

Abstract
Purpose.
To investigate the potential of human trabecular meshwork stem cells (TMSCs) for homing to mouse TM tissue and survival in vivo.
Methods.
Human TMSCs and fibroblasts were labeled with fluorescent membrane dye DiO and injected into normal mouse anterior chamber. Stem cell and TM cell markers were identified by immunofluorescent staining of cryosections or tissue whole mounts. Apoptosis was determined by TUNEL assay. Replicating and inflammatory cells were detected by bromodeoxyuridine (BrdU) incorporation and anti-CD45 staining, respectively. Quantitative RT-PCR detected gene expression of injected cells after isolation by fluorescence activated cell sorting. Intraocular pressure was measured using a TonoLab rebound tonometer.
Results.
Expanded cultures of DiO-labeled TMSCs expressed stem cell markers preferentially in DiO positive cells, demonstrating a slow-cycling, label-retaining stem cell phenotype. DiO-labeled TMSCs injected into the anterior chamber of normal mice localized primarily in TM, remaining in the tissue at least 4 months. Within 1 week, TM-associated TMSCs began expressing TM marker protein CHI3L1. Fibroblasts injected in mouse anterior chamber showed distributed localization in corneal endothelium, lens epithelium, and TM and did not express CHI3L1. Little apoptosis was detected in injected TM tissue and intraocular pressure was not elevated during the experiment. Dividing cells or CD45-staining cells were not detected after TMSC-injection.
Conclusions.
Stem cells isolated from human TM and expanded in vitro exhibit the ability to home to the TM and differentiate into TM cells in vivo. Such cells present a potential for development of a novel cell-based therapy for glaucoma.
Human trabecular meshwork stem cells (TMSCs) injected into the anterior chamber of normal mice can home to the TM without inducing an inflammatory response and increasing intraocular pressure; TMSCs can survive 4 months and differentiate into TM cells in vivo.
Introduction
Glaucoma is a progressive optic neuropathy with loss of retinal ganglion cells and axons, resulting in visual field impairment. Elevated intraocular pressure (IOP) and aging are important risk factors for most forms of glaucoma. The main aqueous outflow pathway of the eye consists of a series of endothelial cell-lined channels in the angle of the anterior chamber comprising the trabecular meshwork (TM), Schlemm's canal, collector channels, and episcleral venous system. Pathological changes in the TM and Schlemm's canal are prime suspects for increased resistance to the aqueous outflow and elevated IOP. It has been suggested that the age- and disease-related decrease of TM cells,1–5 abnormal accumulation of ECM materials, and the appearance of the cross-linked actin networks in the TM cells6–8 contribute to an increased resistance of the aqueous outflow and subsequent increase of IOP.
TM cells in vivo play two primary roles, including secretion of specific enzymes and extracellular matrix, and phagocytosis of debris in the aqueous humor.9 Cell-based restoration of TM function in glaucomatous eyes is a potential therapy not yet explored. We recently described stem cells from adult human TM that can be greatly expanded and maintain the novel ability to differentiate into phagocytic TM cells in vitro.10 In the current study, we examined the ability of these stem cells to home to the TM region and maintain the stem cell characteristics or become functional TM cells without causing IOP increase after transplantation into normal mouse anterior chamber.
Materials and Methods
Materials
Antibodies used include anti-ABCG2 (Chemicon, Billerica, MA), anti-Notch1 (BD Pharmingen, San Diego, CA), anti-MUC1 (Santa Cruz Biotechnology, Santa Cruz, CA), anti-CHI3L1 (R&D Systems, Minneapolis, MN), anti-AQP1 (Santa Cruz Biotechnology), and anti-CD45-PE conjugated (BD Pharmingen). Secondary antibodies used are anti-mouse Alexa-546 or 647, antirabbit or antigoat Alexa-546 (Life Technologies, Carlsbad, CA).
Stem Cell Culture
Stem cells from human trabecular meshwork were isolated and cultured as described before.10 In brief, deidentified human corneas were obtained from the Center for Organ Recovery & Education (Pittsburgh, PA). Cells from the TM were cultured as either explant or dissociated cell culture in stem cell growth medium (SCGM), modified from a corneal endothelial cell culture medium11 that contained reduced serum medium (OptiMEM-1; Life Technologies) supplemented with 5% fetal bovine serum (Hyclone, Logan, UT); 10 ng/mL EGF (Upstate Biotechnologies, New York, NY); 100 μg/mL bovine pituitary extract (Biomedical Technologies, Stoughton, MA); 20 μg/mL ascorbic acid; 200 μg/mL calcium chloride; 0.08% chondroitin sulfate (Sigma-Aldrich, St. Louis, MO); 100 IU/mL penicillin; 100 μg/mL streptomycin and 50 μg/mL gentamicin (Sigma-Aldrich). TMSCs were isolated as clonal cultures as described previously.10
DiO Staining
Cells were prelabeled with membrane dye (Vybrant DiO; Life Technologies) for detection of the stem cell label–retaining ability in vitro or for in vivo experiments to trace transplanted cells. Cells were suspended in Dulbecco's modified Eagle's medium (DMEM)/F-12 at 1 × 106 cells/mL. DiO was added at a dilution of 50 μg/mL at 37°C for 30 minutes. Then the cells were washed twice with DMEM/F-12 and resuspended for further experiments.
Anterior Chamber Cell Transplantation
Female C57BL/6 mice (Charles River Laboratories International, Inc., Wilmington, MA) at 9 weeks of age were used in cell injection experiments. All experimental procedures were reviewed and approved by the University of Pittsburgh Institutional Animal Care and Use Committee and handled according to guidelines provided in the Association for Research in Vision and Ophthalmology Resolution on the Use of Animals in Ophthalmic and Vision Research.
Forty-eight mice were used for anterior chamber injection of TMSCs and fibroblasts. One eye from each mouse was used for the injection and the other eye served as the untreated control. Passage-4 TMSCs cultured in SCGM or passage-8 corneal fibroblasts cultured in DMEM/F12 with 10% FBS were prelabeled with DiO and resuspended in DMEM/F12 at 2.5 × 104 cells/μL for injection. The cells were injected into the mouse anterior chamber following the procedures described by McKenna12 with minor modifications. All mice were anesthetized by intraperitoneal injection of 2 mg of ketamine hydrochloride and 0.2 mg of xylazine (IVX Animal Health, Inc., St. Joseph, MO) in 0.2 mL of Dulbecco's PBS. The eyes were washed with PBS and anesthetized by topical drops of proparacaine HCl (Falcon Pharmaceuticals, Fort Worth, TX). A corneal tunnel was made by inserting a 30-gauge needle in the cornea outside of the pupil area and parallel to the iris. Aqueous humor was allowed to flow out. An air bubble was then introduced into the anterior chamber by injecting a 1.5-μL volume of air with a microsyringe (Hamilton, Reno, NV) fitted with a 33-gauge beveled needle. Next, 5 × 104 TMSCs or fibroblasts in 2 μL of DMEM/F12 were injected with a microsyringe fitted with a 33-gauge blunt needle (Hamilton).
IOP was measured using a rebound tonometer for rodents (TonoLab; Colonial Medical Supply, Franconia, NH) before and after cell injection at 3, 5, 7, 10 days and 2, 3, 4, 6 weeks, and 4 months. All IOP measurements were performed around 10 AM on mice that had been anesthetized by intraperitoneal injection of ketamine and xylazine.
Mice were sacrificed at 1 week, 4 weeks, and 4 months after transplantation. Mouse eyes were enucleated either for histology or for fluorescence activated cell sorting (FACS) to isolate DiO-labeled injected cells. For each cell type, six mice were sacrificed at 1 week and at 4 weeks after injection for immunofluorescent staining of cryosections or whole mounts. Twelve mice were sacrificed at 4 months after injection. Among these, six were used for immunostaining and another six were pooled for FACS.
Before sacrificing at 4 months, in vivo imaging of mouse anterior segments was performed using a high-resolution stereo fluorescence biomicroscope with vertical fluorescence illuminator (Leica MZFLIII; Leica Microsystems Inc., Bannockburn, IL). Mice were anesthetized and immobilized with a three-point stereotactic mouse restrainer as previously described.13 The cornea was focused and images were obtained at a magnification of ×25 with visibility to the anterior chamber, iris, pupil, and lens. The corneal endothelium was visualized by in vivo confocal microscopy (Confoscan 3; Nidek Technologies America, New Orleans, LA) following the in vivo imaging. A 40× objective optically coupled to the cornea with transparent gel (Viscotirs; Novartis Ophthalmics, Duluth, GA) was focused on the corneal endothelium.
Bromodeoxyuridine (BrdU) Incorporation
At 1 week and 4 weeks after cell injection, mice were given BrdU intraperitoneal injection at 50 μg/g body weight, 4 hours before sacrificing, to label actively replicating cells. BrdU-incorporated cells were detected by wholemount staining using BrdU kit (BrdU Labeling and Detection Kit I; Roche Molecular Biochemicals, Indianapolis, IN) following the manufacturer's protocol. Antimouse Alexa-546 secondary antibody was used to bind the BrdU antibody as red to distinguish with DiO labeling.
Histology
TMSCs labeled with DiO were cultured for two more passages in SCGM in 35-mm plastic dishes. Before fixation, cells were cocultured with Hoechst 33342 (Sigma-Aldrich) at the concentration of 5 μg/mL in prewarmed DMEM/F12 with 2% FBS at 37°C for 1 hour. After incubation, cells were rinsed briefly in PBS and fixed for 15 minutes in 4% paraformaldehyde (PFA) at room temperature. Nonspecific binding was blocked with 1% BSA for CHI3L1 staining or 10% heat-inactivated goat serum for others. Cells were incubated overnight at 4°C with primary antibodies. After three washes, antimouse Alexa-647, antirabbit, or antigoat Alexa-546 secondary antibodies and nuclear dye 4′,6-diamidino-2-phenylindole (DAPI 0.5 μg/mL; Roche Molecular Biochemicals) were added and incubated for 2 hours at room temperature. Samples were imaged using a confocal microscope (Olympus FluoView FV1000 confocal microscope; Olympus, Center Valley, PA).
Enucleated mouse eyeballs were fixed in 1% PFA at 4°C overnight followed by either storage at 4°C in 50% glycerol and 50% PBS (v/v) for wholemount staining or frozen at −20°C in optimal cutting temperature (OCT) embedding compound (Tissue-Tek OCT; Electron Microscopy Sciences, Hatfield, PA) and cut into 8-μm thick cryosections on a cryostat for immunofluorescent staining.
Sections were hydrated in PBS and postfixed in 4% PFA for 15 minutes. After washing in PBS twice, sections were incubated with DAPI for 1 hour at room temperature. Samples were photographed to detect the injected green cell localization using a confocal microscope (Olympus FluoView FV1000 confocal microscope; Olympus) equipped with a movable stage for obtaining a whole picture of each single section.
TUNEL assay was performed using a cell death detection kit (In Situ Cell Death Detection Kit, TMR red; Roche Molecular Biochemicals) following the manufacturer's protocol on cryopreserved tissue. Nuclei were stained with DAPI. At least three independent TM from each condition and at least nine sections of each condition were stained and imaged using a confocal microscope.
Wholemount Stain
After fixation, the posterior part of the eyeball, 1.5 mm posterior to the limbus, was removed. The anterior part, including the cornea and the TM, was cut into half for wholemount stain and the cornea was cut in the middle to flatten the tissue. Nonspecific binding was blocked with 10% heat-inactivated goat serum or 1% BSA and antimouse CD16/CD32 Fc γ III/II (BD Pharmingen). Tissue was incubated with primary antibodies overnight at 4°C. Following five washes, tissue was incubated with fluorochrome-conjugated secondary antibodies with DAPI for 2 hours at room temperature prior to confocal imaging.
Confocal Imaging of TM Wholemounts
Images were acquired by sequential scanning to avoid fluorescence crossover on a confocal microscope (Olympus FluoView FV1000 confocal microscope; Olympus). Z-stacks through the tissue were acquired using Java-based image processing software (ImageJ; National Institutes of Health, Bethesda, MD). All image reconstructions were made using microscope automation and image analysis software (MetaMorph, version 7.5.4.0; Molecular Devices, Sunnyvale, CA).
FACS for Isolation of Injected Cells
Four months after injection of DiO-labeled stem cells or fibroblasts, six enucleated mouse eyes of each condition were dissected and the TM including the underlining sclera was cut off using corneal scissors (Ketena, Denville, NJ). The TM was digested in collagenase I (Sigma-Aldrich) at 42 units/TM in DMEM-10% FBS for 45 minutes to 1 hour until almost no visible tissue remained. The digest was filtered through a 70-μm nylon mesh and cells were collected by centrifugation and washed in PBS with 1% BSA. The cells were stained with a cell stain kit (Live/Dead Fixable Aqua Dead Cell Stain Kit; Life Technologies) according to the manufacturer's instructions. FACS was performed on a cell sorter (BD FACS-Aria Cell Sorter; BD Biosciences, San Jose, CA) and live green cells were collected for RNA and qPCR.
Quantitative RT-PCR (qPCR)
Sorted cells were lysed with RLT buffer in an RNA purification kit (RNeasy mini kit; Qiagen, Valencia, CA) and RNAs were isolated following the manufacturer's instructions including treatment with DNase I (Life Technologies) and concentration by ethanol precipitation. cDNAs were transcribed from the RNAs using a reverse transcriptase kit (SuperScript II; Life Technologies). qPCR of cDNAs was performed by direct dye binding (SYBR Green; Applied Biosystems, Foster City, CA) as previously described.14 Primers were used as following: human TATA binding protein (sense, 5′-CCCACAGCCTATTCAGAACACC; antisense, 5′-CAATCCCAGAACTCTCCGAAGC) and mouse TATA binding protein (sense, 5′-AGAACAACAGCCTTCCACCTTATG; antisense, 5′-CAAGTTTACAGCCAAGATTCACGG).13 Amplification of 18S rRNA (sense, 5′- CCCTGTAATTGGAATGAGTCCAC; antisense, 5′-GCTGGAATTACCGCGGCT) was performed for each cDNA (in triplicate) for normalization of RNA content. Relative mRNA abundance was calculated as the Ct for amplification of a gene-specific cDNA minus the average Ct for 18S expressed as a power of 2 (2–ΔΔCt). Three individual gene-specific values thus calculated were averaged to obtain mean ± SD.
Statistical Analysis
All values are presented as mean ± SD. The statistical differences were determined by one-way ANOVA, followed by the Tukey posttest to assess the significance of differences between all groups. Statistical significance was set at P < 0.05.
Results
TMSCs Retain Lipophilic Dye DiO In Vitro
Adult stem cells, which divide more slowly than other somatic populations, can be identified in vivo by retention of a DNA-pulse label.15 DiO, a nontoxic, lipophilic membrane dye, can also be used to distinguish slowly dividing cells from rapidly replicating cells.16 Passage 4 TMSCs labeled with DiO and expanded for another two passages showed heterogeneity of DiO staining (Fig. 1). The DiO positive (green) cells exhibited reduced Hoechst 33342 nuclear dye (blue) consistent with the clearance of this dye in side population cells.10,17 Additionally, these DiO-retaining cells were positive for Notch1 (red); ABCG2 (purple, Figs. 1A–E, arrows); and MUC1 (purple), but not CHI3L1 (red, Figs. 1F–J), a differentiated TM cell marker. In contrast, cells in the same cultures in which DiO staining was reduced as a result of cell division expressed CHI3L1, but not MUC1 (Figs. 1F–J, arrows). These results show a correlation of stem cell markers and xenobiotic dye efflux with DiO-label-retaining by a portion of the TMSC population after expansion culture. These results also confirm that DiO labeling of TSMCs in vitro can be a useful marker of cells with stem cell potential.
Figure 1.

TMSCs are slow-cycling in vitro. Cultured TMSCs were labeled with lipophilic dye DiO (green) and, after two passages, incubated with Hoechst 33342 for 1 hour, followed by staining with Notch1 (red) and ABCG2 (purple, [A–E]) or CHI3L1 (red) and MUC1 (purple, [F–J]). Arrows in (B–E) point to DiO-retaining cells; arrows in (G–J) point to DiO-negative CHI3L1 expression cells. DAPI stains nuclei as blue.
TMSCs Localize to the TM after Anterior Chamber Transplantation
Human TMSCs at passage 4 were injected into the anterior chamber of C57BL/6 wild type mice at 5 × 104 cells per eye. The TMSCs were prelabeled with fluorescent green dye DiO. After 1 week, low magnification fluorescent images (Fig. 2A) revealed the majority of the TMSCs localized in the angle of the eye near the TM. Magnification of that region (Fig. 2A1) confirmed localization of the cells primarily in the TM with some in the iris. This pattern of localization was maintained at 4 weeks (Figs. 2C, 2C1) and 4 months (Figs. 2E, 2E1) after injection with no noticeable loss in DiO-labeled cells in the TM over this time period. Corneal fibroblasts injected in the same manner showed a distinctly different pattern of distribution. Fibroblasts were detected in the TM and iris at 1 week (Fig. 2B1), but the most intense concentrations of labeled cells were observed in the corneal endothelium and lens epithelium (Fig. 2B). Labeled fibroblasts were detected in these tissues throughout the 4-month experiment (Figs. 2D, 2F).
Figure 2.

TMSCs home to the TM region after anterior chamber injection. TMSCs or fibroblasts were prelabeled with DiO (green) and injected into normal mouse anterior chamber. At 1 week, 4 weeks, and 4 months after injection, cryosections of mouse anterior segment were stained with DAPI (blue) for nuclei. (A, C, E) show TMSCs (green) in the TM at 1 week, 4 weeks, and 4 months after injection. (A1, C1, E1) are the magnifications of the boxed region in (A, C, E), respectively. (B, D, F) show fibroblasts (green) in the TM, the corneal endothelium and the epithelium of the lens at 1 week, 4 weeks, and 4 months after injection. (B1, D1, F1) are the magnifications of the boxed region in (B, D, F), respectively. Arrows in (B, D, F) show green fibroblasts localized outside of the TM region.
Anterior Chamber–Transplanted TMSCs Do not Initiate Disorders of the Eye
A major concern in stem cell–based therapy is whether injected cells might disrupt normal tissue function. TMSC- and fibroblast-injected eyes were monitored for transparency, inflammation, and intraocular pressure for 4 months after transplant. At 4 months after cell injection, the eyes with either TMSC or fibroblast injection were quiet with clear corneas and normal anterior chambers as shown in Figures 3A–C. The density and morphology of the corneal endothelial cells of the eyes with TMSC injection (Fig. 3E) and with fibroblast injection (Fig. 3F) at 4 months are the same as those of normal corneas without injection (Fig. 3D). The average levels of IOP before and after cell injection at different time points are shown in Fig. 3G. IOP differences between TMSC- and fibroblast-injected eyes were not statistically significant (P > 0.05) and none of the IOPs of injected eyes differed from that of noninjected controls (P > 0.05).
Figure 3.

The anterior segments of the eyes with cell injection maintain undisturbed. At 4 months after TMSC– or fibroblast–anterior chamber injection, pictures of the anterior segments were taken using a biomicroscope and the corneal endothelium was visualized by a corneal microscope. (A, B, C) show the transparent cornea, iris, pupil, and lens of the normal eye (A), the eye with TMSC-injection (B), or with fibroblast-injection (C). (D, E, F) show the morphology of the corneal endothelium of the normal eye (D), the eye with TMSC-injection (E), or with fibroblast-injection (F). The IOP was measured before and after cell injection at different time points comparing with the normal controls; arrow bars show the SD (G).
TMSCs Do not Elicit Inflammatory Response after Xenotransplantation
Although neither injected TMSCs nor fibroblasts caused IOP increase or resulted in obvious corneal pathology, fibroblasts induced an inflammatory response whereas TMSCs did not. Figure 4 shows that 4 weeks after cell injection, no CD45 inflammatory cells were detected in the TM with TMSC injection (Fig. 4B) while CD45-positive inflammatory cells were detected in the TM with fibroblast injection (Figs. 4C, 4D, arrows). In the control eyes, no detectable CD45 positive cells were detected in the TM (Fig. 4A).
Figure 4.
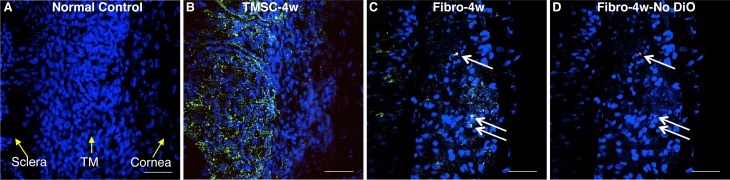
Injection of TMSCs into the anterior chamber does not elicit inflammatory response. DiO- (green) labeled human TMSCs or fibroblasts were injected into normal mouse anterior chamber. At 4 weeks after injection, wholemount staining with anti-CD45 antibody was performed to show CD45 staining on normal mouse TM (A), on the TM tissue after TMSC-injection (B), and on the TM tissue after fibroblast-injection (C). (D) is identical to (C), showing CD45 staining without green channel DiO. Arrows point to CD45+ cells. DAPI stains nuclei as blue. The orientation of the tissue is indicated by yellow arrows in (A). Scale bars: 50 μm.
TMSCs Maintain Viability after Anterior Chamber Transplantation
Although stained cells remain visible in the tissue for 4 months, we considered it is important to determine if the injected human TMSCs actually survived in the mouse TM after xenotransplantation. TUNEL assay was used to detect apoptotic cells in cryosections. Figures 5A–F show that at each time point after anterior chamber injection, green fluorescent TMSCs and fibroblasts were present in the TM tissue. One week after injection, none of the labeled TMSCs exhibited TUNEL staining (red). Some fibroblasts in the TM tissue, however, were apoptotic, staining red (Fig. 5B). In most conditions, apoptotic cells were detected in corneal epithelium showing purple as positive TUNEL stains red while DAPI stains nuclei blue.
Figure 5.

TMSCs maintain viability at least 4 months after anterior chamber injection. TUNEL assay was used to detect apoptotic cells on the cryosections of the eyes with cell injection. TUNEL assay stains apoptotic cells as red and DiO prelabeled cells before injection are green. (A, C, E) show the sections with TMSC-injection at 1 week, 4 weeks, and 4-months after injection. (B, D, F) show the sections with fibroblast injection at 1 week, 4 weeks, and 4 months after injection. The Schlemm's canal and the TM are pointed with arrows. DAPI stains nuclei as blue. Scale bars: 50 μm.
Injected TMSCs Do not Fuse with the Mouse Cells
Previous reports have indicated that stem cells can fuse with cells of the host tissue, thereby gaining expression of different phenotype.18 To rule out that possibility, TM tissue 4 months after injection was dissected and digested with collagenase and the dissociated single cells were stained with dye (Live/Dead dye; Life Technologies). Flow cytometry shows most of the cells were viable with approximately 5% of the cells were DiO stained (Fig. 6A: TMSCs; Fig. 6B: fibroblasts). The DiO-stained live cells (DiO+) and DiO negative live cells (DiO–) from TMSC- or fibroblast-injected eyes were sorted by FACS and quantitative RT-PCR was performed to compare the expression of human TATA-binding protein (hTBP) and mouse TBP (mTBP). Figure 6C shows that TMSCs isolated from the injected tissue expressed approximately a 10:1 ratio of human to mouse TBP whereas the isolated fibroblasts expressed approximately 60:40 ratio of human to mouse TBP. These results suggest that most or all of the TMSCs remained viable and not fused to mouse cells during the 4-month period.
Figure 6.

Injected human TMSCs express human genes only. Human TMSCs or fibroblasts were prelabeled with DiO followed by injection into mouse anterior chamber. 4 months after injection, the TM from TMSC- or fibroblast-injected eyes were polled and digested with collagenase. Viable cells were identified by live/dead dye staining. Flow cytometry shows most cells were alive (A, B) and live human TMSCs (DiO+TMSCs; [A], red square); live mouse cells from human TMSC-injected eyes (DiO-TMSCs; [A], purple square); live human fibroblasts (DiO+Fibro; [B], blue square); and live mouse cells from human fibroblast-injected eyes (DiO-Fibro; [B], green square) were collected by FACS. (C) shows qPCR results comparing the different expression of hTBP and mTBP in the sorted cells.
Some Injected TMSCs Express Differentiated TM Markers
Mouse TM tissue with or without cell injection was stained as tissue wholemounts to detect expression of differentiated TM cell markers. Antimouse AQP1 antibody, which reacts to both human and mouse origins, was used to identify TM cells and corneal endothelial cells (Figs. 7A–E). There were no reactions of normal mouse TM tissue to human-specific antibodies CHI3L1 (Fig. 7F) and MUC1 (Fig. 7K). One week after cell injection, expression of human CHI3L1 (Fig. 7G) and MUC1 (Fig. 7L) was evident in the TM of TMSC-injected eyes. In contrast, few human fibroblasts localized to the TM region expressed CHI3L1 or MUC1 (Figs. 7H, 7M). At 4 weeks and 4 months after cell injection, the number of green TMSCs in the TM region appeared to be increased. Some TMSCs were MUC1 positive, whereas some stained with CHI3L1, a TM cell marker, suggesting differentiation of the injected cells (Figs. 7I, 7N, 7P, 7R). The green fibroblasts in the TM region of fibroblast-injected eyes lacked expression of human CHI3L1 or MUC1 (Figs. 7J, 7O, 7Q, 7S).
Figure 7.

Human TMSCs home to TM region after mouse anterior chamber injection. DiO-labeled human TMSCs or fibroblasts were injected into normal mouse anterior chamber for 1 week, 4 weeks, and 4 months. Wholemount stain was performed to detect the green-injected cells and the expression of AQP1 (red; [A–E]) and human specific antigens CHI3L1 (red; [F–J, P, Q]) and MUC1 (red; [K–O, R, S]) on host TM region. (A, F, K) show staining of the antibodies on normal mouse tissue without cell injection serving as controls. (B, G, L) show staining on TMSC-injected tissue at 1 week. (C, H, M) show staining on fibroblast-injected tissue at 1 week. (D, I, N) show staining on TMSC-injected tissue at 4 weeks. (E, J, O) show staining on fibroblast-injected tissue at 4 weeks. (P, R) show staining on TMSC-injected tissue at 4 months. (Q, S) show staining on fibroblast-injected tissue at 4 months. DAPI stains nuclei as blue. The orientation of the tissue is indicated by yellow arrows in (K, R). Scale bars: 50 μm.
Most Injected TMSCs Are Quiescent In Vivo
Figure 7 indicates that the number of injected TMSCs in the TM tissue at 4 weeks to be greater than that at 1 week. To understand if this increased cell number is due to increased cell division, we used a BrdU incorporation assay to detect actively replicating cells. Four hours before sacrificing, mice were intraperitoneally injected with BrdU. Wholemount tissue staining was performed to detect BrdU incorporating cells. No cells stained in the control mouse TM without BrdU injection (Fig. 8A) nor in the BrdU-injected normal mouse TM without cell transplantation (Fig. 8D), indicating the quiescence of this tissue in adult mouse. There are also no BrdU-labeled cells in the TM after TMSC injection at 1 week (Fig. 8B) and 4 weeks (Fig. 8E). In contrast, a number of BrdU-positive cells were detected in the TM with fibroblast injection at 1 week (Fig. 8C), but almost no BrdU-positive cells were seen in the TM 4 weeks after fibroblast injection (Fig. 8F).
Figure 8.

Mouse TMs with TMSC-injection maintain quiescence. At 1 week and 4 weeks after cell injection, mice were given BrdU intraperitoneal injection 4 hours before sacrificing. Wholemount staining with anti-BrdU antibody on mouse TM tissue was done to detect BrdU-incorporated cells. Human cells were prelabeled with DiO as green and BrdU was stained red. DAPI stains nuclei as blue. (A) shows BrdU stains on normal mouse tissue without BrdU injection and (D) shows BrdU stains on normal mouse tissue with BrdU injection. (B, E) show BrdU staining on TMSC-injected mouse tissue at 1 week and 4 weeks, respectively. (C, F) are BrdU staining on fibroblast-injected mouse tissue at 1 week and 4 weeks, respectively. The orientation of the tissue is indicated by yellow arrows in (D). Scale bars: 50 μm.
Discussion
In the present study, we report that human TMSCs preferentially localize to the TM region and maintain viability for at least 4 months after transplantation into mouse anterior chamber. The cells integrate into the TM, do not elicit inflammatory response, nor do they cause an increase in IOP. An intrinsic property of adult stem cells is to identify and localize in specific tissues where they exhibit tissue-specific differentiation.19–22 The behavior of TMSCs in the anterior chamber is clearly distinct from that of corneal fibroblasts and has all the aspects of a classic homing response typical of adult stem cells. The ability of TMSCs to home to the TM and become differentiated TM cells even in a normal TM without damage is a novel and important observation. It opens a door to explore stem cell-based therapy for glaucoma.
Most stem cells are slow cycling, retaining DNA label over an extended pulse-chase period. This phenomenon is termed “label-retaining cells.”15,23 Lipophilic cell membrane dyes, DiO, DiI,24 or PKH2625 have also been used to identify slow-cycling stem cells. We show here that cloned TMSCs retain DiO during in vitro culture and express stem cell markers Notch1, ABCG2, and MUC1. These cells also exhibit the ability to efflux DNA-binding dye Hoechst 33342, a stem cell–specific cellular function that is widely used as the basis of a technique for isolation of side population stem cells by FACS.17 Hoechst efflux confirms TMSCs as stem cells and the slow-cycling property can be used to explore the location and function of endogenous TMSCs in vivo.
Most importantly, our data provide clear evidence that TMSCs, after anterior chamber injection, are able to home to the TM region (Figs. 2, 5) maintaining viability for at least 4 months (Figs. 5, 6). In contrast, injected fibroblasts attached to a variety of tissues after injection into the anterior chamber. These results support the idea that localization in the TM is not simply a function of the TMSCs being carried passively by aqueous outflow, but a result of a tissue affinity of the TMSCs. TMSCs also appeared in the iris (Figs. 2A, 2E), a tissue with similar developmental origin to TM. Increasingly abundant evidence supports the ability of mesenchymal stem cells to localize and regenerate damaged tissue in vivo (see review articles by Kang et al.21 and by Fong et al.26). We thus believe that in glaucomatous eyes, injected TMSCs may be able to localize to pathological TM and improve aqueous outflow. In glaucomatous eyes, abnormal extracellular matrix of the TM may have effect on stem cells' homing. Our next step is to explore the homing and function of TMSCs in a mouse glaucoma model.
Mesenchymal-like stem cells have been shown with the ability to mediate immunosuppression.13,27–30 Our current study confirms that xenotransplantation of human TMSCs to mouse anterior chamber does not elicit inflammatory response (Fig. 4). It ensures the survival of transplanted stem cells to function in vivo. This observation provides an argument that these stem cells could be tolerated in human allogeneic transplantation. The ability of TMSCs to undergo extensive expansion in vitro makes allogeneic transplantation possible. Since glaucoma has underlying genetic components, it would not seem feasible to do autologous transplantation using the same genetically abnormal cells. The expansion ability of TMSCs provides a possibility to regenerate TM in glaucomatous eyes by allogeneic transplantation of TMSCs without glaucomatous genetic disorders.
Chitinase 3-like 1 (CHI3L1) has been identified as a marker of TM cells10 (Liton PB, et al. IOVS 2009;50:ARVO E-Abstract 4859). CHI3L1 can directly interact with type I collagen that plays an important role in CHI3L1 tissue-remodeling activity.31 Our results demonstrate that a proportion of the transplanted TMSCs expressed the TM marker CHI3L1 (Fig. 7). It clearly indicates that the transplanted TMSCs function in vivo instead of simply attaching to the tissue, which could become a barrier for outflow and cause increase of IOP.
We also show that TMSCs can be expanded in vitro and retain DiO labeling, expressing stem cell markers ABCG2, Notch1, and MUC1. After in vivo injection, the TMSCs home to the TM region and iris while fibroblasts attach to the TM, iris, corneal endothelium, and epithelium of the lens (Fig. 2). The inner layer of the TM, the uveal meshwork, is formed by prolongations of connective tissue arising from the iris and ciliary body stromas,32 which are derived from neural crest cells. Since the TM and the iris have the same origination, it is not surprising that injected TMSCs can home to both TM and iris. We would assume that TMSCs would home to the damaged TM tissue but not the iris if they were introduced into the eye with TM damage. The injected cells do not affect the corneal transparency and do not cause increased intraocular pressure dramatically. Xenotransplantation of TMSCs does not elicit an inflammatory response nor stimulate endogenous TM cell division. The endogenous TM cells in the eyes with the transplanted TMSCs were quiescent with no BrdU incorporation (Fig. 8). Although there were more injected green cells in the TM at 4 weeks than at 1 week after injection, there should not be a concern because IOP was not increased and neither the injected TMSCs nor the endogenous TM cells were actively replicating. Since segmental flow exists in mouse eyes (Swaminathan SS, et al. IOVS 2009;52:ARVO E-Abstract 6620), it is possible that more TMSCs localize in some regions of the TM than other regions. We did not compare the same region of the mouse eyes at different time points. Comparatively, the mouse TM cells in the eyes with the fibroblast-transplantation were stimulated to divide at 1 week represented by BrdU incorporation (Fig. 8C). FACS sorted DiO+ green cells from fibroblast-injected TM tissues expressed both human and mouse TBP (Fig. 6). This phenomenon may be related to the increased dividing of resident mouse cells after fibroblast injection and the dividing cells fused together. Fibroblast transplantation can also result in cell apoptosis as shown in Figure 5 with a positive TUNEL result. Inflammatory response related to fibroblast injection may induce cell apoptosis. This suggests that stem cells are a more appropriate option for cell-based therapy than fibroblasts.
Our demonstration of a resident stem cell population in the TM and their slow-cycling property raises questions as to their roles in vivo. It seems possible that, like dermis, intestine, or corneal epithelium, the TM is a self-renewing tissue, supported by a resident population of stem cells. Such a hypothesis is supported by a recent study of gene expression in TM showing expression of cell cycle and proliferation related genes.33 Future studies elucidating factors controlling TMSC proliferation in vivo might allow development of a pharmacological approach focusing on TM repopulation in eyes with increased intraocular pressure.
An exciting potential application of TMSCs is developing cell-based therapy for glaucoma. The ability of TMSCs introduced into the anterior chamber to home to the TM and adopt a TM phenotype supports the idea that the TM in eyes with high IOP may be restored via such an approach. With the ability to alter the cellular composition of the TM, it will be possible to investigate the mechanism by which aqueous outflow is controlled by the metabolic activity of the TM cells. Information revealed by such studies can point the way to design a cell based-therapy approach to regulate aqueous outflow through the TM.
Acknowledgments
The authors thank Nancy Zurowski for her help in cell sorting, Kira Lathrop for her help in imaging, and James L. Funderburgh for advice and help in preparing the manuscript.
Footnotes
Supported by an anonymous philanthropic donation for stem cells and glaucoma research (YD); NIH Grants EY016415 (James Funderburgh) and P30-EY008098; Eye and Ear Foundation (Pittsburgh, Pennsylvania); and Research to Prevent Blindness.
Disclosure: Y. Du, P; H. Yun, None; E. Yang, None; J.S. Schuman, P
References
- 1. Alvarado J, Murphy C, Polansky J, Juster R. Age-related changes in trabecular meshwork cellularity. Invest Ophthalmol Vis Sci. 1981; 21: 714–727. [PubMed] [Google Scholar]
- 2. Tripathi RC. Pathologic anatomy in the outflow pathway of aqueous humour in chronic simple glaucoma. Exp Eye Res. 1977; 25 (suppl); 403–407. [DOI] [PubMed] [Google Scholar]
- 3. Lutjen-Drecoll E. Morphological changes in glaucomatous eyes and the role of TGFbeta2 for the pathogenesis of the disease. Exp Eye Res. 2005; 81: 1–4. [DOI] [PubMed] [Google Scholar]
- 4. He Y, Leung KW, Zhang YH, et al. Mitochondrial complex I defect induces ROS release and degeneration in trabecular meshwork cells of POAG patients: protection by antioxidants. Invest Ophthalmol Vis Sci. 2008; 49: 1447–1458. [DOI] [PubMed] [Google Scholar]
- 5. Alvarado J, Murphy C, Juster R. Trabecular meshwork cellularity in primary open-angle glaucoma and nonglaucomatous normals. Ophthalmology. 1984; 91: 564–579. [DOI] [PubMed] [Google Scholar]
- 6. Clark AF, Brotchie D, Read AT, et al. Dexamethasone alters F-actin architecture and promotes cross-linked actin network formation in human trabecular meshwork tissue. Cell Motil Cytoskeleton. 2005; 60: 83–95. [DOI] [PubMed] [Google Scholar]
- 7. Read AT, Chan DW, Ethier CR. Actin structure in the outflow tract of normal and glaucomatous eyes. Exp Eye Res. 2006; 82: 974–985. [DOI] [PubMed] [Google Scholar]
- 8. Hoare MJ, Grierson I, Brotchie D, Pollock N, Cracknell K, Clark AF. Cross-linked actin networks (CLANs) in the trabecular meshwork of the normal and glaucomatous human eye in situ. Invest Ophthalmol Vis Sci. 2009; 50: 1255–1263. [DOI] [PubMed] [Google Scholar]
- 9. Buller C, Johnson DH, Tschumper RC. Human trabecular meshwork phagocytosis. Observations in an organ culture system. Invest Ophthalmol Vis Sci. 1990; 31: 2156–2163. [PubMed] [Google Scholar]
- 10. Du Y, Roh DS, Mann MM, Funderburgh ML, Funderburgh JL, Schuman JS. Multipotent stem cells from trabecular meshwork become phagocytic TM cells. Invest Ophthalmol Vis Sci. 2012; 53: 1566–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mimura T, Joyce NC. Replication competence and senescence in central and peripheral human corneal endothelium. Invest Ophthalmol Vis Sci. 2006; 47: 1387–1396. [DOI] [PubMed] [Google Scholar]
- 12. McKenna KC, Xu Y, Kapp JA. Injection of soluble antigen into the anterior chamber of the eye induces expansion and functional unresponsiveness of antigen-specific CD8+ T cells. J Immunol. 2002; 169: 5630–5637. [DOI] [PubMed] [Google Scholar]
- 13. Du Y, Carlson EC, Funderburgh ML, et al. Stem cell therapy restores transparency to defective murine corneas. Stem Cells. 2009; 27: 1635–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Du Y, Funderburgh ML, Mann MM, SundarRaj N, Funderburgh JL. Multipotent stem cells in human corneal stroma. Stem Cells. 2005; 23: 1266–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Blanpain C, Horsley V, Fuchs E. Epithelial stem cells: turning over new leaves. Cell. 2007; 128: 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Krishnamurthy K, Wang G, Rokhfeld D, Bieberich E. Deoxycholate promotes survival of breast cancer cells by reducing the level of pro-apoptotic ceramide. Breast Cancer Res. 2008; 10: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med. 1996; 183: 1797–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rizvi AZ, Swain JR, Davies PS, et al. Bone marrow-derived cells fuse with normal and transformed intestinal stem cells. Proc Natl Acad Sci U S A. 2006; 103: 6321–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reagan MR, Kaplan DL. Concise review: Mesenchymal stem cell tumor-homing: detection methods in disease model systems. Stem Cells. 2011; 29: 920–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu Y, Zhao RC. The role of chemokines in mesenchymal stem cell homing to myocardium. Stem Cell Rev. 2012; 8: 243–250. [DOI] [PubMed] [Google Scholar]
- 21. Kang SK, Shin IS, Ko MS, Jo JY, Ra JC. Journey of mesenchymal stem cells for homing: strategies to enhance efficacy and safety of stem cell therapy. Stem Cells Int. 2012; 2012: 342968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen FM, Wu LA, Zhang M, Zhang R, Sun HH. Homing of endogenous stem/progenitor cells for in situ tissue regeneration: Promises, strategies, and translational perspectives. Biomaterials. 2011; 32: 3189–3209. [DOI] [PubMed] [Google Scholar]
- 23. Cotsarelis G, Cheng SZ, Dong G, Sun TT, Lavker RM. Existence of slow-cycling limbal epithelial basal cells that can be preferentially stimulated to proliferate: implications on epithelial stem cells. Cell. 1989; 57: 201–209. [DOI] [PubMed] [Google Scholar]
- 24. Dembinski JL, Krauss S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin Exp Metastasis. 2009; 26: 611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pece S, Tosoni D, Confalonieri S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010; 140: 62–73. [DOI] [PubMed] [Google Scholar]
- 26. Fong EL, Chan CK, Goodman SB. Stem cell homing in musculoskeletal injury. Biomaterials. 2011; 32: 395–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Beyth S, Borovsky Z, Mevorach D, et al. Human mesenchymal stem cells alter antigen-presenting cell maturation and induce T-cell unresponsiveness. Blood. 2005; 105: 2214–2219. [DOI] [PubMed] [Google Scholar]
- 28. Ren G, Zhang L, Zhao X, et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell. 2008; 2: 141–150. [DOI] [PubMed] [Google Scholar]
- 29. Ghannam S, Bouffi C, Djouad F, Jorgensen C, Noel D. Immunosuppression by mesenchymal stem cells: mechanisms and clinical applications. Stem Cell Res Ther. 2010; 1: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Popp FC, Eggenhofer E, Renner P, et al. Mesenchymal stem cells can affect solid organ allograft survival. Transplantation. 2009; 87: S57–S62. [DOI] [PubMed] [Google Scholar]
- 31. Bigg HF, Wait R, Rowan AD, Cawston TE. The mammalian chitinase-like lectin, YKL-40, binds specifically to type I collagen and modulates the rate of type I collagen fibril formation. J Biol Chem. 2006; 281: 21082–21095. [DOI] [PubMed] [Google Scholar]
- 32. Llobet A, Gasull X, Gual A. Understanding trabecular meshwork physiology: a key to the control of intraocular pressure? News Physiol Sci. 2003; 18: 205–209. [DOI] [PubMed] [Google Scholar]
- 33. Yu M, Sun J, Peng W, et al. Protein expression in human trabecular meshwork: downregulation of RhoGDI by dexamethasone in vitro. Mol Vis. 2010; 16: 213–223. [PMC free article] [PubMed] [Google Scholar]