Email: ehsan.bari@aku.edu.
REFERENCES
- 1.Ahn MS, Jackler RK, Lustig LR. The early history of the neurofibromatosis. Evolution of the concept of neurofibromatosis type 2. Arch Otolaryngol Head Neck Surg. 1996;122:1240–9. doi: 10.1001/archotol.1996.01890230086016. [DOI] [PubMed] [Google Scholar]
- 2.Asthagiri AR, Parry DM, Butman JA, Kim HJ, Tsilou ET, Zhuang Z, et al. Neurofibromatosis type 2. Lancet. 2009;373:1974–86. doi: 10.1016/S0140-6736(09)60259-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brosius S. A history of von Recklinghausen's NF1. J Hist Neurosci. 2010;19:333–48. doi: 10.1080/09647041003642885. [DOI] [PubMed] [Google Scholar]
- 4.Evans DG, Huson SM, Donnai D, Neary W, Blair V, Newton V, et al. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84:603–18. [PubMed] [Google Scholar]
- 5.Evans DG, Huson SM, Donnai D, Neary W, Blair V, Newton V, et al. A genetic study of type 2 neurofibromatosis in the United Kingdom. II. Guidelines for genetic counselling. J Med Genet. 1992;29:847–52. doi: 10.1136/jmg.29.12.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evans DG, Sainio M, Baser ME. Neurofibromatosis type 2. J Med Genet. 2000;37:897–904. doi: 10.1136/jmg.37.12.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans DG. Neurofibromatosis type 2 (NF2): A clinical and molecular review. Orphanet J Rare Dis. 2009;4:16. doi: 10.1186/1750-1172-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feucht M, Griffiths B, Niemüller I, Haase W, Richard G, Mautner VF. Neurofibromatosis 2 leads to higher incidence of strabismological and neuro-ophthalmological disorders. Acta Ophthalmol. 2008;86:882–6. doi: 10.1111/j.1600-0420.2007.01088.x. [DOI] [PubMed] [Google Scholar]
- 9.Han F. Type of mutation in the neurofibromatosis type 2 gene (NF2) frequently determines severity of disease. Am J Hum Genet. 1996;59:331–42. [PMC free article] [PubMed] [Google Scholar]
- 10.Kanter WR, Eldridge R, Fabricant R, Allen JC, Koerber T. Central neurofibromatosis with bilateral acoustic neuroma: Genetic, clinical and biochemical distinctions from peripheral neurofibromatosis. Neurology. 1980;30:851–9. doi: 10.1212/wnl.30.8.851. [DOI] [PubMed] [Google Scholar]
- 11.Mautner VF, Lindenau M, Baser ME, Hazim W, Tatagiba M, Haase W, et al. The neuroimaging and clinical spectrum of neurofibromatosis 2. Neurosurgery. 1996;38:880–5. doi: 10.1097/00006123-199605000-00004. [DOI] [PubMed] [Google Scholar]
- 12.Parry DM, Eldridge R, Kaiser-Kupfer MI, Bouzas EA, Pikus A, Patronas N. Neurofibromatosis 2 (NF2): Clinical characteristics of 63 affected individuals and clinical evidence for heterogeneity. Am J Med Genet. 1994;52:450–61. doi: 10.1002/ajmg.1320520411. [DOI] [PubMed] [Google Scholar]
- 13.Pascual-Castroviejo I, Pascual-Pascual SI, Viaño J, Carceller F, Gutierrez-Molina M, Morales C, et al. Posterior fossa tumors in children with neurofibromatosis type 1 (NF1) Childs Nerv Syst. 2010;26:1599–603. doi: 10.1007/s00381-010-1163-5. [DOI] [PubMed] [Google Scholar]
- 14.Ruggieri M. The different forms of neurofibromatosis. Childs Nerv Syst. 1999;15:295–308. doi: 10.1007/s003810050398. [DOI] [PubMed] [Google Scholar]
- 15.Uppal S, Coatesworth AP. Neurofibromatosis type 2. Int J Clin Pract. 2003;57:698–703. [PubMed] [Google Scholar]