

Abstract
Protein sulfinic acids are formed by the reaction of reactive oxygen species with protein thiols. Sulfinic acid formation has long been considered an irreversible state of oxidation and is associated with high cellular oxidative stress. Increasing evidence, however, indicates that cysteine is oxidized to sulfinic acid in cells to a greater extent, and is more controlled, than first thought. The discovery of sulfiredoxin has demonstrated that cysteine sulfinic acid can be reversed, pointing to a vast array of potential implications for redox biology. Identification of the site of protein sulfinylation is crucial in clarifying the physiological and pathological effects of post-translational modifications. Currently, the only methods for detection of sulfinic acids involve mass spectroscopy and the use of specific antibodies. However, these methodologies are not suitable for proteomic studies. Herein, we report the first probe for detection of protein sulfinylation, NO-Bio, which combines a C-nitroso warhead for rapid labelling of sulfinic acid with a biotin handle. Based on this new tool, we developed a selective two-step approach. In the first, a sulfhydryl-reactive compound is introduced to selectively block free cysteine residues. Thereafter the sample is treated with NO-Bio to label sulfinic acids. This new technology represents a rapid, selective and general technology for sulfinic acid detection in biological samples. As proof of our concept, we also evaluated protein sulfinylation levels in various human lung tumour tissue lysates. Our preliminary results suggest that cancer tissues generally have higher levels of sulfinylation in comparison to matched normal tissues. A new ability to monitor protein sulfinylation directly should greatly expand the impact of sulfinic acid as a post-translational modification.
INTRODUCTION
Reactive oxidant species derived from oxygen or nitrogen (RNOS) were originally notorious for indiscriminately oxidizing various cellular components and for promoting aging and a broad range of pathologies. By contrast, research in the last two decades has shown that low levels of RNOS regulate basic cellular processes including growth, differentiation, and cell migration.1,2 Protein-thiols (SH) are the main target of RNOS-dependent signaling.3 The fine oxidation of specific cysteine (Cys) residues has emerged as a molecular switch for the modulation of protein function and is similar in effect to enzyme-assisted post-translational modifications (PTMs).4 In addition to the well-known disulfide, a variety of products may result from oxidation of thiols, but the most important are sulfenic acids (SOH), sulfinic acids (SO2H), and sulfonic acids (SO3H).5 The development of redox-probes for monitoring RSOH has unequivocally revealed that protein sulfenylation modulates protein activity directly or through the formation of disulfide bonds.6 Persistent lack of efficient tools for tracking SO2H, however, has confined this PTM to a minor role. Since common cellular reductants do not reduce Cys-SO2H, protein sulfinylation was long considered merely a marker of oxidative stress, though mounting evidence indicates that hyperoxidation to SO2H is a more controlled event than previously thought. In fact, increasing number of proteins have been shown to be regulated by selective sulfinylation, including matrilysin, nitrile hydratase, and the Parkinson’s disease protein, DJ-1.7 The best characterized example of modulation of protein activity via sulfinylation, however, occurs in the Peroxiredoxin (Prx) family. Over-oxidation of the catalytic Cys leads to deactivation of peroxidase activity and the formation of high-molecular-weight aggregates, which exhibit molecular chaperone activity.8,9 Prx inactivation is then reversed by Sulfiredoxin (Srx), an ATP-dependent protein that specifically reduces Cys-SO2H in Prxs.10 Furthermore, it has been shown that transient sulfinylation of Prx represents a universal marker for circadian rhythms along all three domains of life.11 The discovery of Srx suggests a more fundamental role for Cys-SO2H, which may constitute an additional layer of redox regulation.12 Finally, in addition to cysteine oxidation by ROS, an enzyme-mediated oxidation has recently emerged. Several plant cysteine oxidases have been identified that can selectively oxidize the penultimate cysteine of transcription factors to SO2H and thereby control the life span of these proteins.13 Accordingly, sulfinylation of specific Cys residue has drawn wide attention as a novel PTM responsible for regulation of protein function. Studies of the role of Cys-SO2H, however, have been hampered by the technically challenging nature of selective assays for such oxoforms, and mass spectroscopy remains the main tool for monitoring this PTM.14 Although SO2H shows higher stability than SOH, mass analyses may introduce a high percentage of artifacts. Moreover, the fact that persulfide modification has the same nominal mass shift of 32 Da raises additional concerns. Antibodies able to detect hyperoxidized forms of specific proteins are known15 but, even without taking in account lack of specificity, are unsuited to global profiling studies.
RESULT AND DISCUSSION
We strongly believe that only the development of chemical probes capable of selectively trapping SO2H will allow a clear elucidation of the role of protein sulfinylation. In this connection, we recently developed chemoselective Sulfinic acid Nitroso Ligation (SNL).16 The addition of SO2H to C-nitroso compounds has been known for more than a century; however, the resulting adduct is base-labile (Figure S1). In order to trap this unstable species, we have incorporated an electrophilic center (Figure 1) in the ortho-position of a nitroso-benzene derivative (1). The transient oxyanion (2) reacts with the ester by intramolecular trans-esterification to form a stable benzisoxazolone (3). Basing our work upon this idea, we have synthesized a class of C-nitroso compounds that show fast reactivity with low molecular weight SO2H. These reagents do not react with other biologically relevant nucleophiles aside from thiols, which, however, do not form stable adducts (Figure S2).
Figure 1.
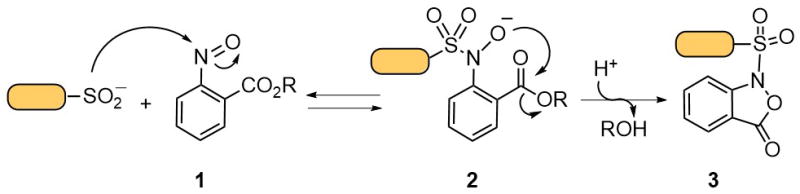
Chemoselective labeling of sulfinic acid with aryl-nitroso compounds.
Using SNL for labeling protein sulfinic acids
Encouraged by these results, we employed SNL to develop chemical probes for detection of protein sulfinylation. First of all, we explored the ability of NO-Ph (Figure 2), the C-nitroso derivative that has shown the best reactivity, to modify SO2H within the double mutant (C64,82S) of the thiol peroxidase Gpx3 from yeast.17 In the presence of H2O2, Gpx3 forms an intramolecular disulfide bond through sulfenylation of catalytic C36, followed by condensation with the resolving C82. Mutation of C82 to serine stabilizes transient Cys36-SOH, allowing its further controlled oxidation to SO2H (see Supporting Information).
Figure 2.
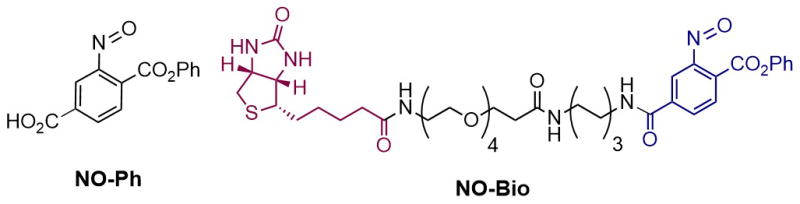
Nitroso probes for selective labeling of protein sulfinic acids.
Incubation of NO-Ph with C64,82S Gpx3-SO2H (22772 Da) yields the expected sulfonamide adduct (22949 Da) as confirmed by ESI-LC/MS analysis (Figure 3A). Our preliminary experiments with small molecules have shown that thiols react with C-nitroso compounds to yield an unstable sulfenamide adduct, which is cleaved by reaction with a second thiol (Figure S2). In order to confirm these results with protein-SH, we treated fully reduced C64,82S Gpx3 (22740 Da) with NO-Ph, followed by incubation with DTT. Surprisingly, ESI-LC/MS revealed the formation of a stable adduct with a mass of 22935 Da (Figure 3B). Alkylation of the Cys residue with N-ethylmaleimide (NEM), conversely, prevented adduct formation (Figure 3C). Even considering that DTT was unable to cleave the sulfenamide formed by the addition of NO-Ph to Cys36, the detected mass increase (Δm = 195) does not correspond to the expected adduct (Δm = 177). Generally, addition of thiol to C-nitroso aryl compound yields an unstable semimercaptale, which can react with a second thiol molecule or undergo rearrangement to form a more stable sulfinamide (Figure S3).18 Spontaneous rearrangement occurs via dissociation of a hydroxyl anion and formation of a cationic nitrenium ion intermediate, which is later hydrolyzed by water. Following the same pathway, the addition of NO-Ph to C64,82S Gpx3-SH would form a sulfinamide adduct with a mass increase of 195 Da, which corresponds exactly to our observations. Acidic environment usually favors semimercaptale rearrangement. With NO-Ph, however, once the benzisoxazolone is generated, the rearrangement appears to occur even at neutral pH, probably because the carboxylate group is much more prone to dissociate from the nitrogen atom than is the hydroxyl anion (Figure S4). The formation of the rearranged sulfinamide would also explain why the adduct was not reduced by DTT. Since sulfinamide formation was never observed with low molecular weight thiols,16 we wondered why the rearrangement occurred with protein-SH. We speculated that, in such cases, the attack of a second thiol molecule on the transient sulfenamide is generally faster than the rearrangement of the latter. Once the sulfenamide is formed, however, the attack of a second Cys-SH would be precluded with the use of C64,82S Gpx3-SH because of steric hindrance. To verify this hypothesis, we tested the reactivity of NO-Ph toward C64S Gpx3, which has both redox-active and resolving cysteines. As expected, the incubation of C64S Gpx3 with NO-Ph exclusively promoted the formation of the internal disulfide (Figure 3D). The presence of the resolving Cys, which can easily interact with the sulfenamide adduct formed with catalytic Cys, prevents the rearrangement of the latter. This result suggests that the formation of stable sulfinamide and disulfide reflects competitive reaction pathways influenced by kinetic factors (Figure S5). As additional proof, we incubated 2-methyl 2-propanethiol with an excess of NO-Ph. In this case, we speculated that, because the interaction of two molecules of thiols would be more hampered for steric hindrance, sulfenamide rearrangement would be facilitated. As anticipated, LC-MS analyses showed formation of the expected sulfinamide adduct (Figure S6).
Figure 3.

ESI-LC/MS spectra of (A) C64,82S Gpx3-SO2H, (B) C64,82S Gpx3-SH, (C) C64,82S Gpx3-S-NEM and (D) C64S Gpx3-SH before and after treatment with NO-Ph. Each Gpx3 form was incubated with NO-Ph (40 equivalt.) for 1 h at room temperature in PBS pH 7.4.
Selective block of free cysteine residues
Sulfenamide rearrangement apparently limits the use of SNL for protein sulfinylation detection. As the experiment with NEM suggests, however, protection of the free cysteines can be employed to prevent formation of non-reducible adducts with hindered thiols. In fact, many chemical methods for detection of specific thiol modifications (e.g., S-nitrosylation) involve selective blocking of reduced thiols.19 The success of these assays relies on the selectivity of the thiol-blocking step and the reagent’s efficiency in fully protecting free thiols without cross-reacting with SO2H. We evaluated the reactivity of several thiol-blocking reagents toward C64,82S Gpx3-SO2H. When we used a large excess of common alkylating agents such as NEM or iodoacetamide (IAM), ESI-LC/MS analyses detected small but significant amounts of alkylated sulfinic acid (Figures S7A and S7B). Although this result may seem unexpected, the reaction of low molecular weight SO2H with Michael acceptors and a-halo carbonyl compounds has been reported.20 Conversely, sulfhydryl reactive compounds that promote mixed-disulfide formations, such as 2,2′-dipyridyl disulfide (DPS) and S-methyl methanethiosulfonate (MMTS), showed no cross-reactivity toward SO2H (Figures S7C and S7D). Next, we examined whether protection of free Cys residues as disulfides was sufficient to prevent cross-reactivity with NO-Ph. After C64S,82S Gpx3-SH was pre-incubated with DPS (Figure S8A) or MMTS (Figure S8B), the excess of thiol-blocking reagents was removed and the protein was incubated with NO-Ph. ESI-LC/MS analyses confirmed that both DPS and MMTS efficiently blocked formation of the sulfinamide adduct with the reduced protein.
Design and synthesis of NO-Bio
Having established that SNL can be efficiently employed for labeling protein SO2H, we designed and synthesized NO-Bio (Figure 2). The new chemical probe combines the C-nitroso warhead (blue) with a biotin handle (violet), which allows detection of protein sulfinylation in biological samples. The synthesis of NO-Bio (Figure S9), which is described in detail in the Supporting Information, involved the coupling of commercially available Biotin-PEG4-NHS with a diamino-linker, N-tert-Butoxycarbonyl-1,6-hexanediamine. The protected amino group was then cleaved by TFA treatment, and the generated primary amine was coupled with the N-succimidyl ester of NO-Ph to yield NO-Bio, which was purified by reverse-phase HPLC.
Development of a chemical approach for protein sulfinic acid detection
As Figure 4A shows, protein sulfinylation could be selectively detected by a two-step method. In the first, a sulfhydryl-reactive compound (DPS or MMTS) is introduced to selectively block free Cys residues. Thereafter the sample is treated with the biotin-tag probe, NO-Bio, to label sulfinic acids. We tested this approach using recombinant DJ-1 as model. The Parkinson’s associate protein has a conserved Cys residue, C106, which is extremely sensitive to oxidative stress and tends to form a stable SO2H. Many studies demonstrate that DJ-1 protects cells against oxidative stress-mediated apoptosis through the formation of C106-SO2H.21,22 In addition, DJ-1 contains two other free Cys residues, C46 and C53, which are not redox-active. Though C53 is not modified by ROS, it is still very reactive toward electrophiles.23 Accordingly, DJ-1 represents an excellent model for testing the selectivity of our strategy. Reduced or oxidized WT DJ-1 was incubated with DPS, following by treatment with NO-Bio. As shown in Figure 4B, mass analysis clearly confirmed the selective modification of the solely oxidized DJ-1. Interestingly, DPS promoted the formation of an internal disulfide between C46 and C53. We speculated that DPS would first react with the highly solvent-exposed C53 to yield a mixed-disulfide. Later, the relatively more deeply buried C46 would attack the active disulfide with consequent generation of an internal disulfide bond (Figure S10). Selective protection of the free Cys can be achieved using MMTS as well (Figure S11). However, our results indicate that MMTS reacts with thiols at a relatively slower rate than does DPS. In fact, small amounts of Cys-SO2H were detected even in the reduced sample, which indicates that C106 was partially oxidized during thiol blocking. Although addition of EDTA in the buffer prevented this unwanted oxidation, we opted to use the more efficient DPS in all subsequent experiments. The biotin handle of the probe allows visualization of the labeled proteins by streptavidin blotting. Therefore, the selectivity of NO-Bio labeling was also confirmed by Western blot analysis. Reduced or oxidized DJ-1 was treated with DPS, followed by incubation with NO-Bio. The reactions were then subjected to SDS-PAGE and analyzed by streptavidin blotting. Treatment of oxidized DJ-1 with NO-Bio afforded selective protein labeling, while DJ-1 was not detected at all by streptavidin blotting in the absence of the oxidant, demonstrating the specificity of our chemoselective approach (Figure S12). NO-Bio showed also higher sensitivity in comparison to a commercially available antibody against hyperoxidized DJ-1 (Figure S13), allowing detection of sulfinylated DJ-1 at relative low concentrations.
Figure 4.

Labeling of DJ-1 sulfinic acid with NO-Bio.
DJ-1 possesses a highly conserved G18 residue, which facilitates the ionization of C106, reduces its pKa, and helps stabilize C106-SO2H. Small changes in this position can drastically influence the oxidative properties of C106.24 For example, the E18D DJ-1 mutant has a lower propensity to form SO2H, but the structurally similar E18N mutant shows an increased oxidation propensity thanks to a strong stabilization of C106-SO2H. We evaluated the sensitivity of NO-Bio, probing the different oxidation propensities of various DJ-1 mutants including C106S DJ-1, which does not contain a redox-active cysteine. Each DJ-1 variant (WT, E18N, E18D and C106S) was exposed to H2O2, treated with DPS, and finally incubated with NO-Bio. Western blot analysis was consistent with expected results (Figure 4c). E18N DJ-1 showed a higher fraction of sulfinylation, even in the absence of H2O2. The sulfinylation level of the E18D mutant exposed to oxidative stress, in contrast, was almost negligible. It is worth noting that C106S DJ-1 treated with H2O2 was not detected by streptavidin blotting, confirming that only C106 is able to form SO2H under relatively mild oxidation conditions.
NO-Bio detects sulfinic acid-modified proteins in cell lysate
Having established the specificity and sensitivity of our two-step approach in homogenous protein solutions, we next investigated whether NO-Bio could detect protein-SO2H in a complex, unfractionated cell lysate. To this end, we tested our optimized chemistry in a whole human cervical cancer (HeLa) cell extract, which was obtained by lysing the cells in modified RIPA buffer containing Catalase and DTT. The reducing lysis buffer prevents further oxidation of SO2H and maintains free Cys in the reduced form, avoiding overestimation of protein sulfinylation. Figure 5A shows an HRP-streptavidin western blot, which indicates that robust levels of protein SO2H can be detected under normal conditions. To demonstrate that DPS efficiently trapped all free thiols and therefore that the streptavidin blot revealed only sulfinylated proteins, we employed iodoacetyl-PEG2-biotin (IAM-Bio). The biotinylated reagent is able to alkylate thiols such as free Cys residues. Pre-treatment of the sample with DPS completely abrogated the IAM-Bio-dependent signal (Figure 5A, Lane 3), which indirectly proved that NO-Bio reacted only with protein SO2H. Next, we determined whether our chemical approach could detect increases in protein sulfinylation in human cell culture. HeLa cells were incubated with increasing amounts of H2O2 for 15 minutes (this time point was chosen after a preliminary time-dependent experiment – Figure S14A), lysed, and then labeled as described above. Western blot analysis showed that the level of protein sulfinylation was increased by H2O2 in a dose-dependent fashion (Figure S14B). Taken together, these results confirm that SO2H is stable enough to be successfully detected in cell lysates and does not require in vivo labeling.
Figure 5.
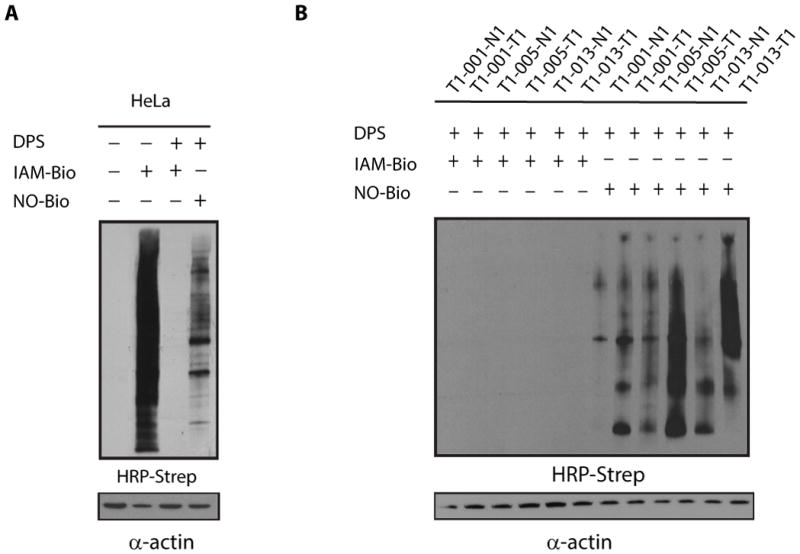
Reactivity of No-Bio in cell lysates - 5 μg protein loaded per lane - (A). Analysis of protein sulfinylation in human lung tumor tissue lysates - 1 μg protein loaded per lane - (B). Legend: T1-001-T1 Papillary adenocarcinoma; T1-001-N1 Matched normal tissue; T1-005-T1 Adenocarcinoma; T1-005-N1 Matched normal tissue; T1-013-T1 Adeno-squamous cell carcinoma; T1-013-N1 Matched normal tissue.
Protein sulfinylation levels in human lung cancer
In order to show that our method can be applied to more complex biological questions, we performed comparative sulfinic acid profiling in human lung tumor tissue. For these experiments, protein sulfinylation was characterized by western blot analysis of whole-cell lysates. Our results showed a highly variable presence of SO2H among the three patient tumor tissue samples (Figure 5B). All three tumor tissue (papillary adenocarcinoma, adenocarcinoma, and adeno-squamous cell carcinoma) exhibited significant increase in the extent of SO2H modifications vs. matched normal tissue. Although the number of samples was too small to draw broad conclusions, these initial observations suggest that elevated levels of SO2H could be used as a cancer marker.
CONCLUSIONS
In summary, sulfinic acid nitroso ligation allows quick conversion of sulfinic acids into sulfonamide adducts. We designed and synthesized a nitroso-based probe, NO-Bio, which can be used to label protein-SO2H. In model protein sulfinic acids, this compound yields stable products. We developed a two-step chemical approach that selectively labels protein sulfinic acid in vitro without cross-reactivity with thiols. Furthermore, NO-Bio was able to detect global increases in protein SO2H modification under oxidizing cellular conditions or in cancer cell lines. To the best of our knowledge, this is the first chemical approach that allows selective protein-sulfinylation detection, making NO-Bio a valuable new tool for monitoring changes in cysteine oxidation and should find a wide variety of applications for the study of biological processes. In addition, the biotin tag provides an opportunity for the enrichment and proteomic analysis of oxidized proteins. These studies are currently underway and will be reported in due course.
METHODS
For protein expression and purification, generation of protein sulfinic acids, synthesis of NO-Bio, screening of the thiol-blocking reagent and mass spectrometry, please see the Supporting Information. Human cancer lung tissue lysates were purchased from Protein Biotechnologies Inc.
General reactivity of recombinant proteins toward NO-Ph (Figure 3)
25 μM of recombinant protein (C64,82S Gpx3-SO2H, C64,82S Gpx3-SH, C64,82S Gpx3-SNEM or C64S Gpx3) was incubated at room temperature in the presence of NO-Ph (500 μM) in 100 mM PBS pH 7.4. After 1 hour, the reaction was quenched by passage through one Micro Bio-Spin P-30 column pre-equilibrated with ammonium bicarbonate (50 mM, pH 8.0) for analysis by ESI-LC/MS.
General SDS-PAGE and Western blot procedures
Protein samples were resolved by SDS-PAGE using Mini-Protean TGX 4–15% Tris-Glycine gels (BioRad) and transferred to a polyvinylidene difluoride (PVDF) membrane (BioRad). After transfer, the PVDF membrane was blocked with 5% BSA in TBST for 1 hour at room temperature. The membrane was washed with TBST (3X) and immunoblotting was performed with the following primary and secondary antibodies at the indicated dilutions: HRP-streptavidin (GE Healthcare, 1:80000), Actin (Santa Cruz Biotechnology, 1:1000), PARK7/DJ-1 (Abcam, 1:1000), oxidized PARK7/DJ-1 (Abcam, 1:1000), rabbit anti-goat IgG-HRP (Invitrogen, 1:2000 – 1:50000), and rabbit anti-mouse IgG-HRP (Invitrogen, 1:20000 – 1:50000). The PVDF membranes were washed with TBST (3X) and developed with ECL Plus chemiluminescence (Pierce) and imaged by film.
NO-Bio labeling of DJ-1 (Figure 4)
Each DJ-1 form (WT, E18N, E18D and C106S) was buffer exchanged using Micro Bio-Spin P-30 column pre-equilibrated with 50 mM HEPES, 100 mM KCl pH 7.4. Three samples of each DJ-1 form (25 μM) were then treated with 5 equivalents of H2O2 or H2O (control samples) in ice for 30 minutes. The reactions were quenched adding 2 mM of DTT. Each sample was incubated for 30 min at room temperature and then passed through one Micro Bio-Spin P-30 column. 40 equivalents of DPS were then added at room temperature. After 1 hour, each sample was quenched by passage through one Micro Bio-Spin P-30 column pre-equilibrated with 100 mM PBS pH 7.4. 10 equivalents of NO-Bio or DMSO (control samples) were then added. After 1 hour, the reactions were quenched adding non-reducing 2x Laemmli buffer. The resulting samples were subjected to SDS-PAGE and Western blot analyses as described above. Equal protein loading was verified by α-DJ1 antibody (Abcam).
Cell Culture
HeLa cell line was obtained from American Type Culture Collection. HeLa cell line were grown in DMEM media supplemented 10% FBS (Invitrogen), 1% penicillin-streptomycin (Invitrogen), 1% of glutagro (Corning) and 1× non-essential amino acids (Invitrogen) at 37 °C in a humidified atmosphere of 5% CO2. For H2O2 stimulation, HeLa cells were plated in a 6-well plate. Once the cells reached 90% of confluence, they were washed with PBS. Cell were exposed to increasing concentration of H2O2 (0.2, 1, 2 mM) for 15 min, and then washed with PBS (3×).
General procedure for lysate preparation
Cell were harvested in modified RIPA buffer [50 mM triethanolamine, pH 7.4, 150 NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 5 mM DTT, 200 U/mL catalase (sigma) and 1x EDTA-free complete mini protease inhibitors (Roche)]. After 20 min incubation on ice with frequent mixing, unlysed cell fragments were removed by centrifugation at 14,000 g at 4 C for 20 min. Protein concentration was measured by BCA assay.
Labeling of protein SO2H in lysate using NO-Bio (Figure 5)
Cell lysate (1 mg/mL) was buffer exchanged using Micro Bio-Spin P-6 column pre-equilibrated with 100 mM PBS, 100 mM NaCl pH 7.4. Free thiols were trapped by incubation with 2 mM of DPS (in DMSO). Final reaction volumes were 0.1 mL, and the samples were incubated for 30 min at r.t. Reactions were quenched by passage through one Micro Bio-Spin P-30 column pre-equilibrated with 100 mM PBS pH 7.4. Protein sulfinylation was assayed adding 250 μM of NO-Ph. After 30 min, reactions were quenched adding non-reducing 2x Laemmli buffer. Three additional controls included untreated lysate, lysate incubated with 250 μM of iodoacetyl-LC-biotin (Thermo) and lysate treated with DPS following by incubation with 250 μM of iodoacetyl-LC-biotin. The samples treated with iodoacetyl-LC-biotin were incubated for 30 min at r.t in the dark. Reactions were quenched via the addition of non-reducing 2x Laemmli buffer. The resulting samples were subjected to SDS-PAGE and Western blot analyses as described above. Equal protein loading was verified by α-actin antibody.
Supplementary Material
Acknowledgments
This work was supported by the Camille Henry Dreyfus Teacher Scholar Award (K.S.C) and the American Heart Association Scientist Development Award (0835419N, K.S.C.).
Footnotes
Supplementary methods, Figures S1–S14. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Holmström KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014;15:411–421. doi: 10.1038/nrm3801. [DOI] [PubMed] [Google Scholar]
- 2.Miller EW, Chang CJ. Fluorescent probes for nitric oxide and hydrogen peroxide in cell signaling. Curr Opin Chem Biol. 2007;11:620–625. doi: 10.1016/j.cbpa.2007.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dickinson BC, Chang CJ. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat Chem Biol. 2011;7:504–511. doi: 10.1038/nchembio.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klomsiri C, Karplus PA, Poole LB. Cysteine-Based Redox Switches in Enzymes. Antioxid Redox Signal. 2011;14:1065–1077. doi: 10.1089/ars.2010.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reddie KG, Carroll KS. Expanding the functional diversity of proteins through cysteine oxidation. Curr Opin Chem Biol. 2008;12:746–754. doi: 10.1016/j.cbpa.2008.07.028. [DOI] [PubMed] [Google Scholar]
- 6.Gupta V, Carroll KS. Sulfenic acid chemistry, detection and cellular lifetime. Biochim Biophys Acta. 2014;1840:847–75. doi: 10.1016/j.bbagen.2013.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lo Conte M, Carroll KS. The Redox Biochemistry of Protein Sulfenylation and Sulfinylation. J Biol Chem. 2013;288:26480–26488. doi: 10.1074/jbc.R113.467738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woo HA, Chae HZ, Hwang SC, Yang KS, Kang SW, Kim K, Rhee SG. Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science. 2003;300:653–656. doi: 10.1126/science.1080273. [DOI] [PubMed] [Google Scholar]
- 9.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 10.Lowther WT, Haynes AC. Reduction of cysteine sulfinic acid in eukaryotic, typical 2-Cys peroxiredoxins by sulfiredoxin. Antioxid Redox Signal. 2011;15:99–109. doi: 10.1089/ars.2010.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edgar RS, Green EW, Zhao Y, van Ooijen G, Olmedo M, Qin X, Xu Y, Pan M, Valekunja UK, Feeney KA, Maywood ES, Hastings MH, Baliga NS, Merrow M, Millar AJ, Johnson CH, Kyriacou CP, O’Neill JS, Reddy AB. Peroxiredoxins are conserved markers of circadian rhythms. Nature. 2012;485:459–464. doi: 10.1038/nature11088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacob C, Holme AL, Fry FH. The sulfinic acid switch in proteins. Org Biomol Chem. 2004;2:1953–1956. doi: 10.1039/b406180b. [DOI] [PubMed] [Google Scholar]
- 13.Weits DA, Giuntoli B, Kosmacz M, Parlanti S, Hubberten HM, Riegler H, Hoefgen R, Perata P, van Dongen JT, Licausi F. Plant cysteine oxidases control the oxygen-dependent branch of the N-end-rule pathway. Nat Commun. 2014;5:3425. doi: 10.1038/ncomms4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Witze ES, Old WM, Resing KA, Ahn NG. Mapping protein post-translational modifications with mass spectrometry. Nat Methods. 2007;4:798–806. doi: 10.1038/nmeth1100. [DOI] [PubMed] [Google Scholar]
- 15.Woo HA, Kang SW, Kim HK, Yang KS, Chae HZ, Rhee SG. Reversible oxidation of the active site cysteine of peroxiredoxins to cysteine sulfinic acid. Immunoblot detection with antibodies specific for the hyperoxidized cysteine-containing sequence. J Biol Chem. 2003;278:47361–47364. doi: 10.1074/jbc.C300428200. [DOI] [PubMed] [Google Scholar]
- 16.Lo Conte M, Carroll KS. Chemoselective Ligation of Sulfinic Acids with Aryl-Nitroso Compounds. Angew Chem Int Ed Engl. 2012;51:6502–6505. doi: 10.1002/anie.201201812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paulsen CE, Carroll KS. Chemical dissection of an essential redox switch in yeast. Chem Biol. 2009;16:217–225. doi: 10.1016/j.chembiol.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 18.Kazanis S, McClelland RA. Electrophilic intermediate in the reaction of glutathione and nitroso arenes. J Am Chem Soc. 1992;114:3052–3059. [Google Scholar]
- 19.Leonard SE, Carroll KS. Curr Opin Chem Biol. 2011;15:88. doi: 10.1016/j.cbpa.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 20.Drabowicz J, Kielbasinski P, Mikolajczyk M. In: Sulphinic Acids, Esters and their Derivatives. Patai S, editor. John Wiley & Sons Inc; New York: 1990. p. 351. [Google Scholar]
- 21.Wilson MA. The role of cysteine oxidation in DJ-1 function and dysfunction. Antioxid Redox Signal. 2011;15:111–122. doi: 10.1089/ars.2010.3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waak J, Weber SS, Görner K, Schall C, Ichijo H, Stehle T, Kahle PJ. Oxidizable residues mediating protein stability and cytoprotective interaction of DJ-1 with apoptosis signal-regulating kinase 1. J Biol Chem. 2009;284:14245–14257. doi: 10.1074/jbc.M806902200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Girotto S, Sturlese M, Bellanda M, Tessari I, Cappellini R, Bisaglia M, Bubacco L, Mammi S. Dopamine-derived quinones affect the structure of the redox sensor DJ-1 through modifications at Cys-106 and Cys-53. J Biol Chem. 2009;287:18738–18749. doi: 10.1074/jbc.M111.311589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blackinton J, Lakshminarasimhan M, Thomas KJ, Ahmad R, Greggio E, Raza AS, Cookson MR, Wilson MA. Formation of a stabilized cysteine sulfinic acid is critical for the mitochondrial function of the parkinsonism protein DJ-1. J Biol Chem. 2009;284:6476–6485. doi: 10.1074/jbc.M806599200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.