

Abstract
14-3-3 isoforms are shown to be upregulated or accumulated in the glial cells of autopsied patient brains affected with progressive multifocal leukoencephalopathy (PML), a demylinating disease caused by JC virus (JCV). The possible involvement of 14-3-3 in JCV tropism, however, has never been examined. To investigate a potential relationship between 14-3-3 isoforms and JCV in vitro, we examined the localization of six 14-3-3 isoforms in human neural progenitors and progenitor-derived astrocytes (PDAs) in cells without JCV exposure. The 14-3-3 zeta isoform was initially localized in the progenitor cytoplasm. When differentiation of progenitors into PDAs was induced, the zeta isoform translocated into the nucleus. However, upon JCV infection, progenitor cells exhibited an uncharacteristic 14-3-3 zeta nuclear presence in the few cells that became infected. JCV-treated PDAs showed elevated levels of 14-3-3 zeta compared to non-infected PDAs. Treatment with TGF-β1, a known stimulant of JCV multiplication, increased the overall number of infected cells and the otherwise absent nuclear presence of 14-3-3 zeta in progenitors. These results suggest that the nuclear presence of 14-3-3 zeta may play a role in JCV infection, and that the isoform may in part determine JCV susceptibility in these cell types.
Introduction
The human polyomavirus, JC virus (JCV), is the etiologic agent of the often fatal demyelinating disease known as progressive multifocal leukoencephalopathy (PML). JCV infects approximately 65% of the human population by early childhood. It persists but remains asymptomatic after its primary infection (Koralnik 2006). PML reactivation occurs during immunocompromised conditions and results in the cytolytic destruction of myelin-producing oligodendrocytes and the degeneration of the myelin sheath in white matter (Enam et al. 2004). The incidence of PML has risen appreciably since the 1980s due in part to the AIDS epidemic.
The principle cytopathological features of PML are evident in permissive astrocytes: infected astrocytes are enlarged with an indiscernible morphology and exhibit hyperchromatic nuclei (Enam et al. 2004; Radhakrishnan et al. 2003). Astrocytes allow for the expression of T-antigen and late gene expression that necessitate a lytic infection. They are not, however, considered a central source of virion production in vivo (Berger and Concha 1995; Del Valle et al. 2000). T-antigen is a viral regulatory protein encoded by the early gene transcripts and the late genes that encode capsid proteins for virion assembly including VP-1, VP-2, and VP-3 (Darbinyan et al. 2002).
Previous studies of JCV tropism in differentiated cell types have focused on the relationship between other fundamental cell-cycle components in an attempt to determine how transcription factors are manipulated to propagate a lytic infection (Bollag et al. 2006; Radhakrishnan et al. 2003). However, the relationship between the ubiquitous and critical regulatory protein 14-3-3 and JCV tropism in permissive cell types has not yet been considered.
Seven 14-3-3 isoforms regulate numerous cellular processes including differentiation, apoptosis, intracellular trafficking, signaling and ion channels (Berg et al. 2003). The cellular balance between these isoforms is considered essential for controlled cellular function at the risk of apoptosis or the manifestation of oncogenic properties (Niemantsverdriet et al. 2008). A few studies have focused on their immunolocalization during development as well as a function of Alzheimer’s disease and various prion diseases, including Creutzfeldt-Jakob disease (Baxter et al. 2002; Umahara et al. 2004).
The isoform zeta is of particular interest as it is heavily involved in Raf-1 activation (RAS/MAPK signaling cascades), which implicates it in neural development and regeneration (Luo et al. 1995; Van Der Hoeven et al. 2000). A recent study also showed that zeta also has a significant neuroprotective effect after NMDA toxicity is introduced in neural progenitors following delta9-tetrahydrocannabinol (THC) treatment (Chen et al. 2007). The zeta isoform’s neuroprotective effect in response to viral toxicity in neural progenitors, however, remains to be studied.
In this study, we first determined the differential localization of six 14-3-3 isoforms - gamma (γ), epsilon (ε), sigma (σ), theta (θ), zeta (ζ) and beta (β) – in neural progenitors and progenitor-derived astrocytes (PDAs). This preliminary study showed an absence of zeta in the nuclear fractions of neural progenitors, although it was present in cytoplasmic fractions of both progenitors and PDAs as well as PDA nuclear fractions. We then studied zeta’s translocation during progenitor differentiation to PDAs. Finally, we examined the potential correlation between the differential localization of the zeta isoform in terms of JCV-infection.
Materials and Methods
Progenitor and PDA cultures
Human CNS multi-potential progenitors were isolated from human fetal brain tissue according to the National Institutes of Health (NIH) guidelines (Messam et al. 2003). Cells were grown on poly-D-lysine-coated plasticware. To induce differentiation of PDAs, the progenitor cell culture medium was replaced with Eagle’s minimal essential medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 50 ug/ml gentamicin, and 100 IU/ml penicillin-streptomycin. Progenitor cultures grown at 10–40% confluence were at least 98% positive for nestin staining and did not express GFAP. Differentiation lasted 27 days, until 80% or more of the cells in culture were positive for glial fibrillary acidic protein (GFAP) by immunostaining. Treatments with TGF-β1 on JCV exposed cultures. Progenitor or PDA cultures were exposed to JCV (Mad-4 variant) at 100 hemagglutination units (HAU)/5 × 105 cells in a minimal covering of appropriate serum-free medium. For induction of TGF-β1 signaling experiments, cultures were treated with 5 ng/ml of recombinant TGF-β1 protein (R&D systems). After overnight JCV exposure, cultures were washed and replenished with appropriate fresh media, as well as TGF-β1 at concentrations identical to that in the original cultures. All JCV-exposed (controls) and JCV-exposed TGF-β1-treated cultures were processed 4 days post JCV exposure.
Nuclear extract and whole-cell extract preparation
Four days after JCV infection, nuclear fractions and whole-cell extracts were separated as previously described (Ravichandran and Major 2008). Protein concentrations were determined by Bio-Rad DC protein assay kit according to the manufacturer’s protocol.
Western Blot
5 to 7μg of protein per lane from nuclear extracts or whole-cell lysates were resolved by SDS-PAGE on 4–12% Bis-Tris NuPage Gels (Invitrogen) and electroblotted onto polyvinylidene difluoride (PVDF) membranes (Millipore). The transblotted membranes were blocked with TBS-TM (TBS-T containing 5% non-fat dry milk) for 2 h at RT, and probed with appropriate primary antibodies (anti-14-3-3 gamma, epsilon, zeta, and theta, Santa Cruz Biotechnology, Santa Cruz, California; anti-14-3-3 beta and sigma, IBL, Minneapolis, Minnesota; anti-VP-1, developed in our laboratory; anti-SV40 T-antigen, Oncogene, Cambridge, Massachusetts; anti-MEK-1, Abcam, Cambridge, Massachusetts; or anti-actin, Sigma-Aldrich, St. Louis, Missouri) in TBS-TM. Bound primary antibodies were detected using either an anti-rabbit or anti-mouse horseradish peroxidase-conjugated secondary included with the SuperSignal West Pico Chemiluminescent substrate kit (Pierce, Rockford, Illinois), according to the manufacturer’s protocol.
Immunofluorescence
Cultured cells were fixed in 4% paraformaldehyde for 15 min and permeabilized with 0.02% Triton for 5 min. After three 1x PBS washes, fixed cells were incubated with mouse monoclonal anti-GFAP (Dako, Glostrup, Denmark), or anti-Nestin (R&D Systems, Minneapolis, Minnesota) at 1:400 as well as rabbit polyclonals against six 14-3-3 isoforms (gamma, epsilon, beta, zeta, sigma, and theta) and VP-1 (monoclonal) at room temperature, 60 min. Three PBS washes followed. Rhodamine- or fluorescence-conjugated anti-rabbit and anti-mouse secondary (Jackson Immunoresearch, West Grove, Pennsylvania) at 1:200 each were incubated on the cells for 60 min at room temperature, followed by three PBS washes. Cells were mounted with a cover slip using Vectashield mounting medium (Vector Laboratories) containing 4′,6 diamidino-2-phenylindole (DAPI) to stain the cellular nuclei blue. A Zeiss Axiovert/Apotome microscope with Axiovision software was used to examine fluorescence.
Results
14-3-3 isoform-specific Western blot detection in neural progenitors and PDAs
The distribution of six 14-3-3 isoforms in progenitors and progenitor-derived astrocytes (PDAs) is shown in Western blots in Figure 1. Epsilon and theta isoforms showed a cytoplasm-specific presence in both cell- types, with no detected presence in cellular nuclei. While epsilon’s localization was similar to that of the theta isoform, it exhibited a slightly more intense presence in the cytoplasm. In contrast, the gamma isoform showed a nucleus-specific presence in both cell types. Sigma and beta isoforms were distributed throughout progenitors and progenitor-derived astrocytes. The sigma isoform, however, was only faintly detected in the progenitor nucleus. Sigma and beta isoforms both showed a more extensive cytoplasm-specific presence in progenitors and PDAs than a nucleus-specific presence. The zeta isoform showed a cytoplasm-specific presence in both progenitors and PDAs, but only appeared in the PDA nuclei (but not in the progenitor nucleus).
Figure 1.
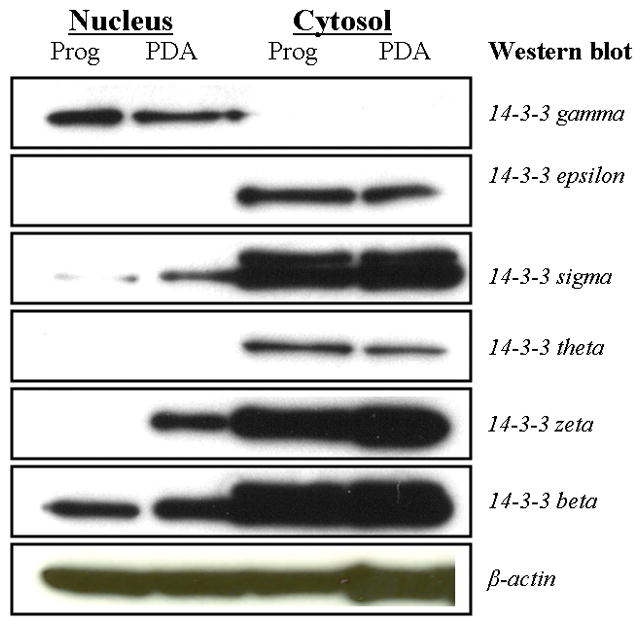
Western blot utilizing six anti-14-3-3 antibodies [anti-gamma (γ), anti-epsilon (ε), anti-sigma (σ), anti-theta (θ), anti-zeta (ζ), and anti-beta (β)] against nuclear and cytoplasmic lysates from human neural progenitor cells and progenitor-derived astrocytes. 7 μg of nuclear and cytoplasmic extracts were resolved on 4–12% gradient gels, transferred to a PVDF membrane, and then probed with isoform-specific antibodies.
14-3-3 zeta showed a gradual increase in nuclear presence as progenitors differentiated into PDAs over a zero to 28 day duration (Figure 2A). 14-3-3 zeta showed no presence in the nucleus within the first two days of differentiation of progenitors into PDAs, but gave a barely detectable appearance in the progenitor-to-PDA nucleus at a week following exposure to astrocytic media (differentiation of progenitors into PDAs). The zeta isoform’s presence progressively increased by day 28. We identified cell type by staining with nestin (for neural progenitors) and GFAP (for PDAs) during this time period (results not shown).
Figure 2.
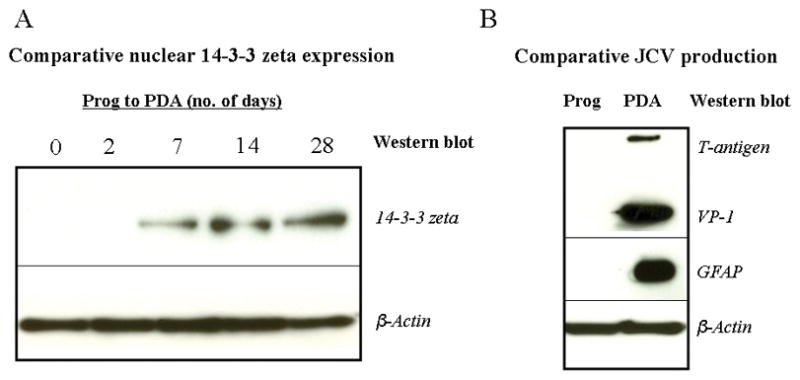
Figure 2A: Comparative Western blots utilizing anti-14-3-3 zeta against nuclear extracts of neural progenitors differentiating into progenitor-derived astrocytes over a 28-day duration. 5 to 7 μg protein equivalent of nuclear extracts were resolved on 4–12% gradient gels, transferred to PVDF membrane, and probed with 14-3-3 zeta antibody. β-actin was a consistent immunoblot loading control for nuclear extracts.
Figure 2B: Comparative Western blots utilizing anti-T-antigen, anti-VP-1, and anti-B-actin against nuclear extracts of progenitors and PDAs to establish astrocytic viral susceptibility and progenitor non-susceptibility through JCV production in this system. As a PDA-specific marker, GFAP level was measured from the total cell lysates. A 5 to 7 μg protein equivalent of nuclear extracts were resolved on 4–12% gradient gels, transferred to PVDF membrane, and probed with appropriate antibodies. β-actin was a consistent immunoblot loading control for nuclear extracts.
Since previous studies have shown that human neural progenitors are not usually permissive to JCV but PDAs are susceptible, a possible comparative relationship may emerge between 14-3-3 zeta’s specific translocation into the nucleus during differentiation and JCV susceptibility (Figure 2B). Western blots shown in Figure 2B indicate that progenitors show no JCV multiplication as measured by T-antigen and VP-1 levels. Infected PDAs showed both TGF-β1 and VP-1 labeling. The differentiation of progenitors into PDAs was further confirmed by the Western blot where the PDA-specific marker, GFAP, was utilized. Beta-actin antibodies were also used as a loading control for Western blots.
14-3-3 zeta immunofluorescent labeling in progenitors and progenitor-derived astrocytes
Interest in the zeta isoform’s translocation into the PDA nucleus and its possible association with JCV-susceptibility led to further confirmation of its unique localization in both cell types by immunofluorescence. The progenitors and PDAs were stained with nestin and GFAP respectively, and approximately 90% were labeled (results not shown). The zeta isoform was found only in the progenitor cytoplasm, as shown in Figure 3A. There exists a distinctive demarcation between the DAPI-stained progenitor nucleus and the cytoplasm-specific 14-3-3 zeta, which is further confirmed by the three-dimensional image (Figure 3B) that shows a conserved nucleus without any 14-3-3 zeta accumulation. This image shows a clear, DAPI-stained nucleus with 14-3-3 zeta in the cytoplasm adjacent to it. In contrast, PDAs show DAPI and 14-3-3 zeta co-localization in the nucleus, as shown in Figure 3C. The three-dimensional image (Figure 3D) also shows a sagittal cross-section of one particular PDA in which this co-localization generates a purple-stained nucleus, surrounded by the 14-3-3 zeta also present in the cytoplasm. For further confirmation, we stained 14-3-3 zeta in PDAs from freshly prepared human fetal brain tissue (rather than differentiating progenitors into PDAs in-vitro), and found the nuclear localization similar to differentiated PDAs (results not shown).
Figure 3.
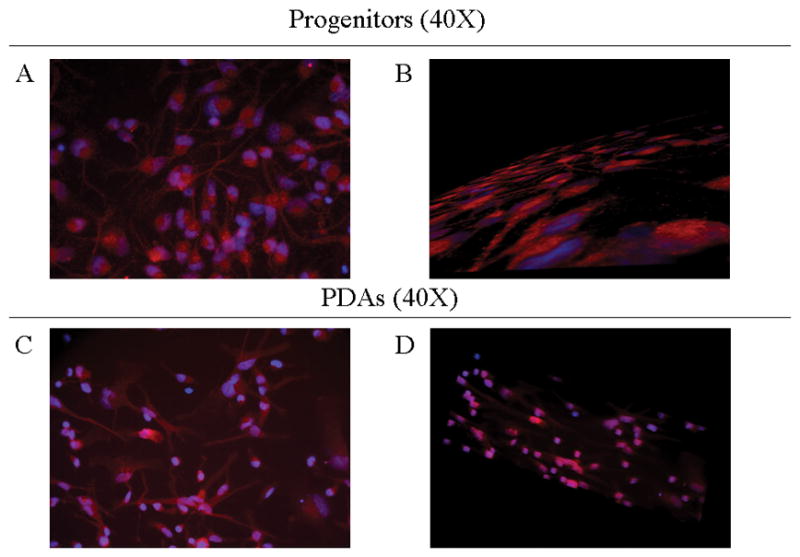
Figure 3A/B: Immunoassaying of progenitor cultures. Progenitor cells that were grown on 4-well chambers were fixed, permeabilized, and then stained with anti-14-3-3 zeta (red, rhodamine) to determine its relative localization to cell nuclei, stained with anti-DAPI (blue). Top (a) shows a 2-dimensional image at 40X, bottom (b) shows a 3-dimensional image manipulated using Apotome at 40X.
Figure 3C/D: Immunoassaying of PDA cultures. PDAs that were grown on 4-well chambers were fixed, permeabilized, and then stained with anti-14-3-3 zeta (red, rhodamine) to show co-localization in cell nuclei, stained with anti-DAPI (blue). Top (a) shows a 2-dimensional image at 40X, bottom (b) shows a 3-dimensional image at 40X.
14-3-3 zeta isoform in TGF-β1 treated, JCV-infected progenitor nucleus
Progenitor cells support only low level JCV activity compared with PDA susceptibility. To determine whether this low level of JCV infection was accompanied by 14-3-3 zeta up- regulation and translocation in the progenitor nucleus, progenitors were exposed to JCV for four days and then stained for VP-1 and 14-3-3 zeta (Figure 4A). The few cells stained for VP-1 exhibited a higher 14-3-3 zeta presence. In an earlier study, JCV-non-susceptible progenitors became susceptible through exposure to TGF-β1, which activates Smad DNA binding proteins that bind to or increase binding to JCV promoter sequences (Ravichandran et al. 2007). Progenitors were exposed to JCV and treated with TGF-β1 to determine whether the increase in VP-1 positive progenitor cells was proportional to 14-3-3 zeta presence in the nucleus. Multiple progenitors were infected after TGF-β1 treatment as measured by VP-1 immunostaining and most appear to indicate 14-3-3 zeta translocation or up-regulation in the nucleus (Figure 4B).
Figure 4.
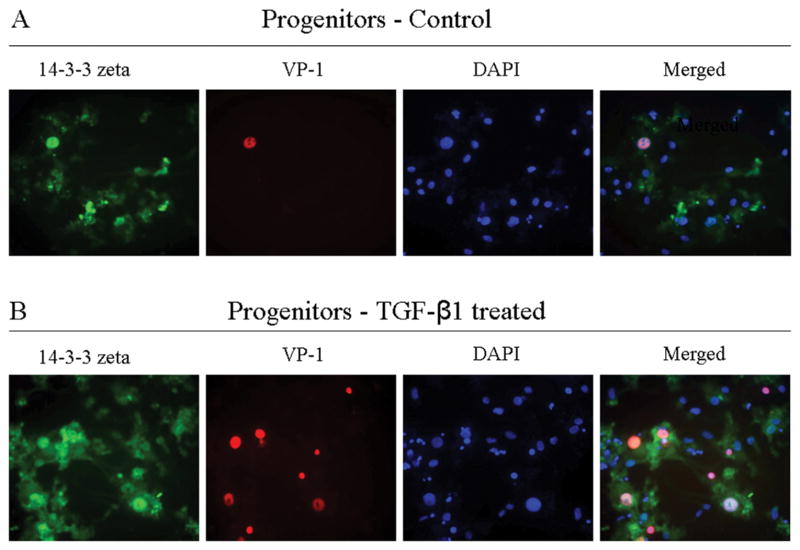
Comparative immunoassaying of progenitor cultures, without exposure to TGF-β1 and with exposure to TGF-β1. Progenitors were infected with JCV in the presence or absence of TGF-β1. Four days post-JCV infection, cells were fixed, permeabilized, and then stained with anti-14-3-3 zeta (green), anti-VP-1 (red) and DAPI (blue). Top row: Control culture; Bottom row: Culture treated with TGF-β1.
JCV-susceptible PDAs were also infected with JCV. Four days post-infection, PDAs were stained for VP-1 (VP-1 staining indicates JCV multiplication) (Figure 5A). Presence of VP-1 in this system accompanied increased 14-3-3 zeta accumulation in the PDA nucleus. TGF-β1 treatment further increased JCV multiplication in PDAs as measured by VP-1 immunostaining. JCV-infected PDAs showed more VP-1 positive cells and an accompanying increase in 14-3-3 zeta nuclear accumulation. In PDAs treated with TGF-β1, however, 14-3-3 zeta showed greater accumulation (Figure 5B). 14-3-3 zeta antibodies appear to accumulate to a significant extent in the PDA nucleus and to a lesser amount in the cytoplasm.
Figure 5.
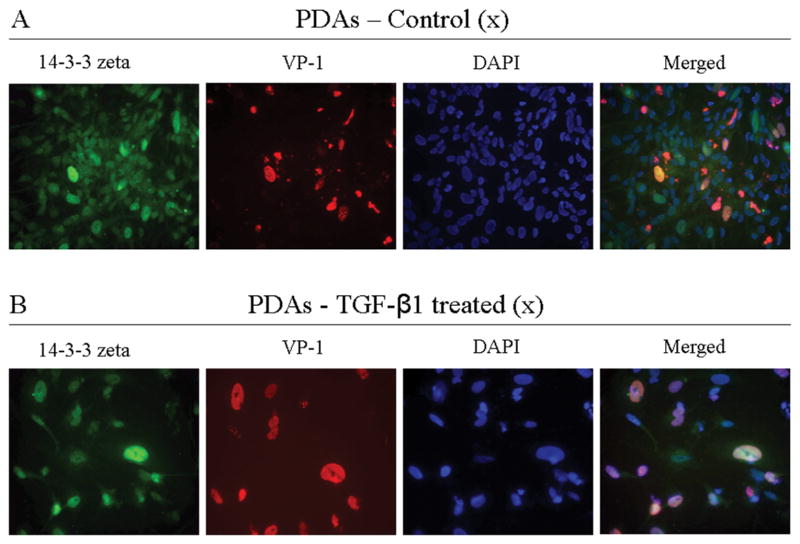
Comparative immunoassaying of PDA cultures, without exposure to TGF-β1 and with exposure to TGF-β1. PDAs were infected with JCV in the presence or absence of TGF-β1. Four days post-JCV infection, cells were fixed, permeabilized, and then stained with anti-14-3-3 zeta (green), anti-VP-1(red), and DAPI (blue). Top row: Control culture; Bottom row: Culture treated with TGF-β1.
In order to further examine the nature of JCV infection and its relationship with 14-3-3 zeta’s nuclear presence, PDAs were untreated or treated with TGF-β1, exposed or unexposed to JCV, and then subjected to Western blot to determine 14-3-3 zeta’s localization (Figure 6). 14-3-3 zeta was barely visible in untreated PDAs without virus exposure. Infected PDAs treated with TGF-β1 without virus exposure showed an increased presence of 14-3-3 zeta. However, 14-3-3 zeta was present in the nucleus in TGF-β1 untreated PDAs with virus exposure to a greater extent than both cells without virus exposure. Infected PDAs treated with TGF-β1 showed the highest accumulation, producing a clear band. Beta-actin antibodies were used as a loading control for Western blots.
Figure 6.
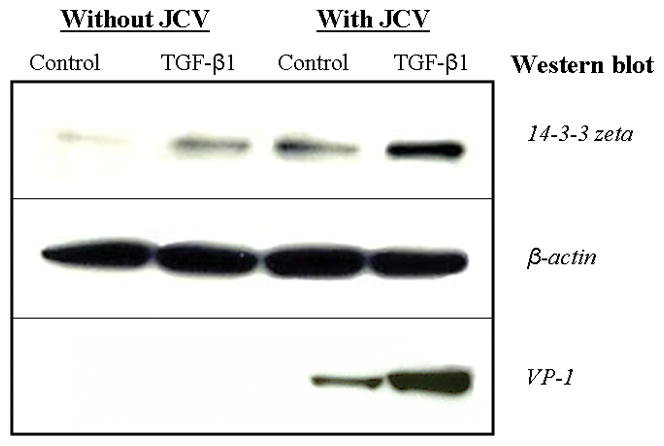
Western blot utilizing anti-14-3-3 zeta; PDAs were either infected with JCV or non-infected, in the presence or absence of TGF-β1. After five days, cells were harvested and nuclear extracts were separated. A 5 to 7 μg protein equivalent of nuclear extracts were resolved on 4–12% gradient gels, transferred to PVDF membrane, and probed with the appropriate antibodies as shown. Control culture unexposed to JCV (lane 1), TGF-β1 treated culture unexposed to JCV (lane 2), control culture exposed to JCV (lane 3), and TGF-β1 treated culture exposed to JCV (lane 4). β-actin was used as a loading control.
Discussion
A recent study showed that autopsied brains from PML patients exhibited dense 14-3-3 immunoreactivity in astrocytes and oligodendrocytes (Kawamoto et al. 2004). Data suggested that 14-3-3 immunoreactivity may be upregulated in glial cells in PML patients, implying that 14-3-3 accumulation may be associated with PML pathogenesis. Close association between 14-3-3 isoforms and PML therefore necessitates additional study of their potential implication in JCV.
PML’s etiologic agent, JC virus, persists principally in oligodendrocytes. (Enam et al. 2004; Radhakrishnan et al. 2003). Neural progenitor cells, however, are only JCV semi-permissive although they have been shown to be transfected successfully in vitro (Hou et al. 2006).
To characterize the potential involvement of these essential isoforms in the development of JCV, we first chose to determine the localization of six isoforms present in the mammalian nervous system in both neural progenitors and PDAs. Proper cellular control is largely determined by the internal balance of these isoforms; therefore identifying their normal states in comparison to their infected states is critical. The localization of these isoforms in cytoplasmic or nuclear cellular compartments indicated that these isoforms were not distributed indiscriminately, with the possible exception of the sigma isoform. The temporal relationship between the differentiation of progenitors into PDAs and 14-3-3 zeta’s translocation from the cytoplasm to the nucleus may indicate the importance of this isoform in development and possibly in viral susceptibility.
The potential role of 14-3-3 zeta in JC viral production is further supported by the introduction of virus in neural progenitors following TGF-β1 treatment, which has previously been shown to increase JCV VP-1 expression by 200% (Ravichandran et al. 2007). Following treatment, immunofluorescence indicated the translocation of 14-3-3 zeta into the infected progenitor nucleus where it was absent prior to transfection. In addition, the accumulation of zeta within the infected PDAs also increased following TGF-β1 treatment.
14-3-3 zeta may influence the degree to which JCV manifests itself within permissive cell types by controlling regulatory proteins and transcription factors responsible for viral assembly. Transcriptional activation and overexpression of the zeta isoform, for instance, has been strongly implicated in a pathway involved in reactivation of Epstein-Barr virus, along with S-adenosylhomocysteine hydrolase and PLA2 (Maas et al. 2006). In addition, 14-3-3 zeta has been shown to associate with nonstructural NS2 proteins of mouse minute virus that are involved in viral DNA amplification and virion production in vivo. The isoform’s established role in viral reactivation suggests that its translocation to the nucleus may be due to a transcriptional mechanism that dictates permissiveness to infection.
This isoform, however, has also been defined previously by its neuroprotective behavior under various conditions (Chen et al. 2007; Murphy et al. 2008), and it is therefore reasonable to assert that its translocation into the nucleus during infection may be partly due to its role in preventing cellular stress or injury. It is also necessary to consider the coexistence of 14-3-3 zeta and VP-1 in both neural progenitors and PDAs. The 14-3-3 protein family’s intrinsic role in cell signaling may also control fundamental aspects of viral replication. 14-3-3 zeta is known to have cruciform-binding activity, hence making it a possible modulating factor in initiating viral transcription in the nucleus (Zannis-Hadjopoulos et al. 2008). This is particularly intriging because the level of JCV activity is known to depend principally upon the nucleotide sequence of the viral regulatory region and the interaction of this promoter sequence with host transcription factors (Ravichandran and Major 2008).
In conclusion, we present here the distinct cellular translocation of 14-3-3 isoforms, notably 14-3-3 zeta, in JCV non-susceptible progenitors and JCV-susceptible PDAs. The specific coexistence of JC viral protein VP-1 and 14-3-3 zeta in the JCV-infected nuclei indicate the potential role of 14-3-3 zeta for JCV tropism, although further study is required.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institute of Neurological Disorders and Stroke, National Institutes of Health. We thank Soren Lowell (NINDS/NIH) and Blanche Curfman (NINDS/NIH) for their constructive criticism.
References
- Baxter HC, Fraser JR, Liu WG, Forster JL, Clokie S, Steinacker P, Otto M, Bahn E, Wiltfang J, Aitken A. Specific 14-3-3 isoform detection and immunolocalization in prion diseases. Biochem Soc Trans. 2002;30(4):387–91. doi: 10.1042/bst0300387. [DOI] [PubMed] [Google Scholar]
- Berg D, Holzmann C, Riess O. 14-3-3 proteins in the nervous system. Nat Rev Neurosci. 2003;4(9):752–62. doi: 10.1038/nrn1197. [DOI] [PubMed] [Google Scholar]
- Berger JR, Concha M. Progressive multifocal leukoencephalopathy: the evolution of a disease once considered rare. J Neurovirol. 1995;1(1):5–18. doi: 10.3109/13550289509111006. [DOI] [PubMed] [Google Scholar]
- Bollag B, Kilpatrick LH, Tyagarajan SK, Tevethia MJ, Frisque RJ. JC virus T′135, T′136 and T′165 proteins interact with cellular p107 and p130 in vivo and influence viral transformation potential. J Neurovirol. 2006;12(6):428–42. doi: 10.1080/13550280601009553. [DOI] [PubMed] [Google Scholar]
- Brockhaus K, Plaza S, Pintel DJ, Rommelaere J, Salomé N. Nonstructural proteins NS2 of minute virus of mice associate in vivo with 14-3-3 protein family members. J Virol. 1996;70(11):7527–34. doi: 10.1128/jvi.70.11.7527-7534.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Lee CT, Errico SL, Becker KG, Freed WJ. Increases in expression of 14-3-3 eta and 14-3-3 zeta transcripts during neuroprotection induced by delta9-tetrahydrocannabinol in AF5 cells. J Neurosci Res. 2007;85(8):1724–33. doi: 10.1002/jnr.21304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darbinyan A, Darbinian N, Safak M, Radhakrishnan S, Giordano A, Khalili K. Evidence for dysregulation of cell cycle by human polyomavirus, JCV, late auxiliary protein. Oncogene. 2002;21(36):5574–81. doi: 10.1038/sj.onc.1205744. [DOI] [PubMed] [Google Scholar]
- Del Valle L, Croul S, Morgello S, Amini S, Rappaport J, Khalili K. Detection of HIV-1 Tat and JCV capsid protein, VP-1, in AIDS brain with progressive multifocal leukoencephalopathy. J Neurovirol. 2000;6(3):221–8. doi: 10.3109/13550280009015824. [DOI] [PubMed] [Google Scholar]
- Enam S, Sweet TM, Amini S, Khalili K, Del Valle L. Evidence for involvement of transforming growth factor beta1 signaling pathway in activation of JC virus in human immunodeficiency virus 1-associated progressive multifocal leukoencephalopathy. Arch Pathol Lab Med. 2004;128(3):282–91. doi: 10.5858/2004-128-282-EFIOTG. [DOI] [PubMed] [Google Scholar]
- Hou J, Seth P, Major EO. JC virus can infect human immune and nervous system progenitor cells: implications for pathogenesis. Adv Exp Med Biol. 2006;577:266–73. doi: 10.1007/0-387-32957-9_19. [DOI] [PubMed] [Google Scholar]
- Kawamoto Y, Akiguchi I, Kovacs GG, Flicker H, Budka H. Increased 14-3-3 immunoreactivity in glial elements in patients with multiple sclerosis. Acta Neuropathol. 2004;107(2):137–43. doi: 10.1007/s00401-003-0785-z. [DOI] [PubMed] [Google Scholar]
- Koralnik IJ. Progressive multifocal leukoencephalopathy revisited: Has the disease outgrown its name? Ann Neurol. 2006;60(2):162–73. doi: 10.1002/ana.20933. [DOI] [PubMed] [Google Scholar]
- Luo ZJ, Zhang XF, Rapp U, Avruch J. Identification of the 14.3.3 zeta domains important for self-association and Raf binding. J Biol Chem. 1995;270(40):23681–7. doi: 10.1074/jbc.270.40.23681. [DOI] [PubMed] [Google Scholar]
- Maas D, Maret C, Schaade L, Scheithauer S, Ritter K, Kleines M. Reactivation of the Epstein-Barr virus from viral latency by an S-adenosylhomocysteine hydrolase/14-3-3 zeta/PLA-2 dependent pathway. Med Microbiol Immunol. 2006;195(4):217–23. doi: 10.1007/s00430-006-0022-1. [DOI] [PubMed] [Google Scholar]
- Messam CA, Hou J, Gronostajski RM, Major EO. Lineage pathway of human brain progenitor cells identified by JC virus susceptibility. Ann Neurol. 2003;53(5):636–46. doi: 10.1002/ana.10523. [DOI] [PubMed] [Google Scholar]
- Murphy N, Bonner HP, Ward MW, Murphy BM, Prehn JH, Henshall DC. Depletion of 14-3-3 zeta elicits endoplasmic reticulum stress and cell death, and increases vulnerability to kainate-induced injury in mouse hippocampal cultures. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05447.x. [DOI] [PubMed] [Google Scholar]
- Niemantsverdriet M, Wagner K, Visser M, Backendorf C. Cellular functions of 14-3-3 zeta in apoptosis and cell adhesion emphasize its oncogenic character. Oncogene. 2008;27(9):1315–9. doi: 10.1038/sj.onc.1210742. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan S, Otte J, Enam S, Del Valle L, Khalili K, Gordon J. JC virus-induced changes in cellular gene expression in primary human astrocytes. J Virol. 2003;77(19):10638–44. doi: 10.1128/JVI.77.19.10638-10644.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran V, Jensen PN, Major EO. MEK1/2 inhibitors block basal and transforming growth factor 1beta1-stimulated JC virus multiplication. J Virol. 2007;81(12):6412–8. doi: 10.1128/JVI.02658-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran V, Major EO. DNA-binding transcription factor NF-1A negatively regulates JC virus multiplication. J Gen Virol. 2008;89(Pt 6):1396–401. doi: 10.1099/vir.0.2008/000059-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umahara T, Uchihara T, Tsuchiya K, Nakamura A, Iwamoto T, Ikeda K, Takasaki M. 14-3-3 proteins and zeta isoform containing neurofibrillary tangles in patients with Alzheimer’s disease. Acta Neuropathol. 2004;108(4):279–86. doi: 10.1007/s00401-004-0885-4. [DOI] [PubMed] [Google Scholar]
- Van Der Hoeven PC, Van Der Wal JC, Ruurs P, Van Dijk MC, Van Blitterswijk J. 14-3-3 isotypes facilitate coupling of protein kinase C-zeta to Raf-1: negative regulation by 14-3-3 phosphorylation. Biochem J. 2000;345(Pt 2):297–306. doi: 10.1042/0264-6021:3450297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zannis-Hadjopoulos M, Yahyaoui W, Callejo M. 14-3-3 cruciform-binding proteins as regulators of eukaryotic DNA replication. Trends Biochem Sci. 2008;33(1):44–50. doi: 10.1016/j.tibs.2007.09.012. [DOI] [PubMed] [Google Scholar]