

Abstract
Neuromyelitis optica (NMO) is an autoimmune demyelinating condition of the central nervous system often associated with aquaporin-4 (AQP4) autoantibodies manifesting as severe optic neuritis and long segment myelitis with tendency to relapse. Seronegative patients and who do not meet the NMO criteria are classified as having NMO Spectrum Disorder (NMOSD), but are treated identically to clinically definite NMO. Acute relapse is treated with intravenous methylprednisolone for 5 days with or without subsequent treatment with plasma exchange (PE). This must be followed by oral steroid to prevent rebound worsening and further relapse. For relapse prevention, immunosuppressive agents that have been found to be effective are azathioprine, rituximab, mycophenolate mofetil, methotrexate, and mitoxantrone; although none of which have been validated in randomized, controlled trial. Some patients do relapse with monotherapy, and switching to more effective agent or use of combination therapy is beneficial in such situation. There is no consensus about the duration of preventive therapy, but generally 2-3 years of relapse-free period is considered the minimum, taking into account the risks of long-term toxicity of these agents.
Keywords: Aquaporin-4, drug therapy, immunosuppression, neuromyelitis optica
Introduction
Neuromyelitis optica (NMO) is an inflammatory demyelinating disease of the central nervous system, with a predilection for the optic nerves and spinal cord. NMO was initially considered to be a monophasic disease associating paraplegia, due to severe myelitis, and blindness, due to severe optic neuritis. However, recent studies have shown that in more than 80% of cases, NMO is a relapsing disease.[1,2] It is associated with autoantibodies to aquaporin-4 (AQP4) water channels in 50-70% of patients.[3] This signifies NMO is a B-cell-mediated disease, and thus, different from multiple sclerosis (MS). Although it affects all races and ages, NMO is more common than MS in Afro-Caribbean population (NMO:MS ratio is 1:7) of the French West Indies[4] than in the Caucasian population of France (ratio is 1:400).[1] In 2006, revised criteria for NMO were proposed [Table 1] including, in addition to the two major symptoms (myelitis and optic neuritis), any two of the following three criteria: Extended myelitis on spinal cord magnetic resonance imaging (MRI), normal brain MRI at onset, and positive anti-AQP4 antibodies.[5] NMO-IgG seropositive patients with a history of optic neuritis or transverse myelitis who do not meet full clinical criteria are classified as having NMO Spectrum Disorder (NMOSD), but are treated identically to clinically definite NMO.
Table 1.
Revised criteria for NMO (Wingerchuk 2006)

The available data appear to show that NMO is more severe than MS,[1,4,6] for example, about 25-30% die after a mean of 5 years from onset. It also has a high early morbidity as compared to MS because of severely disabling relapses, for example, about half of the patients develop significant walking difficulties at a mean time from onset of 7 years and many patients become dependent on wheelchairs. Similarly, visual impairment is also common, with blindness affecting at least one eye in 60-70%, at a mean time from onset of 5 years. The disabilities in NMO result from accumulating damage during acute attacks, rather than from a supervening progressive course, which is the usual case in MS.[7]
All these suggest that the most effective therapeutic option in NMO is immunosuppressive rather than immunomodulatory drugs[8] and prevention of relapse is a therapeutic priority.
In this article, we are providing a review of existing therapeutic strategies for patients with NMO and for that we would like our readers to go through the case vignette given below.
Case Vignette
A 38-year-old lady presented with acute onset paraplegia with urinary retention and sensory level at D4 dermatome. There was no preceding history of any infection or recent vaccination. She did not have any history of joint pain, skin rash, or any systemic illness in the past. There was no previous history of any neurological disease. Her investigations including complete blood count, erythrocyte sedimentation rate, blood sugar, blood urea, serum creatinine, liver function, urinalysis, and skiagram of chest were normal. The serology for human immunodeficiency virus (HIV), hepatitis B surface antigen (HBsAg), and anti-hepatitis C virus (anti-HCV) antibody were negative. The collagen profile including antinuclear antibody (ANA), anti-double stranded deoxyribonucleic acid (anti-dsDNA), and ANA profile were negative. The visual evoked potential showed prolonged P100 latencies on both sides with normal amplitude. MRI spine showed hyperintense lesion in T2-weighted image, suggesting long segment myelitis extending from upper border of D2 to lower border of D6 vertebrae [Figure 1]. The serum anti-AQ4 antibody was positive and the MRI brain was normal. She was treated with intravenous methylprednisolone 1,000 mg/day for 5 days and improved with this treatment and returned to her normal activities after 2 months of her illness. She was treated with oral prednisolone 1 mg/kg/day. After 3 months, she again had a relapse and became bedbound with retention of urine with long segment myelitis at dorsal level. She received pulse methylprednisolone with improvement of her weakness. Afterwards she was treated with a combination of azathioprine and oral prednisolone. She is relapse-free for last 1 year.
Figure 1.
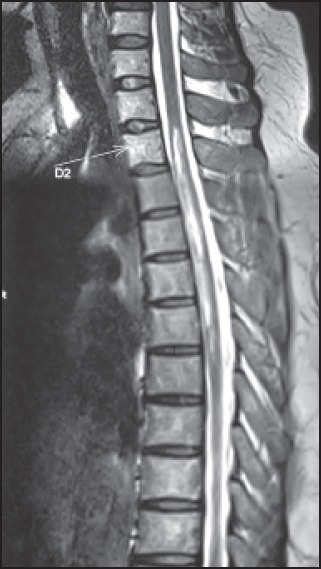
Longitudinally extensive transverse myelitis extending from upper border of D2 to lower border of D6 vertebrae
The aforesaid case depicts a typical NMO and its acute management and maintenance therapy for prevention of future relapse. There are no prospective randomized clinical trials offering class I evidence to guide therapy.[9] Therefore, treatment decisions are largely guided by case series and expert opinions. Treatment for NMO includes management of acute attacks to promote recovery, prevention of NMO exacerbations (by long-term maintenance immunosuppression), prevention and monitoring of adverse effects, and decisions regarding switching therapy due to breakthrough disease or lack of tolerability.
Treatment of Acute NMO Events
In an acute setting, that is, in the initial presentation or during exacerbation of NMO, the primary aim of treatment is to minimize the irreversible damage to the nervous system and to quickly restore neurologic function. To achieve this, it is important to initiate the therapy as early as possible. This requires urgent reporting of relapses by the patient. Each patient needs to be counseled and educated about the disease and its management strategies. The standard treatment for an acute attack of myelitis or optic neuritis is with high dose intravenous methylprednisolone at a daily dose of 1,000 mg for 3-5 days. This gives some recovery to most of the patients. For those who show no response after 5 days therapy or poor and inadequate response after 7-10 days therapy with IV methylprednisolone, plasma exchange (PE) to be started quickly. Typically, five cycles of PE, each removing a total of 1.0-1.5 volumes of circulating plasma are used. It has been seen that marked improvement can occur after several weeks of PE. PE has been demonstrated to be effective in a randomized, double-blind clinical trial in patients with severe demyelinating disease.[10,11,12]
Intravenous immunoglobulin (IVIg) therapy has not been reliably demonstrated to be effective in the acute treatment of NMO exacerbations. In a placebo-controlled, randomized study in severe optic neuritis, Noseworthy et al., (2001) did not demonstrate any positive effect in terms of visual recovery in the IVIg treated group. However, it should be noted that this study evaluated the effect of IVIg in patients with a recent residual visual deficit rather than in patients at the very acute phase.[13] There are, however, several case reports available favoring role of IVIg in prevention of NMO relapse,[14,15] warranting further trial of this agent in acute therapy.
Following a successful treatment of an acute attack, oral steroid needs to be continued for prevention of further relapse. Continuation of oral steroid also prevents the rebound worsening after improvement of acute attack in patients with monophasic illness (seronegative). It is thus advisable to continue oral prednisolone for 2-6 months with gradual tapering.
Preventive Therapy: General Principles
Patients who have the risk of relapse should receive long-term immunosuppression following treatment of an acute attack. AQP4-positive patients and those who fulfill the NMO criteria need long-term therapy. Patients who have had only a single attack of longitudinally extensive transverse myelitis (LETM) or optic neuritis (ON) (bilateral or severe) but who are antibody negative need long-term treatment only if they relapse. The agent for long-term use needs to be chosen carefully considering the effectiveness as well as short- and long-term side effects. Consideration must also be given to age, associated medical conditions, functional status, access to and cost of agent, and response to previous preventive therapies. Immunumodulators used as disease-modifying agent in MS have not been found to be effective in NMO. Several series have reported poor efficacy or harmful effects of these agents, including beta-interferons,[8,16,17] natalizumab,[18,19] and fingolimod.[20]
Immunosuppressive agents have been found effective in multiple studies in NMO are azathioprine, rituximab, mycophenolate mofetil, methotrexate, prednisone, and mitoxantrone [Table 2]. Till date no randomized controlled trials of preventive agents for NMO have been published mostly due to the relative rarity of the disease.
Table 2.
Agents used for NMO treatment
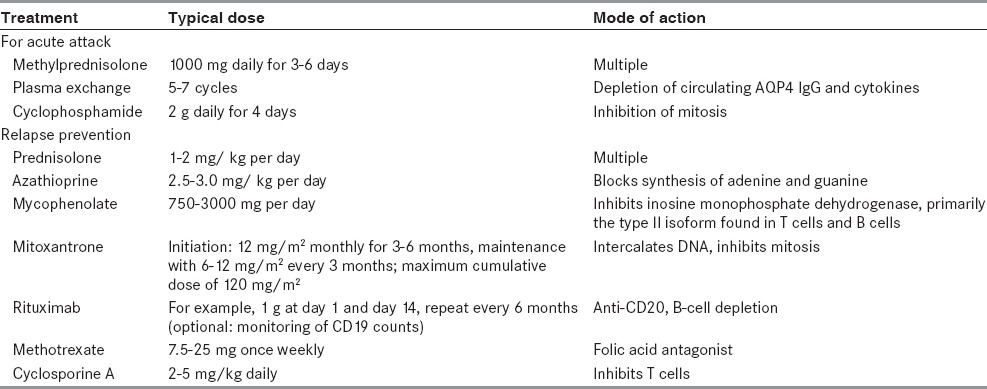
The duration of preventive treatment in NMO has not been adequately studied. This is because our knowledge about the disease is only a decade old and the natural history of NMO is relatively unpredictable. It has been seen that relapses occur in clusters even after long period of remission. Absence of new clinical relapses during a long period of preventive therapy (> 2 years) is generally viewed as probable treatment success. Weinshenker and colleagues suggested that NMO-IgG seropositive patients who present with a first ever attack of LETM should be treated with immunosuppression for 5 years. Although, there is no consensus about the duration of preventive therapy, it is generally decided based on the potential benefits of therapy during a period of higher relapse risk (the first 2-3 years after presentation) against the risks of long-term toxicity of the agent, particularly treatment-related malignancy. While deciding to stop treatment, the clinician must discuss the matter with the patient taking into account the duration of treatment, history of relapse (frequency, severity, and recovery), treatment toxicity (actual or potential), and other factors (e. g., a woman's desire to become pregnant).
Corticosteroid
Generally oral steroid is continued after the treatment of an acute attack, then its dose is slowly reduced once the steroid-sparing immunosuppressive therapy becomes effective, thereby reducing the risks of long-term side effects of steroid. Daily or alternate day regimen of oral prednisolone is commonly used at a starting dose of up to 1 mg/kg. The first-line steroid-sparing agents are introduced while prednisolone is still continued in the same dosage. The prednisolone is usually reduced to a maintenance dose over 6 months while steroid-sparing agents are taking effect. After this period if the patient remains stable, a further tapering of the dose of prednisolone may be tried. However, owing to the risk of relapse which makes patient severely disable as against other antibody-mediated disease like myasthenia gravis, it is difficult to recommend withdrawal of steroid in NMO. Many NMO patients are steroid dependent and it has been seen that maintaining with a low dose of prednisolone (10-20 mg daily or an equivalent alternate-day regimen) is effective in preventing attack. Monitoring NMO-IgG antibody titer may prove to be useful for monitoring individual patients, particularly if tested at times relevant to treatment changes and during relapse, to compare with levels during stable periods. However, antibody titers do not predict disease severity or individual patient thresholds for relapse.
Azathioprine
It is a first-line, steroid-sparing agent. It is generally started along with prednisolone immediately following treatment of an acute attack. It is generally started with small dose of 25 mg/day and slowly increased over weeks to a target maintenance dose of 2.5 mg/kg, which can be increased further to 3 mg/kg. It is recommended to measure thiopurine methyltransferase (TPMT) level before starting the treatment; a low level of this enzyme in blood is a contraindication to azathioprine. It is also recommended to monitor blood count and liver function test during treatment to prevent toxicity. An elevated mean corpuscular volume or lymphopenia indicate that the treatment is at a therapeutic level and, if not present, may suggest the need to increase the dose.
In United States, about 11% of population has been found to have reduced TPMT activity leading to azathioprine toxicity.[21] On this basis, it is recommended to test for TPMT activity before starting azathioprine in any new patient. Those with mutations affecting TPMT activity may be very sensitive to azathioprine-induced gastrointestinal adverse effects and excessive immunosuppression.[22] For heterogygotes patients with low-normal TPMT activity, one should consider an alternative treatment option; and if azathioprine is used, these patients require more frequent blood count monitoring and a lower dosage for effective immunosuppression. Homozygotes with low TPMT activity should avoid azathioprine. These patients should receive of one of the other treatments discussed below.
Azathioprine was first used by Mandler and colleagues in a prospective study of seven patients of NMO in 1998. After 18 months of treatment with 75-100 mg of azathioprine and 10 mg of prednisone daily, each patient improved clinically and there were no new neurologic symptoms, exacerbations, or serious adverse events.[23] Subsequently, a number of studies have shown beneficial effects of azathioprine in prevention of NMO attack. Bichuetti et al., (2010) from Brazil reported that azathioprine plus prednisone led to stable disability scores and decrease in the annualized relapse rate (ARR) from 2.1 to 0.6 in 25 NMO patients.[24] Similar results were also reported in 28 NMO patients from Iran by Sahraian et al. (2010).[25]
In 2011, Costanzi et al., published the largest series of 99 NMO patients treated with azathioprine over a 15-year period.[26] While 86 patients fulfilled Wingerchuk NMO criteria, the remaining cases were AQP4 autoantibody seropositive cases with limited forms of NMO. Among the 70 patients who had been followed for at least 1 year, the ARR decreased when treated with azathioprine either with or without prednisone from 2.20 to 0.52 relapses/year over median treatment duration of 22 months. The reduction in ARR was less robust in those taking less than 2 mg/kg/day, (pretreatment ARR 2.09 versus on-treatment ARR 0.82 relapses/year). The mean Expanded Disability Status Scale (EDSS) and mean visual outcome scores (3.5 and 2, respectively) were stable during treatment.
The common side effects of azathioprine include nausea, diarrhea, elevated transaminases, and leukopenia. Other rare side effects are bone marrow suppression, fatigue, hair loss, and hepatotoxicity. Long-term use of azathioprine has been associated with myelotoxicity in up to 10% of patients.[27] An increased risk of lymphoma has also been observed in patients with inflammatory gastrointestinal disease on azathioprine.[28]
Rituximab
It is monoclonal antibody directed against the CD20 antigen, an epitope that is expressed on the B-cell lineage from pre B through mature B-cells, but absent on plasma cells.
The first study was conducted by Cree et al., (2005) who used rituximab in eight patients with severe NMO refractory to a variety of immunosuppressive and immunomodulatory therapies.[29] The dosage used was 375 mg/m2 weekly infusion for 4 weeks followed by two weekly 1,000 mg infusions if there was return of CD19 + B-cells in the peripheral blood. Six of the eight patients remained relapse-free during an average follow-up of 12 months. The median attack rate declined from 2.6 attacks/patients/year to zero on rituximab.
In a retrospective review of 25 NMO patients from seven tertiary centers in US, Jacob et al., (2008) reported the experience of rituximab where other treatments were not working effectively to reduce attacks.[30] Patients of relapsing NMO (n = 23) or NMO-IgG seropositive LETM (n = 2) were included in the study. Each of them received at least one dose of rituximab, and was followed for at least 6 months after being treated with rituximab. They tried two regimens: 375 mg/m2 weekly for 4 weeks (18 patients), and 1,000 mg infused twice with 2 weeks between doses (four patients). The median annualized pretreatment relapse rate declined from 1.7 to zero at a median posttreatment follow-up of 19 months. EDSS scores stabilized or improved in 80% of the patients. There were several adverse events during the treatment and follow-up periods. One patient died following a severe relapse. This individual had also developed recurrent Clostridium difficile colitis and a urinary tract infection prior to relapsing. Another patient developed fatal septicemia related to a urinary tract infection. Additionally, three patients developed new or reactivated infections (herpes simplex, herpes zoster, and a cutaneous fungal infection). Finally, one patient with pre-existent seborrheic dermatitis experienced worsening of the condition.
In another retrospective review of 23 NMO patients treated with rituximab, Bedi et al., (2011) reported that the median relapse rate declined from 1.87 relapses/patient/year to zero during median follow-up 32.5 months.[31] The median EDSS declined from 7.0 before treatment to 5.5 after treatment. Seventeen of the patients remained relapse-free during the observation period and the remaining six patients each had only one relapse. The investigators stated that relapses appeared to be attributable to unplanned prolongation of the interval between rituximab infusions. Adverse events occurred in seven of 23 patients and included recurrent herpes zoster, a urinary tract infection, two mild respiratory infections, fatigue, transient leukopenia, and transient transaminase elevation. The favorable outcome in this series could be because of different treatment regime used in this series (19 patients received 1,000 mg biweekly every 6 months).
In a 2-year prospective open label study, Kim et al., (2011) treated 30 patients with rituximab, 24 of whom failed to respond to other therapies, with either 375 mg/m2/week for 4 weeks or 1,000 mg biweekly infusions and then re-dosed upon reconstitution of CD27+ memory B-cells.[32] Twenty-eight of the 30 patients had reduction in relapse rate; the mean ARR declined from 2.4 to 0.3 over 24 months; 70% were relapse-free on treatment. The EDSS score declined for all but a single patient. AQ4 antibody levels also declined. In contrast to previous studies, maintenance rituximab therapy was provided upon the reappearance of peripheral CD27+ memory B-cells rather than CD19 cells. CD27+ B-cells are markers of antigen-specific memory B-cells that differentiate into antibody producing cells upon re-exposure of the antigen.[33] The most common adverse events in this study occurring during the initial infusion were transient hypotension and transient flu-like symptoms; approximately 40% of patients developed at least one mild infection during the course of treatment.
Rituximab induces B-cell activating factor (BAFF), which is thought to be an explanation for occasional reports of transient exacerbation following use of rituximab. However, the role of BAFF in the pathogenesis of NMO is still not clear. One study has reported AQP4-IgG titers and CD19 + B-cell counts rise before relapse and fall with remission, whereas another study suggested that the suppression of disease activity by rituximab correlates with the extent of B-cell depletion, but not with serum AQP4-IgG titer or serum levels of BAFF or a proliferation-inducing ligand. Thus, till date the search is still on for a definitive biomarker of disease activity in rituximab-treated individuals.
The most common infusion related adverse effect of rituximab is an allergic response. This can be prevented with prior use of methylprednisolone (125 mg intravenously 30 min before), diphenhydramine (25-50 mg oral dose), and/or acetaminophen (650 mg oral dose). The most common non-infusion related adverse events among all patients treated with rituximab have been infections. There are some rare reports of progressive multifocal leukoencephalopathy (PML)[34] in patients receiving either concomitant or sequential immunosuppressive drugs. Although the estimated risk of PML in all patients is now estimated at 1:25,000; there has not been a single PML case reported in association with rituximab use for NMO or MS.
Methotrexate
Methotrexate is an inhibitor of dihydrofolate reductase and folate-dependent enzyme necessary for purine and thymidylate synthesis. The specialists and primary care physicians usually have experience in using this drug by treating more common conditions such as psoriasis and rheumatoid arthritis. It is usually started with an initial maintenance dose of 15 mg once weekly (starting 7.5 mg in week 1 and increasing by 2.5 mg per week), with folate supplementation. If relapses occur, the dose is increased by 2.5 mg/week to a maximum of 25 mg weekly. However, it is not suitable for women of childbearing age and may lower the sperm count in men; and therefore it is recommended stopping it 3 months before trying to conceive. It has been used in relapse prevention in NMO, although there are only few reported series.[35,36,37] Minagar and Sheremata (2000) used methotrexate in eight NMO patients in conjunction with oral prednisolone.[35] Four were treated weekly with combined 50 mg of methotrexate intravenously and oral prednisone 1 mg/kg/day. Four others were treated with intravenous methylprednisolone (1 g/day for 10 days) and cyclophosphamide (8 mg/kg/day for 10 days as a loading dose followed by a 700 mg/m2 maintenance dose q4 weeks), three of whom were later switched to methotrexate plus prednisone after treatment failure. Each of the seven methotrexate patients subsequently stabilized, as evidenced by unchanged or reduced EDSS scores. In a recent study by Ramanathan et al., (2014) methotrexate was found to be safe and efficacious as a single long-term immunosuppressive therapy along with low dose corticosteroids.[37] They followed nine patients for a median of 62 months. While five patients were started on methotrexate as an initial long-term immunosuppressant strategy, three patients were initially treated with pulse cyclophosphamide followed by methotrexate as a preplanned step-down strategy and one patient was started on azathioprine prior to methotrexate. No patient had side effects requiring change in methotrexate therapy. While five patients had stabilization of EDSS, one patient had a small increase in EDSS due to concomitant illness, other three patients (33%) had methotrexate treatment failure evidenced by worsening EDSS and ongoing relapses while on methotrexate, mandating a change in methotrexate therapy. Average ARR in the entire group comparing 18 months prior versus 18 months after methotrexate treatment was reduced by an absolute value of 64% (3.11 vs 1.11).
Mycophenolate mofetil
Mycophenolate mofetil is an effective alternative and may be quicker acting than azathioprine. Mycophenolic acid is the active metabolite that is a reversible inhibitor of inosine monophosphate dehydrogenase, and thereby hinders de novo synthesis of guanosine nucleotides, thus suppressing lymphocyte proliferation. Though developed for transplant rejection (cardiac, liver, and renal), mycophenolate is used in a variety of autoimmune conditions. It is generally started with 500 mg daily in week 1, and then increased to 500 mg twice daily in week 2, then 1 g morning and 500 mg evening in week 3, and 1 g twice daily thereafter. It is relatively contraindicated in pregnancy because of an increased risk of first trimester pregnancy loss and of congenital malformations. There are very limited studies on mycophenolate in NMO/NMOSD available in the literature. Jacob et al., (2009) reported a retrospective case series of 24 patients, of whom 15 patients met NMO diagnostic criteria and nine patients were seropositive NMOSD.[38] Seven were treatment-naive, while the remainder had used various other immunosuppressive or immunomodulatory therapies. The median dose of mycophenolate was 2,000 mg/day, ranging from 750 to 3,000 mg/day. Patients had a median follow-up of 27 months after beginning mycophenolate treatment. At last follow-up, 19 patients continued treatment; two had discontinued mycophenolate — one received rituximab (personal preference) and the other one died. In those patients who continued treatment, the ARR declined from 1.28 to 0.09 relapses/year. Disability remained relatively unchanged. Six patients (25%) experienced adverse effects including headache, constipation, bruising, anxiety, hair loss, diarrhea, and leukopenia.
Mitoxantrone
Mitoxantrone inhibits topoisomerase II, suppresses lymphocyte and macrophage development, and inhibits B-cell activation. Kim et al., (2011) reported efficacy of mitoxantrone for a series of 20 NMO spectrum patients.[39] These patients were treated with mitoxantrone (three to six monthly cycles of 12 mg/m2 followed by 6-12 mg/m2 maintenance doses up to a maximum dose of 120 mg/m2) for an average of 17 months. The study showed a reduction in ARR (2.8 before treatment to 0.7 after treatment) and mean EDSS score (5.6 to 4.4). In another small case series, four of five patients with NMO showed clinical benefit and three of five patients became relapse-free. A significant decline in left ventricular ejection fraction was observed in one patient after a cumulative dose of 72 mg/m2. Mitoxantrone-related leukemia, a serious consequence of treatment, has not been reported in any NMO patient, probably owing to the low number of patients treated with mitoxantrone to date.
Combination Therapies
Combination therapy with cytotoxic, immunomodulatory, and B-cell-depleting therapies is being used for treatment of many autoimmune disorders such as rheumatoid arthritis. Primarily because of rarity of the condition, the cost of therapy, and the risk of infectious complications; the combination therapy has not been tested in NMO. Till date, the use of combination therapy in NMO has been limited to oral corticosteroids (prednisolone or prednisone) plus immunosuppressive agents such as azathioprine and cyclosporine. The combination has demonstrated a reduction in ARR and EDSS. Other effective combinations are intermittent PE with immunosuppressant, or intermittent IVIg in combination with immunosuppressant. Prospective studies comparing combination therapy, sequential therapy, and induction therapy will be needed to balance benefits and risks.
Selection of Therapies
While choosing an agent, consideration must be given about the efficacy as per available clinical data, toxicity profile, and the cost of therapy. Owing to the more-extensive availability of clinical data; azathioprine, mycophenolate mofetil, and rituximab tend to be the most-recommended first-line therapies for NMO prophylaxis. However, considering the cost of therapy and the potential side effect of severe infection, rituximab is to be used with caution. Second-line therapies include methotrexate, mitoxantrone, and cyclosporine. Due to the potential toxicity of mitoxantrone and the limited clinical data on methotrexate and cyclosporine, physicians should consider restricting their use to refractory cases [Table 3].
Table 3.
Therapy of acute attack and relapse prevention

In patients with severe onset of NMO/NMOSD use of therapeutic agents which have already had reported success in reducing relapses like rituximab is justified. It is generally considered that such patients are more vulnerable to further severe attacks. In patients with mild onset, given the perceived need for long-term immunosuppression in patients with NMO/NMOSD, and given the excellent safety profile of azathioprine and methotrexate, these can be used as initial therapy. Elderly patients, being perhaps more susceptible to side effects of chronic immune suppression, may be good candidates for initial therapy with methotrexate.
The NMO exacerbation does not increases in pregnancy, but increases in the postpartum period and in the year following childbirth. Azathioprine, mycophenolate, and methotrexate are pregnancy category D or X (D - adverse effects on fetus in human studies and X - adverse effects on fetus in human and animal studies) and should not be continued during pregnancy. Although rituximab and prednisone are pregnancy class C (adverse effects on fetus in animal studies without good data from human studies), the absence of an increase in ARR during pregnancy makes continued therapy during pregnancy questionable unless the patient displays evidence of renewed disease activity. Owing to increased relapse rate following delivery, rapid introduction of prophylactic therapy is warranted. However, the benefits of these therapies should be balanced against the benefits of breastfeeding. Till date, evidence comes from only one case report.[40]
Rituximab is also preferred for treatment failure after initial attempts with agents such as mycophenolate mofetil, azathioprine, and methotrexate. It has been proven in several series that who failed with therapies before rituximab, stabilization ensued after changing to this monoclonal antibody. Head-to-head trials of rituximab versus other existing agents (like azathioprine, mycophenolate mofetil, or methotrexate) for treatment of NMO/NMSD are deemed to be difficult to perform due to the rarity of the disorders, the long-term follow up that is needed, and the severe consequences of treatment failure among other factors. However, controlled trials are badly needed.
Symptom Management
Management of general symptom is out of scope of this article, however, following symptoms deserve discussion in respect to NMO.
Patients of NMO/NMOSD sometime develop refractory and unexplained vomiting and hiccups. These symptoms might suggest a brainstem relapse, and an MRI of brain is justified.
Transverse myelitis-associated pain is more common than MS and does not respond to tricyclics, gabapentin, or pregabalin. It typically starts as recovery begins from the acute attack and can continue for years. It sometime becomes severe and disabling and affects the quality of life. It requires involvement of experts of pain management.
Tonic spasms from transverse myelitis attacks are also more common than in MS. This usually improves with anticonvulsants. A small dose of carbamazepine is often very effective. Alternatives such as oxcarbazepine, lamotrigine, gabapentin, or pregabalin can help.
Investigational Agents
The understanding of the NMO pathogenesis has led to the use of agents, some of which are being used in different other autoimmune disorders. These are targeting:
-
(i)
Complement,
-
(ii)
Interleukin (IL)-6 receptor, and
-
(iii)
Granulocytes.
The monoclonal antibody targeting C5 complement, eculizumab, which is approved for use in paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome, has recently been tested in NMO in an open-label trial. In 14 patients, eculizumab significantly reduced attack frequency, and stabilized or improved neurological disability measures. The prohibiting cost of the agent is a limiting factor for its use.[41]
Several case reports showed reduced relapse rate in NMO patients treated with tocilizumab, a humanized murine anti-IL-6 receptor monoclonal antibody. Another anti-IL-6 receptor monoclonal antibody, SA237, has a fourfold greater duration of action than tocilizumab.
Sivelestat, an inhibitor of neutrophil elastase, and second-generation antihistamines cetirizine and ketotifen, which have eosinophil-stabilizing actions, have also been thought to prevent NMO relapse due to the role of granulocytes in the pathogenesis of NMO. Agents that block CD19 and tumor necrosis factor (TNF) have the potential effects in modifying the disease course in NMO. Some of these agents are currently under active investigation. Future potential treatments include humanized anti-CD20 monoclonal antibodies (e. g., ofatumumab and ocrelizumab), modulation of Th17 lymphocytes, glutamate receptor and BAFF, AQP-4 binding protective antibodies, etc.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Collongues N, Marignier R, Zephir H, Papeix C, Blanc F, Ritleng C, et al. Neuromyelitis optica in France: A multicenter study of 125 patients. Neurology. 2010;74:736–42. doi: 10.1212/WNL.0b013e3181d31e35. [DOI] [PubMed] [Google Scholar]
- 2.Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome) Neurology. 1999;53:1107–14. doi: 10.1212/wnl.53.5.1107. [DOI] [PubMed] [Google Scholar]
- 3.Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, et al. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet. 2004;364:2106–12. doi: 10.1016/S0140-6736(04)17551-X. [DOI] [PubMed] [Google Scholar]
- 4.Cabre P, Gonzalez-Quevedo A, Bonnan M, Saiz A, Olindo S, Graus F, et al. Relapsing neuromyelitis optica: Long term history and clinical predictors of death. J Neurol Neurosurg Psychiatry. 2009;80:1162–4. doi: 10.1136/jnnp.2007.143529. [DOI] [PubMed] [Google Scholar]
- 5.Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66:1485–9. doi: 10.1212/01.wnl.0000216139.44259.74. [DOI] [PubMed] [Google Scholar]
- 6.de Seze J, Lebrun C, Stojkovic T, Ferriby D, Chatel M, Vermersch P. Is Devic's neuromyelitis optica a separate disease? A comparative study with multiple sclerosis. Mult Scler. 2003;9:521–5. doi: 10.1191/1352458503ms947oa. [DOI] [PubMed] [Google Scholar]
- 7.Wingerchuk DM, Pittock SJ, Lucchinetti CF, Lennon VA, Weinshenker BG. A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology. 2007;68:603–5. doi: 10.1212/01.wnl.0000254502.87233.9a. [DOI] [PubMed] [Google Scholar]
- 8.Papeix C, Vidal JS, de Seze J, Pierrot-Deseilligny C, Tourbah A, Stankoff B, et al. Immunosuppressive therapy is more effective than interferon in neuromyelitis optica. Mult Scler. 2007;13:256–9. doi: 10.1177/1352458506070732. [DOI] [PubMed] [Google Scholar]
- 9.Sato D, Callegaro D, Lana-Peixoto MA, Fujihara K Brazilian Committee for Treatment and Research in Multiple Sclerosis. Treatment of neuromyelitis optica: An evidence based review. Arq Neuropsiquiatr. 2012;70:59–66. doi: 10.1590/s0004-282x2012000100012. [DOI] [PubMed] [Google Scholar]
- 10.Weinshenker BG, O’Brien PC, Petterson TM, Noseworthy JH, Lucchinetti CF, Dodick DW, et al. A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann Neurol. 1999;46:878–86. doi: 10.1002/1531-8249(199912)46:6<878::aid-ana10>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 11.Watanabe S, Nakashima I, Misu T, Miyazawa I, Shiga Y, Fujihara K, et al. Therapeutic efficacy of plasma exchange in NMO-IgG-positive patients with neuromyelitis optica. Mult Scler. 2007;13:128–32. doi: 10.1177/1352458506071174. [DOI] [PubMed] [Google Scholar]
- 12.Bonnan M, Valentino R, Olindo S, Mehdaoui H, Smadja D, Cabre P. Plasma exchange in severe spinal attacks associated with neuromyelitis optica spectrum disorder. Mult Scler. 2009;15:487–92. doi: 10.1177/1352458508100837. [DOI] [PubMed] [Google Scholar]
- 13.Noseworthy JH, O’Brien PC, Petterson TM, Weis J, Stevens L, Peterson WK, et al. A randomized trial of intravenous immunoglobulin in inflammatory demyelinating optic neuritis. Neurology. 2001;56:1514–22. doi: 10.1212/wnl.56.11.1514. [DOI] [PubMed] [Google Scholar]
- 14.Bakker J, Metz L. Devic's neuromyelitis optica treated with intravenous gamma globulin (IVIG) Can J Neurol Sci. 2004;31:265–7. doi: 10.1017/s0317167100053932. [DOI] [PubMed] [Google Scholar]
- 15.Okada K, Tsuji S, Tanaka K. Intermittent intravenous immunoglobulin successfully prevents relapses of neuromyelitis optica. Intern Med. 2007;46:1671–2. doi: 10.2169/internalmedicine.46.0217. [DOI] [PubMed] [Google Scholar]
- 16.Shimizu J, Hatanaka Y, Hasegawa M, Iwata A, Sugimoto I, Date H, et al. IFNbeta-1b may severely exacerbate Japanese optic-spinal MS in neuromyelitis optica spectrum. Neurology. 2010;75:1423–7. doi: 10.1212/WNL.0b013e3181f8832e. [DOI] [PubMed] [Google Scholar]
- 17.Uzawa A, Mori M, Hayakawa S, Masuda S, Kuwabara S. Different responses to interferon beta-1b treatment in patients with neuromyelitis optica and multiple sclerosis. Eur J Neurol. 2010;17:672–6. doi: 10.1111/j.1468-1331.2009.02897.x. [DOI] [PubMed] [Google Scholar]
- 18.Barnett MH, Prineas JW, Buckland ME, Parratt JD, Pollard JD. Massive astrocyte destruction in neuromyelitis optica despite natalizumab therapy. Mult Scler. 2012;18:108–12. doi: 10.1177/1352458511421185. [DOI] [PubMed] [Google Scholar]
- 19.Kleiter I, Hellwig K, Berthele A, Kümpfel T, Linker RA, Hartung HP, et al. Neuromyelitis Optica Study Group. Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch Neurol. 2012;69:239–45. doi: 10.1001/archneurol.2011.216. [DOI] [PubMed] [Google Scholar]
- 20.Min JH, Kim BJ, Lee KH. Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Mult Scler. 2012;18:113–5. doi: 10.1177/1352458511431973. [DOI] [PubMed] [Google Scholar]
- 21.Weinshilboum RM, Sladek SL. Mercaptopurine pharmacogenetics: Monogenic inheritance of erythrocyte thiopurine methyltransferase activity. Am J Hum Genet. 1980;32:651–62. [PMC free article] [PubMed] [Google Scholar]
- 22.Priest VL, Begg EJ, Gardiner SJ, Frampton CM, Gearry RB, Barclay ML, et al. Pharmacoeconomic analyses of azathioprine, methotrexate and prospective pharmacogenetic testing for the management of inflammatory bowel disease. Pharmacoeconomics. 2006;24:767–81. doi: 10.2165/00019053-200624080-00004. [DOI] [PubMed] [Google Scholar]
- 23.Mandler RN, Ahmed W, Dencoff JE. Devic's neuromyelitis optica: A prospective study of seven patients treated with prednisone and azathioprine. Neurology. 1998;51:1219–20. doi: 10.1212/wnl.51.4.1219. [DOI] [PubMed] [Google Scholar]
- 24.Bichuetti DB, Lobato de Oliveira EM, Oliveira DM, Amorin de Souza N, Gabbai AA. Neuromyelitis optica treatment: Analysis of 36 patients. Arch Neurol. 2010;67:1131–6. doi: 10.1001/archneurol.2010.203. [DOI] [PubMed] [Google Scholar]
- 25.Sahraian MA, Moinfar Z, Khorramnia S, Ebrahim MM. Relapsing neuromyelitis optica: Demographic and clinical features in Iranian patients. Eur J Neurol. 2010;17:794–9. doi: 10.1111/j.1468-1331.2009.02928.x. [DOI] [PubMed] [Google Scholar]
- 26.Costanzi C, Matiello M, Lucchinetti CF, Weinshenker BG, Pittock SJ, Mandrekar J, et al. Azathioprine: Tolerability, efficacy, and predictors of benefit in neuromyelitis optica. Neurology. 2011;77:659–66. doi: 10.1212/WNL.0b013e31822a2780. [DOI] [PubMed] [Google Scholar]
- 27.Gisbert JP, Gomollon F. Thiopurine-induced myelotoxicity in patients with inflammatory bowel disease: A review. Am J Gastroenterol. 2008;103:1783–800. doi: 10.1111/j.1572-0241.2008.01848.x. [DOI] [PubMed] [Google Scholar]
- 28.Kandiel A, Fraser AG, Korelitz BI, Brensinger C, Lewis JD. Increased risk of lymphoma among inflammatory bowel disease patients treated with azathioprine and 6-mercaptopurine. Gut. 2005;54:1121–5. doi: 10.1136/gut.2004.049460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cree BA, Lamb S, Morgan K, Chen A, Waubant E, Genain C. An open label study of the effects of rituximab in neuromyelitis optica. Neurology. 2005;64:1270–2. doi: 10.1212/01.WNL.0000159399.81861.D5. [DOI] [PubMed] [Google Scholar]
- 30.Jacob A, Weinshenker BG, Violich I, McLinskey N, Krupp L, Fox RJ, et al. Treatment of neuromyelitis optica with rituximab: Retrospective analysis of 25 patients. Arch Neurol. 2008;65:1443–8. doi: 10.1001/archneur.65.11.noc80069. [DOI] [PubMed] [Google Scholar]
- 31.Bedi GS, Brown AD, Delgado SR, Usmani N, Lam BL, Sheremata WA. Impact of rituximab on relapse rate and disability in neuromyelitis optica. Mult Scler. 2011;17:1225–30. doi: 10.1177/1352458511404586. [DOI] [PubMed] [Google Scholar]
- 32.Kim SH, Kim W, Li XF, Jung IJ, Kim HJ. Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch Neurol. 2011;68:1412–20. doi: 10.1001/archneurol.2011.154. [DOI] [PubMed] [Google Scholar]
- 33.Wingerchuk DM, Weinshenker BG. White matter disease: Optimizing rituximab therapy for neuromyelitis optica. Nat Rev Neurol. 2011;7:664–5. doi: 10.1038/nrneurol.2011.154. [DOI] [PubMed] [Google Scholar]
- 34.Carson KR, Focosi D, Major EO, Petrini M, Richey EA, West DP, et al. Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: A Review from the Research on Adverse Drug Events and Reports (RADAR) Project. Lancet Oncol. 2009;10:816–24. doi: 10.1016/S1470-2045(09)70161-5. [DOI] [PubMed] [Google Scholar]
- 35.Minagar A, Sheremata WA. Treatment of devic's disease with methotrexate and prednisone. Int J MS Care. 2000;2:43–9. [Google Scholar]
- 36.Kitley J, Elsone L, George J, Waters P, Woodhall M, Vincent A, et al. Methotrexate is an alternative to azathioprine in neuromyelitis optica spectrum disorders with aquaporin-4 antibodies. J Neurol Neurosurg Psychiatry. 2013;84:918–21. doi: 10.1136/jnnp-2012-304774. [DOI] [PubMed] [Google Scholar]
- 37.Ramanathan RS, Malhotra K, Scott T. Treatment of neuromyelitis optica/neuromyelitis optica spectrum disorders with methotrexate. BMC Neurol. 2014;14:51. doi: 10.1186/1471-2377-14-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jacob A, Matiello M, Weinshenker BG, Wingerchuk DM, Lucchinetti C, Shuster E, et al. Treatment of neuromyelitis optica with mycophenolate mofetil: Retrospective analysis of 24 patients. Arch Neurol. 2009;66:1128–33. doi: 10.1001/archneurol.2009.175. [DOI] [PubMed] [Google Scholar]
- 39.Kim SH, Kim W, Park MS, Sohn EH, Li XF, Kim HJ. Efficacy and safety of mitoxantrone in patients with highly relapsing neuromyelitis optica. Arch Neurol. 2011;68:473–9. doi: 10.1001/archneurol.2010.322. [DOI] [PubMed] [Google Scholar]
- 40.Ringelstein M, Harmel J, Distelmaier F, Ingwersen J, Menge T, Hellwig K, et al. Neuromyelitis optica and pregnancy during therapeutic B cell depletion: Infant exposure to anti-AQP4 antibody and prevention of rebound relapses with low-dose rituximab postpartum. Mult Scler. 2013;19:1544–7. doi: 10.1177/1352458513498125. [DOI] [PubMed] [Google Scholar]
- 41.Pittock SJ, Lennon VA, Mckeon A, Mandrekar J, Weinshenker BG, Lucchinetti CF, et al. Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: An open-label pilot study. Lancet Neurol. 2013;12:554–62. doi: 10.1016/S1474-4422(13)70076-0. [DOI] [PubMed] [Google Scholar]