

Abstract
Understanding the biological features of cancer is the basis for designing efficient anti-cancer nanomedicines. On one hand, important therapeutic targets for anti-cancer nanomedicines need to be identified based on cancer biology, to address the unmet medical needs. On the other hand, the unique pathophysiological properties of cancer affect the delivery and interactions of anti-cancer nanomedicines with their therapeutic targets. This review discusses several critical cancer biological properties that challenge the currently available anti-cancer treatments, including cancer heterogeneity and cancer stem cells, the complexcity of tumor microenvironment, and the inevitable cancer metastases. In addition, the biological bases of the enhanced permeability and retention (EPR) effect and tumor-specific active targeting, as well as the physiological barriers for passive and active targeting of anti-cancer nanomedicines are covered in this review. Correspondingly, possible nanomedicine strategies to target cancer heterogeneity, cancer stem cells and metastases, to overcome the challenges related to tumor passive targeting and tumor penetration, and to improve the interactions of therapeutic payloads with the therapeutic targets are discussed. The focus is mainly on the designs of polymeric anti-cancer nanomedicines.
Keywords: Nanomedicines, polymer conjugates, tumor microenvironment, cancer heterogeneity, cancer stem cells, metastasis, EPR effect, targeting
Introduction
The last several decades witnessed a largely improved understanding of the biological processes of tumorigenesis, malignant transformation and tumor progression, as well as rapidly developing anti-cancer therapies that improved cancer patients' survival. However, the achievements hardly changed the fact that cancer remains the top cause of morbidity and mortality (Jemal et al., 2010). The therapeutic agents employed in traditional chemotherapies or novel molecularly targeted therapies include small molecule toxic agents, natural or chemical compounds, antibodies, proteins, and peptides. They exert anticancer effects via different mechanisms, but they usually have common weaknesses: once administered into the human body their poor solubility and/or unfavorable physicochemical properties result in poor pharmacokinetics, difficult access to the tumor site, inadequate entry into cancer cells, as well as nonspecific toxicities to normal organs. To address these problems, nanosized materials have been developed as drug carriers and hold great potential to improve the delivery and anti-cancer effects of the currently available therapeutics (Wagner et al., 2006; Heidel & Davis, 2011; Canal et al., 2011). Examples of the nanocarriers under development are based on liposomes, polymer conjugates (Kopeček & Kopečková, 2010), micelles (Matsumura & Kataoka, 2009), polymersomes, polymeric nanoparticles, as well as inorganic nanoparticles (Cho et al., 2008; Lammers et al., 2008).
The advantages of applying nanosized drug carriers to cancer therapies over small molecule therapeutics, as demonstrated in polymer-bound drugs or other nanoformulations, include preferential accumulation of the drug at the tumor site by enhanced permeability and retention (EPR) effect (Shiah et al., 2001a), increased active accumulation of the drug at the tumor site by specific targeting (Omelyanenko et al., 1998b), active uptake by pinocytosis or receptor-mediated endocytosis (Liu et al., 2009), controlled drug release, the ability to incorporate multifunctional components (Canal et al., 2011), long-lasting circulation in the blood stream (Shiah et al., 2001a), decreased nonspecific toxicity of the conjugated drug (Kopeček & Kopečková, 2010), immunoprotecting and immunomobilizing activities (Říhová et al., 1988, 2001), and modulation of the cell signaling pathways (Minko et al., 1999, 2000; Nishiyama et al., 2003; Malugin et al., 2006). A growing number of nanosized drug delivery systems have shown improved therapeutic index in animal models and have entered clinical trials for further validations in human (Northfelt et al., 1998; O'Brien et al., 2004; Kopeček & Kopečková, 2010; Heidel & Davis, 2011). The scientific bases of this drug delivery concept have been confirmed, with several formulations receiving FDA approval, while the translations to clinical use have been slow compared to the recent burst of preclinical research in this area.
As the knowledge of cancer biology and pathophysiology increased, important reasons that render the clinical translation more difficult have been identified: first, more anatomical and physiological barriers have been realized which may impede the efficient delivery of nanocarriers (Lammers, 2010; Jain & Stylianopoulos, 2010); second, cancer is not a single disease, but a collection of diseases with intratumoral and intertumoral heterogeneity.
Drug delivery applications compose major aspects of nanomedicine, which refers to the use of nanostructured materials in medical applications. This review will mainly describe the current achievements in the field of anticancer nanomedicines, especially polymer anti-cancer therapeutics, the biological factors that influence nanocarrier behavior, challenges as well as possible strategies to overcome these challenges. In particular, recent research and clinical experiences reveal that the presence of cancer stem cells, the complicated cancer cell-microenvironment interactions, and the high incidences of metastases are several critical biological features responsible for the failure of current anti-cancer therapies. The biological bases as well as possible nanomedicine strategies to target these features will be discussed.
The biological features of cancer
Heterogeneity of cancer cells
It has long been acknowledged that the intratumoral biological and functional heterogeneity exists among cancer cells (Shackleton et al., 2009; Dick, 2009). This notion, brought up decades ago (Furth & Kahn, 1937; Dick, 2008; Clevers, 2011), still remains a great challenge for the development of effective anti-cancer therapies that can both lead to tumor regression and prevent relapse and metastasis. While this heterogeneity may arise from the diverse genetic changes acquired by different cancer cells, as demonstrated by the traditional clonal evolution model; the cancer stem cell (CSC) hypothesis was recently revived and captured researchers' attention as an alternative model to explain the cancer cell heterogeneity (Reya et al., 2001; Shackleton et al., 2009; Dick, 2009; Rosen & Jordan, 2009; Clevers, 2011). Both tumor models give us valuable insight into the design of anti-cancer treatments.
The CSC model proposes that the tumor development is similar to that of normal organs, with stem-like cancer cells at the top of the hierarchy to maintain tumor growth and progression (Shackleton et al., 2009; Dick, 2009). Only the CSCs, with the ability to self-renew and differentiate, have the tumorigenic potential and are able to generate phenotypically heterogeneous tumor cell populations that resemble the original organizations of the parent tumor (Figure 1A). On the other hand, the clonal evolution model states that cancer cells randomly acquire genetic changes, triggered by both intrinsic and extrinsic environmental factors, and develop into heterogeneous subclones with genetic differences (Shackleton et al., 2009; Dick, 2009). The key word of this model, and the key difference from the CSC model, is “random” or “stochastic”, which suggests that all cancer cells have equal potential to be tumorigenic (Figure 1A).
Figure 1.
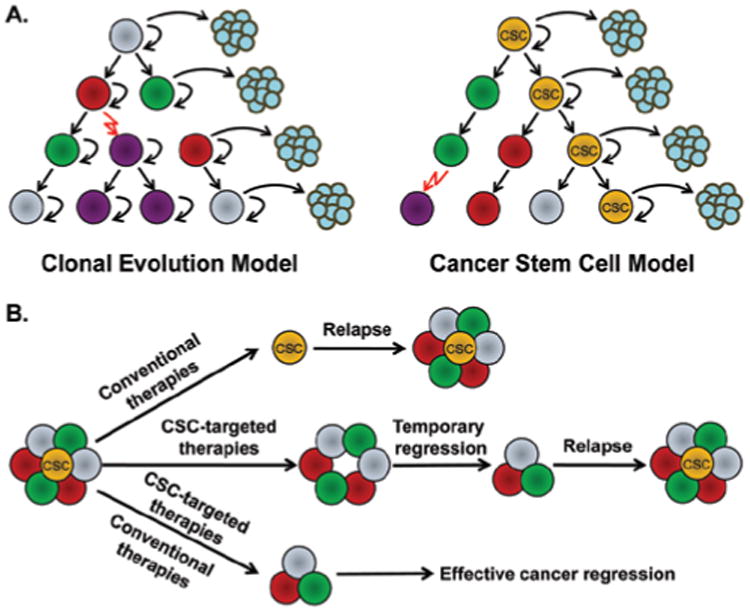
(A) Two models of tumor heterogeneity. The left cartoon shows the clonal evolution model: cancer cells are phenotypically heterogeneous, but they acquire the tumorigenic ability randomly, with the equal potential to generate new tumors; the right cartoon illustrates the CSC model: only a subset of cancer cells (CSC) have the ability to self-renew and form new tumors, whereas most other cancer cells are depleted of this ability. (B) The rationale for the development of anti-cancer therapeutics based on CSC model. Conventional therapies kill non-CSCs but fail to eliminate CSCs, resulting in relapse; CSC-targeted therapies inhibit CSCs and lead to temporary cancer regression, but may not stop the generation of new CSCs from bulk cancer cells; the combination of conventional and CSC-targeted therapies will effectively target both bulk cancer cells and CSCs, leading to effective cancer regression. Adapted with permission from Reya et al. (2001).
Actually the CSC concept is not truly novel, but has a history of experimental explorations as long as the clonal evolution model (Dick, 2009; Clevers, 2011). The earliest tumor cell transplantation assay can be traced to 1937, when a single mouse tumor cell was found to be able to generate a new tumor in the recipient mice (Furth & Kahn, 1937). Around the mid-1900s, researchers have already noticed that not every single tumor cell possesses this tumorigenic capacity and the frequency of the tumor-initiating cells is relatively low (Bruce & Van Der Gaag, 1963). Subsequent research identified tumorigenic cancer cells within the undifferentiated areas that give rise to cancer cells in well-differentiated areas using radiolabeling approach, correlating tumor development with the hierarchical development of normal tissues (Pierce & Wallace, 1971). Interestingly, from the 1970s cancer research focused more on the clonal evolution concept (Nowell, 1976). This was due to the breakthrough in the discovery of oncogenes and tumor suppressor genes and probably to technical limitations in isolating and studying stem-like cells within tumors. Although considerable progress has been achieved in treating cancers based on clonal evolution theory in the last several decades, treatment failures are still inevitable.
The CSC hypothesis excited cancer researchers again since 1990s (Bonnet & Dick, 1997; Al-Hajj et al., 2003). Generally, researchers utilized FACS (Fluorescent Activated Cell Sorting) combined with limiting dilution transplantations into highly immunodeficient mice to assess the tumor engraftment capacities of the different cell subpopulations with different cell marker combinations sorted from primary tumors (Shackleton et al., 2009). Cancer cells containing enriched CSC population would show significantly enhanced tumorigenicity than the other subsets of sorted cancer cells. Using these techniques, Dick et al. identified a subset of leukemic stem cells (Bonnet & Dick, 1997). Similarly, Clark et al. identified breast CSCs, the first type of CSCs found in solid tumors (Al-Hajj et al., 2003). Then a series of studies revealed the presence of CSCs in other solid tumors including brain (Singh et al., 2004), colon (Ricci-Vitiani et al., 2007; O'Brien et al., 2007), and prostate cancers (Wang & Shen, 2011; Collins & Maitland, 2009).
Since CSCs share a variety of similar functional properties with normal tissue stem cells, it is reasonable to hypothesize that CSCs, similar to normal stem cells, are resistant to traditional chemo- or radiotherapies that are designed to target rapidly dividing cancer cells (Dean et al., 2005; Tang et al., 2007; Dick, 2009). Indeed, experimental and clinical studies support the intrinsic resistance of CSCs. For example, in breast cancer patients following standard chemotherapy, CD44 + CD24low CSCs within the patient residual tumor samples did not decrease but instead expanded to a larger proportion, resulting in higher tumorigenicity (Yu et al., 2007). Similarly, resistance of leukemic stem cells to Imatinib was observed in chronic myeloid leukemia (CML) patients (Oravecz-Wilson et al., 2009). The mechanisms of resistance include the quiescent status, overexpression of drug efflux pumps (Hirschmann-Jax et al., 2004), enhanced DNA damage repair (Bao et al., 2006), and anti-apoptotic machinery (Dean et al., 2005). Consequently, if current anti-cancer therapies can shrink the tumor mass efficiently, resistant CSCs are still left surviving; these CSCs have the ability to regenerate tumor populations following treatment, resulting in lethal recurrence and metastasis (Dick 2009; Clevers, 2011). Overall, the CSC theory provides at least critical, if not all, reasons to the current anti-cancer therapy failures. Effective anti-cancer treatments definitely require the elimination of the CSCs (Dick, 2009).
To be noted, nowadays the CSC and clonal evolution models are not being considered mutually exclusive (Shackleton et al., 2009; Dick, 2009). Based on recent evidence, CSCs may evolve by clonal evolution; cancers that follow CSC model may also undergo clonal evolution. The spontaneous inter-conversion between CSC and non-CSC phenotypes has been observed under culture conditions (Gupta et al., 2011) as well as under certain microenvironmental influences (Iliopoulos et al., 2011). Some data showed that the epigenetic difference not only correlates with the expression of CSC phenotypes, but also is responsible for drug resistance (Shackleton et al., 2009; Sharma et al., 2010). For instance, the up-regulation of histone demethylase KDM5A contributes to the epidermal growth factor receptor (EGFR) inhibitor–tolerant state (Sharma et al., 2010). However, genetic differences are still commonly seen among the cancer cell subclones. Even though the strongest evidence of the CSC model is found in hematopoietic malignances, the development of drug resistance is observed by the generation of new clones with the mutation in the drug target, as seen in the emergence of Imatinib resistance in CML (Shah et al., 2007). Despite of all these seemingly controversial evidences, the CSC model is meaningful; even genetically identical cancer cells can hold different differentiation status in a hierarchy (Clevers, 2011). Therefore, to be able to target all cancer cells, it is necessary to develop therapeutics that target both bulk tumor cells and CSCs, or inhibit the transition from non-CSCs to CSCs (Figure 1B) (Gupta et al., 2011).
Besides the issues discussed above, many unanswered questions and challenges related to the CSC model remain (Reya et al., 2001; Shackleton et al., 2009; Dick, 2009; Rosen & Jordan, 2009; Clevers, 2011). For instance, does the CSC model apply to all tumor types? What is the frequency of CSCs? What is the origin of CSCs? How to improve the accuracy of the in vivo xenograft transplantation assay? Are there unique and reliable biomarkers for CSCs? No doubt that further investigations are necessary, but these questions do not discount the therapeutic significance of the CSC model. It promises a distinct perspective in developing anti-cancer therapies based on the newly discovered properties of CSCs, which are not successfully targeted by the traditional therapeutics.
Tumor microenvironment
By viewing cancer as complex “organ-like structures”, the tumor microenvironment cannot be ignored when discussing anti-cancer therapeutics and drug delivery. In the past decade, numerous studies have indicated the importance of tumor microenvironment in cancer growth, progression, and metastasis (Friedl & Alexander, 2011; Hanahan & Coussens, 2012). Cancer cells are embedded in unique extracellular matrix (ECM) and are surrounded by various tumor stromal cells. The whole tumor is in constant remodeling through the reciprocal communications between cancer cells and the various tumor microenvironment components by cell-cell interactions, cell-matrix interactions as well as via secreted growth factors and/or cytokines. We shall not review all aspects of this field, but focus on those closely relevant to the development and delivery of nanomedicines.
Tumor angiogenesis, regulated by many pro- and anti-angiogenic signals, is a complex but important phenomenon. Patterns of tumor angiogenesis include not only the vessel sprouting from existing blood vessels (similar as in normal tissues), but also other tumor-specific patterns such as novel blood vessel formation from cells recruited from the bone marrow, and the differentiation of CSCs into endothelial-like cells (Friedl & Alexander, 2011; Hanahan & Coussens, 2012; Ricci-Vitiani et al., 2010; Wang & Oliver, 2010). Both the endothelial cells and the perivascular supporting cells grow abnormally under the persistent stimulation from abnormally activated growth signals (Jain & Stylianopoulos, 2010; Hanahan & Coussens, 2012; Roberts & Palade, 1997). These phenomena eventually result in leaky and tortuous tumor vasculatures, with large sized interendothelial junctions up to several hundred nanometers associated with irregular blood flow (Roberts & Palade, 1997; Jain, 1987; Hashizume et al., 2000; Jain et al., 2002). In addition, the lymphatic system inside the tumor, especially near the center of the tumor, is impaired and does not drain the fluid efficiently (Leu et al., 2000; Padera et al., 2004). These phenomena comprise the major physiological bases of the enhanced permeability and retention (EPR) effect (Maeda et al., 2000). On the other hand, despite the defects in tumor lymphatics flow, lymphatic vessels at the tumor periphery are still able to mediate cancer metastasis (Padera et al., 2002; Jain et al., 2007).
Besides the angiogenic vascular cells (including endothelial cells and pericytes), tumor stromal cells recruited into the tumor microenvironment also include tumor-associated fibroblastic cells and infiltrating immune cells (including tumor-associated macrophages, lymphocytes, neutrophils, etc) (Friedl & Alexander, 2011; Hanahan & Coussens, 2012). All three types of tumor stromal cells get involved in supporting the cancer cells in various ways and contribute to the core hallmarks of cancer. Moreover, the stromal cells influence the deposition of the ECM components, growth factors and cytokines, and vice versa (Friedl & Alexander, 2011; Hanahan & Coussens, 2012).
Importantly, the interplay of different cell and non-cell components create unique intratumoral fluid physical dynamics and chemical properties that are different from the normal tissue (Jain & Stylianopoulos, 2010; Minchinton & Tannock, 2006). In normal organs, the interstitial fluid pressure (IFP) is lower than the intravascular pressure (IVP), allowing ready perfusion of the tissue; while in tumor, the leakage of the tumor vasculature together with the inefficient drainage of lymphatic vessels result in interstitial hypertension (Figure 2) (Jain, 1998; Heldin et al., 2004). The blood flow in the tortuous tumor blood vessels is found to be slower than normal, thus leaving poorly perfused areas (Leunig et al., 1992; Yuan et al., 1994). In addition, the tumor vasculature is not uniformly permeable. In some cases such as desmoplasia, the highly proliferating cancer cells and stromal cells compress the tumor blood vessels, causing blood vessel collapse (Padera et al., 2004). Altogether, the elevated IFP, heterogeneity of vascular permeabilities, irregular blood flow, and dense ECM components render the uniform delivery of therapeutics extremely difficult (Figure 2).
Figure 2.
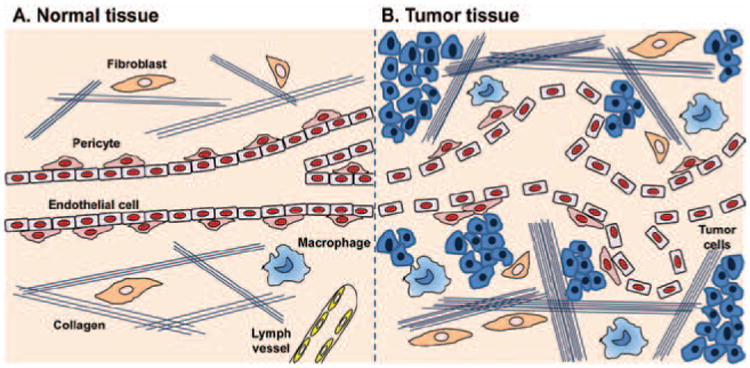
The structural differences between normal and tumor tissue microenvironment. (A) Normal tissues have blood vessels with tightly aligned endothelial cells and pericytes. The extracellular matrix is comprised of a loose network of collagen fibers and a few fibroblasts and macrophages. Lymph vessels are present. (B) Tumor tissues contain leaky and tortuous blood vessels with irregular blood flow. Pericytes covering outside the tumor vasculatures are still present. The tumor extracellular matrix is much denser than normal tissues, with thicker network of collagen fibers, more fibroblasts and macrophages. In addition, tumors usually lack functional lymph vessels. All the above features contribute to the increased interstitial fluid pressure (IFP). Adapted with permission from Heldin et al. (2004).
Metastasis
The most striking difference between “benign” and “malignant” tumors is the ability to metastasize. Clinically, most cancer patients suffer deaths from relapse and metastasis, not from the primary tumor. However, therapeutics efficiently preventing and targeting metastasis are very limited. This is partially due to the complex and yet not-well-understood mechanism of metastasis, as well as the difficulty to access all macro- and micro-scopic metastatic sites. Efforts have been made to understand the genetic and molecular processes contributing to metastasis; the macroscopic patterns including the steps, timing and the preferential sites of metastasis are also under investigation (Friedl & Alexander, 2011; Nguyen et al., 2009; Klein, 2009; Spano & Zollo, 2012).
Metastatic lesions form only when the cancer cells successfully survive through the following steps: local invasion, intravasation, survival in the circulation, extravasation, and finally colonization (Chambers et al., 2002). Different processes are governed by different metastatic gene patterns. For example, genes promoting metastasis initiation usually mediate cancer cell motility, epithelial-mesenchymal transition (EMT) and ECM degradation, such as TWIST1, SNAI1, SLUG, etc (Yang and Weinberg, 2008). Once in circulation, cancer cells need to infiltrate distant organs by extravasation and surviving in the newly invaded sites. These “metastasis progression genes” include PTGS2, EREG, MMP1, CCL5 targets, etc (Nguyen et al., 2009). Finally, the “metastatic virulence genes” confer activities that reinitiate cell growth and colonization (Kang et al., 2003; Mundy, 2002). Although numerous genes have been identified, the expression patterns of most genes are heterogeneous across different tumor types and metastatic sites. It is also difficult to dissect the genes that are most crucial and determining as therapeutic targets. It is well known that the extracellular proteases, matrix metallo-proteinases (MMPs), play a key role in ECM degradation, thus facilitating cancer cell migration and invasion (Wolf et al., 2007). However, the enthusiasm to develop MMP inhibitors as antimetastatic therapeutics was diminished by their failure to prevent cancer cell invasion (Bacac & Stamenkovic, 2008; Sorbera et al., 1999). Blocking protease activities resulted in the conversion of mesenchymal (fiber-based) cell migration to ameoboid movement (Wolf et al., 2003). Furthermore, to achieve clinically effective prevention of metastasis, MMPs need to be inhibited by therapeutic quantities of MMP inhibitors all across the tumor microenvironment and even other tumor seeding tissues. Thus, it is not quite practical to develop drug delivery systems for MMP inhibitors.
The heterogeneity of cancer metastasis was demonstrated by the clinically observed preferential colonization to diverse organ sites and variable temporal courses for different tumor types. For example, breast cancer principally metastasizes to bone, lungs, liver and brain (Kouros-Mehr et al., 2008; Hess et al., 2006); prostate cancer metastasis is largely confined to bone (Edlund et al., 2004); while colorectal and pancreatic cancers mainly metastasize to liver and lungs (Hess et al., 2006). These patterns can be influenced by many factors such as the circulation patterns, the specific affinity to certain organs for cancer cell retention and the specific interactions between metastatic niche and cancer cells. Another observation is the different kinetics of metastasis among different tumor types. For instance, lung cancers develop detectable metastasis in distant organs just within months of diagnosis (Feld et al., 1984), but breast cancer metastasis is often detected after years of primary tumor remission (Schmidt-Kittler et al., 2003).
Another challenge of targeting metastasis is the recent observation that metastasis may develop in parallel with the development of the primary tumor, starting to disseminate tumor cells very early (Klein, 2009). Based on this metastasis model, it is crucial to identify the “metastasis founder cells” that disseminate early to multiple distant sites. Again, the CSCs are hypothesized to consist of “metastasis founder cells” (Brabletz et al., 2005). However, only a few data support this hypothesis so far and further validation is needed in different tumor models, most importantly in patients.
Targetability of anti-cancer nanomedicines
Passive targeting (EPR effect)
Most nanomedicines developed to date mainly exploit the EPR effect to achieve improved tumor delivery of macromolecules than small molecule drugs (Taurin et al., 2012). The EPR phenomenon of macromolecules was first discovered and reported by Matsumura and Maeda (1986). Subsequently, this phenomenon was observed in many other nanosized therapeutic formulations, including polymer-drug conjugates (Seymour et al., 1995; Fang et al., 2003; Noguchi et al., 1998), liposomes (Campbell et al., 2002), micelles (Matsumura & Kataoka, 2009), and proteins such as IgG (Matsumura & Maeda, 1986). As mentioned in section “Tumor microenvironment”, EPR effect is largely a tumor vasculature-dependent phenomenon; both anatomical and pathophysiological properties of the tumor vasculature are responsible. Anatomically, most tumors are well vascularized with high density; usually, tumor angiogenesis is extensively induced when tumors reach the size larger than 0.8–1 mm (Folkman, 1995; Fang et al., 2011). Moreover, rapid growth of blood vessels leads to irregular vascular alignment and defects of the junctions between endothelial cells, creating large fenestrations that allow the transport of large-sized molecules (Roberts & Palade, 1997; Jain, 1987; Hashizume et al., 2000; Jain et al., 2002). Physiologically, tumors also overexpress many permeability-enhancing factors including, but not limited to, vascular endothelial growth factors (VEGF) (Leung et al., 1989), bradykinin (Maeda et al., 1988), nitric oxide (NO) (Wu et al., 1998), peroxynitrite, and prostaglandins (PGs) (Fang et al., 2011). On the other hand, the enhanced retention is due to the defective lymphatic clearance of macromolecules than of small molecules (Matsumura & Maeda, 1986). In addition, the slower venous return promotes the accumulation of large molecules inside the tumor (Jain, 1988).
As observed in numerous experiments assessing tumor accumulation of macromolecules, the extent of the EPR effect is largely a size-based phenomenon. Although the vascular permeability to individual nanosized drug carriers or proteins decreases as the size increases, the overall tumor accumulation of the drug carriers is enhanced with the increased macromolecule size, as long as the size is below the size limit of the endothelial fenestrations (Yuan et al., 1995; Seymour et al., 1995; Shiah et al., 2001a). This is due to the prolonged circulation time and relatively longer retention time of the larger-sized molecules inside the tumor. Thus, for nanomedicines with the same architecture, the larger the molecular weight, the higher the tumor accumulation. This phenomenon has been observed, for example, for N-(2-hydroxypropyl) methacrylamide (HPMA) copolymer-based drug conjugates in tumor-bearing mice (Shiah et al., 2001a). But other features, such as conformation or surface charge also play considerable roles in the extent of tumor accumulation, since these factors may change the electrostatic interactions between drug carriers and vascular cells, or change the whole biocompatibility and reticuloendothelial system clearance rate of the nanomedicines (Nugent & Jain, 1984; Pluen et al., 1999; Campbell et al., 2002). For example, drug carriers with positive charges bind readily to vascular endothelial surfaces since the vascular surface is negatively charged. This may reduce the plasma half life and tumor accumulation (Campbell et al., 2002). On the other hand, liver and spleen takes up the negatively charged carriers faster than neutral ones (Li & Huang, 2008).
Although the EPR effect favors tumor accumulation, there are still other physiological barriers that hinder the overall tumor accumulation, especially the homogeneous distribution of the nanomedicines. Previous experiments in mice with large-sized tumors have shown that following the intravenous injection of Evans blue, the dye accumulated mainly in the well-perfused tumor periphery, but little in the necrotic or poorly-perfused areas (Matsumura & Maeda, 1986; Fang et al., 2011). Although this is a good illustration that EPR effect is a vascular-dependent phenomenon, it also reveals that EPR effect is not evident in all tumor areas (Taurin et al., 2012). Moreover, the EPR effect may become very weak in some types of tumors, such as pancreatic and metastatic liver cancers, which possess low vascular densities and poor perfusion. To achieve homogeneous accumulation, nanomedicines need to move deeply into the whole tumor tissue after extravasation. However, the transport of nanomedicines can be largely impeded by the elevated interstitial pressure and the dense tissues composed of pericytes, stromal cells and different ECM components between vasculature and tumor cells (Jain & Stylianopoulos, 2010).
Active targeting
The term “active targeting” in tumor-targeted nanomedicines refers to the incorporation of targeting ligands that can specifically recognize and bind to the tumor cells, mediate nanomedicine-cell interactions, thus enhancing the therapeutic efficacy. To ensure the tumor targeting specificity, the targeting ligands chosen usually specifically bind to the receptors or antigens that are overexpressed at the tumor sites, but rarely expressed on normal cells (Allen, 2002). Antibodies (Omelyanenko et al., 1998a,b; Liu et al., 2009), antibody fragments (Lu et al., 1999; Hongrapipat et al., 2008a; Johnson et al., 2009; Colombo et al., 2010), peptides (Nguyen et al., 2010; Ruoslahti, 2012), as well as natural ligands to the cell surface receptors (such as certain carbohydrates) (Seymour et al., 2002) can be employed as targeting moieties for nanomedicines. The most evident and commonly acknowledged advantage of active targeting, compared to the non-targeted formulations, is to improve the cell biorecognition and intracellular uptake of nanomedicines via receptor mediated endocytosis (Lammers et al., 2012). After all, most of the small molecule drugs need to be internalized into the cells to exhibit therapeutic effects. So far, the targeting moieties have been used to either directly target the cancer cells; or to target the tumor vascular endothelial cells and indirectly inhibit cancer cell growth by deprivation of the oxygen and nutrients carried by tumor vasculatures (Neri & Bicknell, 2005; Arap et al., 1998).
Despite of numerous preclinical experiences of actively targeted nanomedicines, few are in clinical use right now except for several antibody-based nanomedicines such as Zevalin (Wiseman et al., 1999) and Bexxar (Horning et al., 2005). The possible reasons for the slow clinical translation of actively targeted nanomedicines may include: (1) the advantage of active targeting is limited by the above mentioned physiological barriers before the targeted nanomedicine can reach the target cancer cells, such as high IFP, presence of pericyte and fibroblast cell layers between endothelial and cancer cells, and high cellular densities (Kwon et al., 2012); (2) the targeted nanomedicines tend to stick to the first receptors they find and stop further tumor penetration. This “binding-site barrier” effect may impede the deep tissue penetration (Juweid et al., 1992).
Anti-cancer nanomedicines in preclinical/clinical development
Among the efforts to improve the therapeutic index of the anti-cancer therapies, nanomedicines are promising strategies. Thus far, around 40 nanomedicine formulations have been in routine clinical uses for treatments of diverse diseases, with fewer than 10 products for cancer treatment (Duncan & Gaspar, 2011; Taurin et al., 2012). Moreover, several dozens of nanomedicine formulations have entered clinical trials currently for evaluations in human patients; the majority of the products are for cancer treatment (Wagner et al., 2006). Most of the formulations under clinical development have been liposomes or poly(ethylene glycol) modified (PEGylated) liposomes, polymer-drug conjugates, polymer-protein conjugates, polymeric micelles for small molecule drug delivery, and nanoparticles for diagnostic purposes (Heidel & Davis, 2011; Cho et al., 2008; Lammers et al., 2008; Canal et al., 2011; Kopeček & Kopečková, 2010; Matsumura & Kataoka, 2009). In particular, polymer-drug conjugates in recently ongoing clinical trials have been mainly based on polyHPMA (i.e., HPMA copolymer-DACH (diaminocyclohexane) platinum conjugate) (Nowotnik and Cvitkovic, 2009), PEG (i.e., PEG-irinotecan) (Vergote et al., 2010), or poly(glutamic acid) (i.e., PGA-paclitaxel) (O'Brien et al., 2008) backbones. Some formulations have shown promising anti-cancer efficacy and safety profiles as compared to free drug. For example, HPMA copolymer-DACH platinum conjugate showed significantly higher delivery of the active platinum to tumor cells as well as to the target DNAs of tumor cells than free oxaliplatin. It showed superior safety profile and comparable or better efficacy in recurrent ovarian cancer patients in European phase I/II trials (Nowotnik & Cvitkovic, 2009). PEG-irinotecan exhibited significantly prolonged circulation half-life and lower plasma peak concentration, which may contribute to its reduced toxicity than free irinotecan. It showed clinical responses in 50% of patients in a phase II study in patients with platinum-resistant/refractory breast cancer and is being tested in phase III trials (Vergote et al., 2010). Despite of this, the translation of nanomedicines to real clinical use is still relatively slow as opposed to the rapidly growing preclinical research. A large number of the candidate nanomedicine formulations were discontinued at later stage clinical trials due to insufficient efficacies.
Even the approved nanomedicines do not always provide better overall survival benefits than the small molecule chemotherapeutic interventions based on the clinical experiences (Northfelt et al., 1998; O'Brien et al., 2004; Gill et al., 1996). Only under limited indications (for certain types or stages of cancer), nanomedicines showed mildly prolonged overall survivals compared to standard chemotherapy (Gordon et al., 2001; Gradishar et al., 2005). For example, for metastatic breast cancer patients the overall survival changed from 46.7 weeks for those receiving standard paclitaxel treatment to 56.4 weeks for those receiving Abraxane treatment (Gradishar et al., 2005). In addition, nanomedicines still cannot prevent lethal cancer relapses, which account for large parts of cancer deaths. The long-term deposition of residual nanocarriers in certain organs also potentially leads to nanomedicine-related side effects. These experiences with current nanomedicines indicate that further improvements on the chemical and biological aspects of nanomedicine designs are needed.
Design considerations of current anti-cancer nanomedicines
Strategies to target the cancer cell heterogeneity
Cancer stem cell targeted nanomedicines
Although the biology of CSCs is not fully understood, the CSC concept adds new dimensions to the design of anti-cancer therapeutics. Several attempts to develop CSC-targeted nanomedicines have been reported. But strictly speaking, most of the currently reported designs are not really specific to CSCs; instead, they are therapies to which the resistant CSCs are sensitive. The strategies reported so far can be summarized into the following aspects: (1) employing agents targeting the key stem cell related properties especially self-renewal and differentiation ability; (2) targeting of CSCs mediated by the CSC-specific markers; (3) inhibiting CSCs by overcoming multidrug resistance and sensitizing CSCs to conventional therapies; (4) destruction of CSC-supporting microenvironment.
The search for effective anti-CSC agents has been an essential task, since the traditional small molecule chemotherapeutics or radiotherapies are not the ideal agents due to CSCs' intrinsic resistance. The modulators of several important signaling pathways regulating critical stem cell properties such as self-renewal and differentiation, including hedgehog (Bar et al., 2007; Karhadkar et al., 2004; Thayer et al., 2003), notch (Wang et al., 2009), and wnt pathways (Teng et al., 2010), have come into researchers' sights. Most of these agents have already been developed as a new class of molecularly targeted anti-cancer agents, based on the findings that these stem cell related pathways are aberrantly up-regulated in certain cancers. As the concept of CSCs was gaining acceptance, these pathways have been further correlated with CSC properties, cancer progression, recurrence and metastasis. Most of these small molecule pathway inhibitors have the same drawbacks as other traditional anticancer drugs, such as poor solubility, low stability and nonspecific toxicity. Therefore, macromolecular drug delivery systems carrying the CSC-targeted agents have been developed to improve their anti-CSC behaviors.
Such examples include the recent development of the HPMA copolymer based-drug conjugate incorporating the hedgehog pathway inhibitor cyclopamine (Zhou et al., 2012). Initial studies have shown that the conjugate (P-GFLG-cyclopamine; P is the HPMA copolymer backbone; GFLG a cathepsin B sensitive spacer) inhibited the sphere forming ability as well as the growth of the prostate CSC-enriched population in a prostate cancer cell model. Furthermore, P-GFLG-cyclopamine or free cyclopamine treatments led to significantly decreased percentages of viable CD133+ prostate CSC-enriched population within the whole cancer cell population, in contrast to slight cytotoxicities on bulk cancer cells. On the contrary, docetaxel treatment caused significant bulk cancer cell deaths; however, docetaxel could not decrease the fraction of CD133+ prostate CSCs (Figure 3A) (Zhou et al., 2012). The selective cytotoxic effects of cyclopamine and docetaxel on CSC and non-CSC subpopulations of prostate cancer cells suggest a promising combination macromolecular therapeutic strategy that is comprised of two HPMA copolymer-based conjugates containing cyclopamine and docetaxel respectively (Figure 3B).
Figure 3.
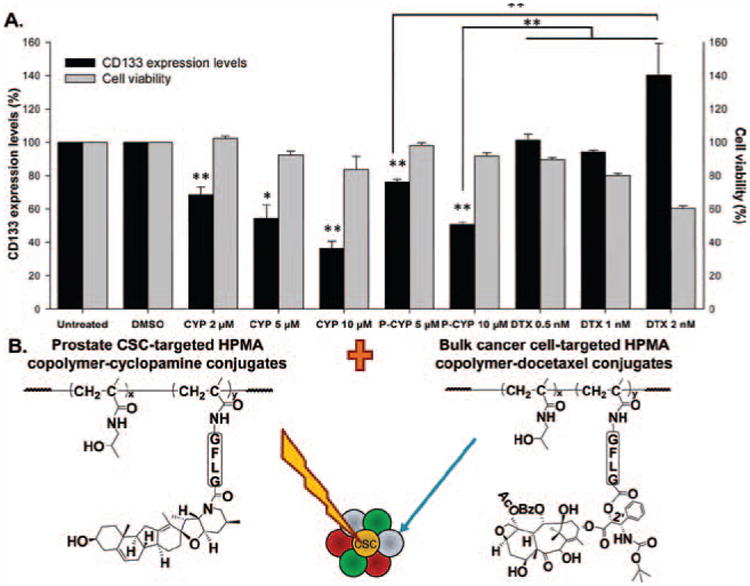
(A) Summary of changes on CD133+ prostate CSCs and whole cell viabilities following HPMA copolymer-cyclopamine conjugate, free cyclopamine or docetaxel treatments on RC-92a/hTERT prostate cancer cells in vitro (Zhou et al., 2012). Black columns: CD133 expression level (%); gray columns: Cell viability (%). The data are presented as mean ± SD of the experiments done in triplicate. *p < 0.05; **p < 0.01. Vehicle (DMSO) treated and untreated cells were used as controls. (B) The scheme of the combination HPMA copolymer-based macromolecular therapeutics for improving the treatment of prostate cancer, by targeting both bulk cancer cells and prostate CSCs. Adapted with permission from Zhou et al. (2012).
In another example, mesoporous silica nanoparticles loaded with γ-secretase inhibitors (GSI) for notch signaling have shown improved tumor growth inhibition. Although the direct anti-CSC effects were not evaluated in this case, such design has the potential to suppress CSCs (Mamaeva et al., 2011). Another formulation, nanoparticle loaded curcumin as a hedgehog inhibitor (NanoCurc™) was developed and showed inhibition of the anchorage-independent growth and reduction of CD133+ CSCs in brain tumor models (Lim et al., 2011). For comprehensive evaluations of these nanomedicines, an important future direction is to assess the normal stem cell (NSC) related toxicities since all these agents target the pathways, which most probably are present in NSCs. However, to which extent these pathways are responsible for NSC maintenance has not been clearly stated yet. Some studies have shown that GSI can cause intestinal cell metaplasia, indicating stem cell related adverse effect (Milano et al., 2004); but some studies found that the inhibition of hedgehog pathway did not affect hematopoietic stem cell (HSC) maintenance and functions (Hofmann et al., 2009; Gao et al., 2009); other studies showed the opposite effect of PI3K/mTOR pathway on leukemia stem cells and HSCs, opening the possibility to specifically target CSCs while sparing NSCs (Yilmaz et al., 2006; Zhang et al., 2006).
High throughput small molecule screening could contribute to the identification of more specific anti-CSC agents. In one study, the antibiotic salinomycin was found to have specific toxicity towards breast CSCs but not the bulk tumor cells by high throughput cytotoxicity screening on both EMT induced CSCs and bulk tumor cells (Gupta et al., 2009). However, the anti-CSC mechanism was not clear. As the screening efficiency and the reliability of the CSC model develop, more molecules specific to CSCs may be discovered and developed into anti-cancer therapeutics.
Active targeting approaches have also been attempted in the design of CSC-targeted nanomedicines. Several surface markers have been commonly used for the identification of CSCs, including CD44, CD133, and certain integrins. Therefore, in some studies, targeting moieties were incorporated to specifically target these biomarkers. The examples are hyaluronan-drug conjugates (Banzato et al., 2008; Choi et al., 2010) and anti-CD133 antibody targeted nanoparticles (Bourseau-Guilmain et al., 2012). The main advantage of this actively targeted therapeutics is to enhance the cellular uptake of nanomedicines into CD44+ or CD133+ cancer cells, thus improving the cell killing effects. However, this approach should be generalized with caution. Current CSC studies found that these biomarkers could not represent the very specific CSC population; furthermore, CSCs may present heterogeneous biomarkers or marker combinations in different cancers and even different subtypes of the cancer from the same tissue origin (Shackleton et al., 2009; Clevers, 2011). In addition, simply directing the therapeutics to the targeted putative CSCs may not result in sensitization of these CSCs to the carried drugs.
A reported example to sensitize the resistant CSCs to conventional radiation therapy is to combine the gold nanoshell-mediated hyperthermia with radiation therapy (Atkinson et al., 2010). As evaluated in both the triple-negative breast cancer syngeneic mouse model and xenograft model, the breast CSC population expanded after ionizing radiation. However, following combination of hyperthermia at 42°C and ionizing radiation, the percentage of breast CSCs, the mammosphere forming ability, and the in vivo tumorigenicity (by limiting dilution transplantation) in the residual tumors significantly decreased compared to the radiation only-treated tumors and mock-treated tumors (Figure 4) (Atkinson et al., 2010).
Figure 4.
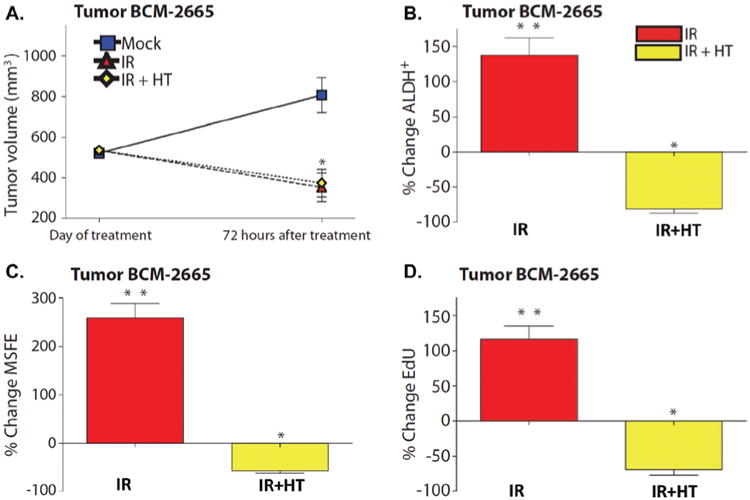
The antitumor and anti-CSC effect of the thermal enhancement with gold nanoshells in combination with radiation in the BCM-2665 breast cancer model. (A) Tumor volume changes following radiation (IR, red triangle) and the combination of radiation and hyperthermia (IR + HT, yellow diamond). (B) The changes in the percentages of ALDH + breast CSCs following IR (red) and IR + HT (yellow) treatments. (C) The changes in mammosphere forming efficiency (MSFE) following IR (red) and IR + HT (yellow) treatments. (D) EdU incorporation following IR (red) and IR + HT (yellow) treatments (*p < 0.05; **p < 0.001). The % changes in B–D were normalized to the mock treatment. Adapted with permission from Atkinson et al. (2010).
Besides the nanocarrier designs, comprehensive evaluations on the CSC properties with and without treatment are of great importance for the development of CSC-targeted nanomedicines. Although considerable nanomedicines were designed aiming at inhibiting CSCs, only few of them actually test directly their inhibitory effects on the CSCs (Vinogradov and Wei, 2012). The development of the standard efficacy evaluation procedures in parallel with the nanomedicine design would be a future direction to push the effective anti-CSC therapeutics into real practice.
Overcoming multidrug resistance
A hurdle to cancer cures is the acquired resistance of cancer cells to various therapies. Besides the CSC concept that offers new insights to the ways to overcome cancer resistance towards traditional therapies, the other mechanisms of cancer multidrug resistance (MDR) have long been studied and developed as targets for nanomedicines. Several known reasons responsible for MDR include the overexpression of drug efflux pumps commonly characterized by ATP-binding cassette family such as P-glycoprotein (P-gp), detoxification processes, enhanced DNA repair, or the imbalance of pro- and antiapoptotic pathways (Dean et al., 2005; Szakács et al., 2006; Broxterman et al., 2009).
Thanks to the unique cellular uptake patterns of nanomedicines by pinocytosis or receptor-mediated endocytosis, the nanocarrier-bound drugs are transferred into the cells inside vesicular compartment, instead of through diffusion, thus circumventing the interactions with the various drug efflux pumps on the cell surfaces. In addition, the drug transport through the endocytotic pathway helps to create a drug concentration gradient decreasing from the perinuclear to the region near the cell membrane, further reducing the chances of its interactions with the efflux pumps (Omelyanenko et al., 1998a,b). The advantages of HPMA copolymer-bound doxorubicin (DOX) (P-GFLG-DOX) over free DOX have been demonstrated in mice models of both DOX sensitive and resistant human ovarian cancer xenografts. Although free DOX could not inhibit the tumor growth in resistant tumors and only shrunk the tumor size three times in sensitive tumors, P-GFLG-DOX led to 18 times and 28 times reduction in tumor size in resistant and sensitive tumors, respectively (Figure 5) (Minko et al., 2000). P-GFLG-DOX not only overcame the MDR by avoiding the P-gp mediated drug efflux, but also modulated other cell responses at the molecular level, including the down-regulation of antiapoptotic Bcl-2 family expressions, suppression of drug detoxification related genes encoding glutathione and UDP, and inhibition of DNA repair mechanisms (Minko et al., 2000, 2001).
Figure 5.
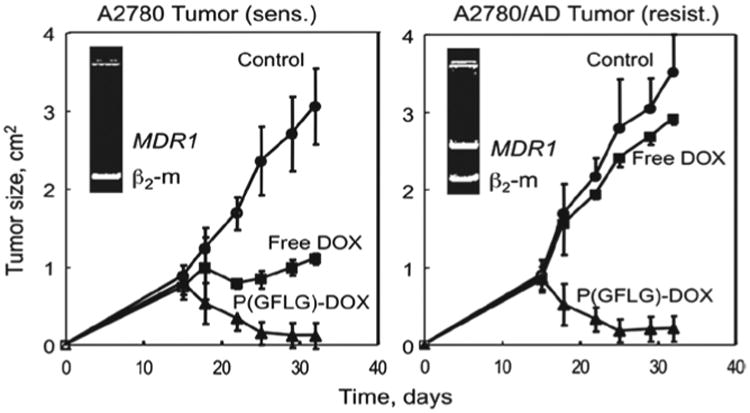
The effect of i.p. administered HPMA copolymer-DOX conjugate and free DOX on the growth of both sensitive (left) and resistant (right) human ovarian carcinoma xenografts in mice. Triangles: HPMA copolymer-DOX conjugate; squares: free DOX; circles: control. The data are presented as mean ± SE (Minko et al., 2000). Reprinted with permission from Kopeček & Kopečková (2010).
Biologically, the inhibition of the drug efflux pump functions can be achieved at the gene, molecular and protein levels by RNA interference, monoclonal antibodies to P-gp or small molecule inhibitors of the drug efflux transporters (Goda et al., 2007; Nadali et al., 2007; Ren et al., 2008; Tsuruo et al., 1981). Nanomedicines have a large potential to increase the tumor accumulation and reduce the nonspecific toxicities of these agents.
Employing molecularly targeted therapeutics to target the hallmarks of cancer
The cancer cell killing effect of the first-generation traditional chemotherapeutic agents utilizes the rapid replication of cancer cells. In the last several decades, extensive advances have been made in nanomedicines delivering these chemotherapeutics. However, the nonspecific drug action mechanism still does not ensure adequate antitumor efficacy and safety. On the other hand, novel anti-cancer therapeutics, so called molecularly targeted therapeutics have been developed (Sharkey & Goldenberg, 2006; Collins & Workman, 2006). This kind of therapeutics exploits agents that specifically target the key molecular events deregulated in cancer. Elements involved in the extensive proliferating signaling, evasion of growth suppression signaling, resistance of cell deaths, replicative immortality, angiogenesis, invasion and metastasis activating pathways, deregulation of cancer cell energetics, avoidance of immuno protection and tumor-promoting inflammation can all be the anticancer therapeutic targets (Hanahan & Weinberg, 2000; Hanahan & Weinberg, 2011). Nanomedicine designs have also been extended to this area. The molecularly targeted therapeutics can take advantages of the macromolecular carrier system to improve their circulation time and stability, avoid degradation in the body and achieve favorable cellular and subcellular entry into the site of action (Davis, 2009; Heidel & Davis, 2011).
Several monoclonal antibodies specific for certain overexpressed cancer cell surface markers have been developed as molecularly targeted therapeutics, such as Trastuzumab targeting HER2 (Pegram et al., 2000) and Rituximab targeting CD20 (Blanco et al., 2011). Besides specific binding, the antibody-cell surface receptor interactions induce further intracellular signaling events that eventually inhibit cancer cell growth or trigger apoptosis. Effective nanomedicines can be developed based on these molecular events by modifying the antibodies. For example, the antibody fragments derived from Trastuzumab can be attached to the nanoparticles containing chemotherapeutics, both as active targeting moieties and exerting their therapeutic effects (Colombo et al., 2010). Another interesting nanomedicine design exploited the unique apoptotic induction effect by the crosslinking of CD20-bound antibodies on the surfaces of malignant B cells (Wu et al., 2010, 2012). Instead of using anti-CD20 and secondary antibodies, the crosslinking was efficiently mediated by the recognition of a pair of pentaheptad peptides (CCE and CCK), which form antiparallel coiled-coils. Raji B cells were first exposed to the anti-CD20 Fab'-CCE construct and allow the binding of Fab' fragments to the noninternalizing antigens CD20. Then the administration of HPMA copolymers containing multiple copies of complementary CCK peptides as grafts led to the crosslinking of the CD20 surface antigens by the formation of coiled-coil heterodimers. This system showed efficient malignant B cell depletions in vivo (Wu et al., 2012).
Strategies to target metastases
The design of nanomedicines can be varied to target different stages of metastases, from cancer cell invasion, intravasation, circulation, and extravasation to seeding and growing in the secondary metastatic sites.
To date, a number of important molecular pathways responsible for cancer cell invasion and dissociation from the bulk tumor have been identified. Therapeutics have been developed to target these pathways, including the modulators of cell adhesion proteins, protease inhibitors, ion or water channel blockers, and the inhibitors for certain transcription factors or signaling molecules (Veiseh et al., 2011). Blocking the cancer cell invasion locally would potentially reduce the possibility of cancer metastasis in distant organs. To increase the access of small molecule therapeutics to the tumor cells, nanomedicines have been employed. For example, liposomal nanoparticles incorporated with anti-RhoA siRNA were used for treating breast cancer; reduced cancer cell invasion was observed as demonstrated in the migration assay (Gao et al., 2010). In another case, the delivery of anti-PAR-1 siRNA by liposomes to melanoma cells led to significant inhibition of lung metastases, indicating the prevention of melanoma cell invasion (Villares et al., 2008).
In addition to directly treating invasive tumors, imaging of the cancer invasive front by nanoparticles could aid to achieve a more complete surgical removal of the whole tumor. Nguyen et al. and Olson et al. linked the protease activatable cell penetrating peptides (ACPPs) to the surface of dendrimers (ACPPDs) dually labeled with Cy5 and gadolinium (Gd) (Nguyen et al., 2010; Olson et al., 2010). The ACPPs, once cleaved in the invasive tumor microenvironment by MMPs, would be activated and exhibit cell penetrating functions. This led to the significantly increased uptake of the dendrimers into tumor cells. Thus, the tumor margins were marked by the internalized dendrimers under pre-operative MRI and intraoperative optical imaging (Figure 6). The proportion of residual tumor cells was reduced by 90% following this imaging guided surgery than by the standard surgery.
Figure 6.
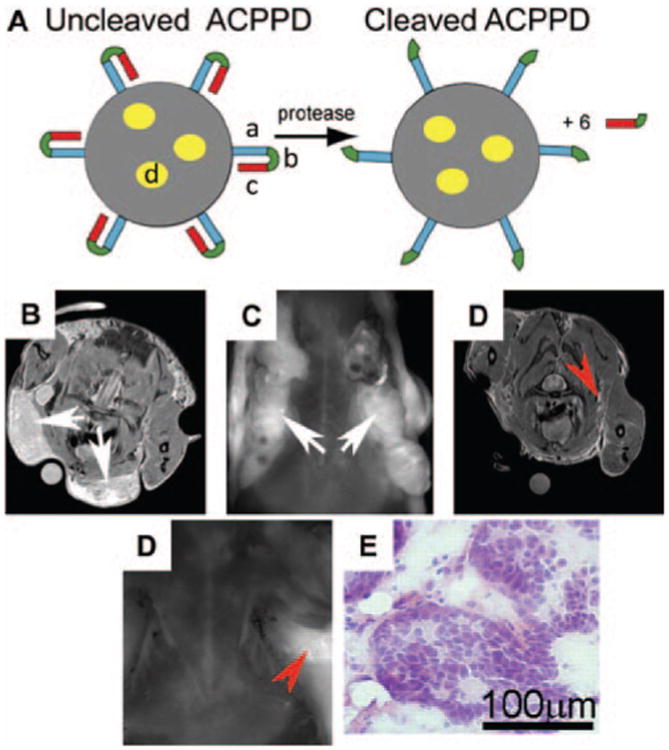
(A) The structure of Activatable Cell Penetrating Peptide-conjugated Dendrimer (ACPPD). Multiple ACPPs are covalently attached via the polycationic segments (a) to the dendrimer (gray circle). Yellow ovals (d) demonstrated the payloads such as Cy5 and/or Gd. Upon exposure to MMP-2 or -9 in tumor microenvironment, the linkers (b) are cleaved and polyanions (c) are released, leaving the cationic dendrimers for cell entry. (B–E) The enhanced uptake of Gd and Cy5 dually labeled ACPPDs in regions with high MMP activities and infiltrative tumors. (B–C) Axial MR and fluorescence images of a transgenic PyMT mouse before surgery (white arrows: the tumor burdens); (D–E) Axial MR and fluorescence images of the same mouse after surgical removal of tumor under white light. Red arrows: regions of residual hyperintensity on MR and fluorescence imaging; (F) Regions of hyperintensity on MRI were removed under fluorescence imaging and stained with hematoxylin/eosin to verify the presence of tumor. Adapted with permission from Olson et al. (2010).
On the other hand, once the metastatic tumors in the secondary organs are established, nanomedicines can be exploited as a multifunctional platform to achieve both passive and active targeting to improve the therapeutic effect of the free drug. Due to the favorable metastatic niches in certain organs, life-threatening metastases are prone to take place in organs such as bone, liver, brain, lung or lymph nodes (Schroeder et al., 2012). Therefore, the tissue specific properties can be exploited for efficient targeting. For instance, to treat breast cancer bone metastasis, an HPMA copolymer conjugate containing the chemotherapeutic agent paclitaxel (PTX) as well as bone-targeting moiety alendronate (ATN) was designed (Miller et al., 2011). Paclitaxel is cytotoxic to rapidly dividing cancer cells; in addition, it exhibits antiangiogenic effects at low dose (Wang et al., 2003). Alendronate is an aminobisphosphonate that prevents the bone breakdown. It can be used as bone-targeting moiety due to its strong binding to bone mineral hydroxyapatite (Pan et al., 2008). The in vivo evaluation in a murine syngeneic model of breast adenocarcinoma showed the greatest metastatic tumor growth inhibition, decreased microvessel density and increased circulating endothelial cell apoptosis of the HPMA copolymer-PTX-ATN conjugate, compared with the combination of free PTX and ATN, or PTX alone (Figure 7). In another study to treat primary and metastatic ovarian cancer, a PEG based polymer-drug-peptide conjugate was constructed containing three components - an anticancer drug camptothecin (CPT), a therapeutic peptide BH3 that suppresses antiapoptotic defense, and a synthetic analog of luteinizing hormone-releasing hormone (LHRH) as tumor targeting moiety. Although the metastatic cancer cells found in malignant ascites were more resistant and aggressive than the primary tumor cells, this conjugate was effective to inhibit the resistant cancer cell growth and suppress the development of metastatic malignant ascites (Chandna et al., 2010).
Figure 7.
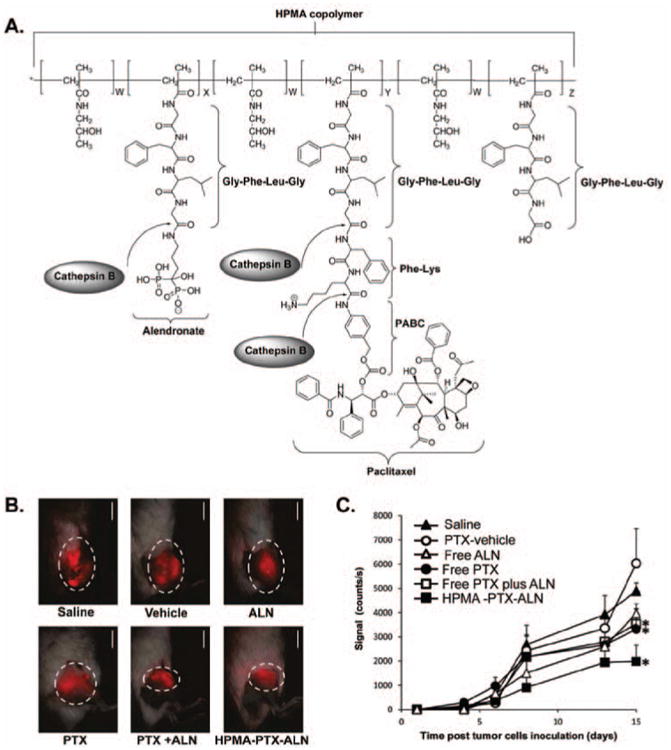
Inhibition of metastatic 4T1 mammary adenocarcinoma in the tibia by HPMA copolymer-PTX-ALN conjugate. (A) Chemical structure of the conjugate and the release mechanism of PTX and ALN from the conjugate. (B) Fluorescent images of 4T1 mCherry tumors in the tibia following single, combination of free drugs, and HPMA copolymer-PTX-ALN conjugate treatments on day 15. The treatments were administered i.v. every other day. Scale bar represents 15 mm. (C) Antitumor efficacies of HPMA copolymer-PTX-ALN conjugate (closed squares), free PTX plus ALN (open squares), free PTX (closed circles), and free ALN (open triangles) as compared to vehicle (open circles) and saline (closed triangles) controls. Y-axis represents the fluorescent intensities of the tumors as measured quantitatively by intravital noninvasive fluorescence imaging. The data are presented as mean ± SE. PTX, paclitaxel; ALN, alendronate. Adapted with permission from Miller et al. (2011).
The lymphatic system and pro-lymphangiogenesis factors always promote the lymph node metastasis, which is very detrimental to cancer patients (Alitalo et al., 2005; Tammela & Alitalo, 2010; Wang & Oliver, 2010). Lymph node targeted anti-cancer nanomedicines would be an indispensible direction against tumor metastasis. Several anti-lymphangiogenetic drugs have been developed with the potential to suppress tumor metastasis through lymphatic vasculatures (Chandna et al., 2010). However, homing of the therapeutics to lymph nodes is the prerequisite for efficient treatment. Interestingly, some properties of nanoparticles have been linked with the preferential targeting to lymph nodes following systemic administration (Hsu & Juliano, 1982; Nahrendorf et al., 2011; Harisinghani et al., 2003). Selected nanoparticles in the circulation were found to bind to the leukocyte surface receptors, facilitating their transport to lymph nodes and accompanied with leukocyte homing as a part of normal immune process (Raz et al., 1981). Furthermore, several modifications such as carbohydrates or immunoglobulin G coating of the nanoparticles were found to enhance their accumulation in lymph nodes (Tassa et al., 2011). Based on these properties, iron oxide nanoparticles (Combidex®), which are biocompatible and non-toxic, have been used in clinics for the imaging of lymph node metastasis (Harisinghani et al., 2003).
Specific targeting moieties for lymphatic system homing have also been identified (Laakkonen et al., 2004). For example, a homing peptide LyP-1, selected by phage display, can specifically bind to tumor cells in the hypoxic regions, tumor-associated macrophages as well as the endothelial cells of tumor lymphatic vasculatures in certain tumors, but not to normal endothelial cells. Moreover, LyP-1 exhibited proapoptotic and cytotoxic activities to these malignant tumor and endothelial cells. Recent studies using LyP-1 conjugated liposomes showed the potential of the actively targeted nanomedicines to treat and suppress the lymph mode metastatic tumors (Yan et al., 2011; Yan et al., 2012).
Combination therapy
Anti-cancer combination therapies refer to both the combination of different anti-cancer drugs and the combination of different treatment modalities such as surgery, chemotherapy, and radiotherapy. The therapeutics should be combined in the ways that ultimately achieve synergistic anti-cancer effect better than either type of the therapies alone, or reduce the adverse effects while maintaining the therapeutic effect, or achieve both. Notably, the nanosized drug carriers present the targetability to tumor site, the ability to reduce free drug side effects and the capacity to incorporate multifunctional components. These properties make nanomedicines well suited for the development of improved anti-cancer combination therapies. Several possible strategies of combination therapies will be discussed below, mainly those using polymer-based macromolecular carriers, especially HPMA copolymer conjugates as examples.
Combination of the polymer-bound anti-cancer therapeutics
HPMA copolymer-based linear drug carriers allow the introduction of different functional groups to the side chains of the conjugate, thus facilitating the attachment and delivery of different types of therapeutics in one polymer construct to the tumor simultaneously. An example for this type of combination therapy is the development of an HPMA copolymer conjugate containing both ALN and a potent anti-angiogenic agent TNP-470, for the treatment of osteosarcoma (Segal et al., 2009). By delivering two therapeutic agents to the tumor site at the same time and location, the conjugate showed synergistic anti-cancer effect by inhibiting the proliferation of osteosarcoma cells, as well as anti-angiogenic effects by inhibiting the vasculature formation and reducing vascular permeability. In addition to the significant tumor growth inhibition by 96%, this macromolecular delivery approach dramatically reduced the adverse effects of the free TNP-470.
The combination of chemotherapeutic agents is also of much clinical values since it is hardly possible to treat certain advanced cancers without combination therapy. Although the macromolecular combination chemotherapeutics is not available in clinical practice so far, researches have been attempting to address this need. For example, Mayer and coworkers have developed and evaluated liposomes co-loaded with both irinotecan and floxuridine, or both daunorubicin and cytarabine (Tardi et al., 2007; Tardi et al., 2009); they are currently trying to translate the combination formulations into clinical use. Vicent, Duncan and coworkers designed an HPMA copolymer conjugate containing DOX and anti-hormonal agent AGM (aminoglutethimide) linked through lysosomally degradable linkers. It showed synergistic therapeutic effect in vitro in the hormone dependent breast cancer cell model (Greco et al., 2007). Lammers et al. evaluated another combination, an HPMA copolymer-gemcitabine-doxorubicin conjugate in vivo, demonstrating the enhanced apoptosis induction and anti-angiogenic effects of the combination conjugate than the conjugates containing either single agent alone (Lammers et al., 2009). For further development of the combination nanomedicines with multiple drugs in the same carrier, it is critical to control the content of each drug as well as the incorporated drug ratios for optimal therapeutic effect.
The synergistic effect of the two different anti-cancer therapeutic agents could also be observed when incorporated separately into different nanosized drug carriers. This has been demonstrated in the HPMA copolymer-based combination chemotherapy and photodynamic therapy (PDT) (Shiah et al., 1999; Shiah et al., 2000). To this end, mesochlorin e6 mono(N-2-aminoethylamide) (Mce6) was chosen as the photosensitizer that is activated by a specific wavelength of light to generate singlet oxygen. Although the clinical application of PDT using low molecular weight photosensitizers was limited due to the severe nonspecific side effects to normal tissues, the HPMA copolymer-bound Mce6 plus light reduced the side effects significantly, while still remaining comparable antitumor effect. Furthermore, the combination of HPMA copolymer-GFLG-DOX conjugate and HPMA copolymer-GFLG-Mce6 conjugate showed improved antitumor response than either chemotherapy or PDT alone in mice bearing human ovarian carcinomas (Shiah et al., 1999, 2000).
The selection of combination strategies (different drugs on one drug carrier or a combination of two drug carriers containing one drug each) is affected by a collection of issues regarding to the drug properties, mechanism and site of drug action, drug release, biodistribution/pharmacokinetics, as well as the ease to control the synthesis and scale-up. If the two drugs have different physicochemical properties and pharmacokinetics, the body and subcellular distributions may be different between the cases of the conjugate containing both drugs and the two conjugates each containing one drug, thus, leading to different anti-cancer efficacies. As demonstrated in some cases such as the combination of AGM (aminoglutethimide) and DOX, only when conjugating the two drugs to one HPMA copolymer carrier, synergistic anti-cancer effect was observed and higher efficacy was shown as compared to the combination of two conjugates (Greco et al., 2007). While in other cases, the combination of two conjugates was able to induce synergistic anti-cancer effect (Hongrapipat et al., 2008a,b). Therefore, it would be important to evaluate thoroughly the combination effects case by case for the choice of combination patterns, for example, by analyzing the combination index (CI) (Hongrapipat et al., 2008a,b). However, from the synthetic point of view, the conjugate properties and the incorporated drug amounts is more controllable when using the combination of two conjugates than the conjugate containing two types of drugs. In addition, the drug release kinetics might be altered when the two types of drugs attach to the same polymer (Vicent et al., 2005).
The heterogeneity of tumor cells as well as the tumor microenvironment renders the treatment of tumors more difficult by single type of nanomedicine. Therefore, another advantage of the combination of multiple macromolecular drug carriers is the possibility to target distinct tumor populations to improve the antitumor effects. Several directions worth to consider include: first, based on the current findings of the biological importance of CSCs, it is conceivable to develop nanomedicines comprised of two entities: one targeting CSCs and the other targeting bulk tumor cells (see Figure 3); second, to address the problem of heterogeneous tumor penetration of passive targeted nanomedicines, it would be possible to combine the nanomedicines that are designed to preferentially localize in tumor vessel-rich areas with those preferentially targeting poorly vascularized hypoxic tumor regions (Gupta et al., 2011; Laakkonen et al., 2004).
Polymer-bound anti-cancer therapeutics combined with other treatment modalities
Surgery, radiotherapy, hyperthermia, immunotherapy, etc. are all important anti-cancer approaches. These modalities are usually combined with chemotherapy in clinical practice. The combination of macromolecular chemotherapeutics with the above mentioned modalities might provide unique benefits for cancer treatment. As an example, the investigation of “carrier-based radio-chemotherapy”, which refers to the combination of radiotherapy with macromolecular chemotherapy, is briefly discussed here. The traditional small molecule-based chemotherapy enhances the therapeutic effect of the simultaneously applied radiotherapy; however, usually it is not as well tolerated as when used alone (Maduro et al., 2003). In contrast, when employing polymeric carriers for the delivery of chemotherapeutic agents, the therapeutic index for radiochemotherapy could be increased, due to the reciprocal enhancement of the chemo- and radiotherapy (Lammers, 2010). As early as a decade ago, radiotherapy was found to improve the tumor accumulation, even the tumor penetration of polymeric drug conjugates or other nanosized drug carrier systems, for example, poly(l-glutamic acid)-paclitaxel conjugate (Li et al., 2000), PEGylated DOX-loaded liposome (Davies et al., 2004), and HPMA copolymer-drug conjugate (Lammers et al., 2007). This could be due to the increased VEGF and/or basic fibroblast growth factor (bFGF) expression within the tumor which increase the vascular permeability, the loosening of tumor tissues by the reduced tumor cell density and the decrease of IFP, induced by radiotherapy (Lee et al., 1995; Chung et al., 2006; Peschke et al., 1999). Meanwhile, given the fixed dosing frequencies of the chemo- and radiotherapies, the prolonged and enhanced accumulation of the macromolecular chemotherapeutics at the tumor site in turn prolonged the interaction periods during which the chemo- and radiotherapy are both exposed at the tumor tissue (Lammers, 2010).
Strategies to improve passive targeting
The currently developed tumor-targeted macromolecular therapeutics largely depends on the EPR effect to achieve enhanced delivery to the tumor site. However, the EPR effect is often more pronounced in animal models than in real patients. The nanomedicines are usually evaluated in mice when reaching certain tumor sizes, but the tumors in mice are much larger in terms of the size and weight percent of total body weight than those found in patients. In addition, the inoculated tumor cells grow much faster in mice models than the development of tumors in patients, resulting in different tumor pathophysiologies. For example, the inadequately developed tumor vessels tend to be more leaky and poorly differentiated, covered with fewer pericytes in mice models than in humans (Lammers et al., 2012). Therefore, observing favorable tumor accumulation in mice does not ensure the same tumor-tropic effect of the identical nanomedicine formulation in patients. The extent of tumor accumulation of the macromolecular drug carriers must be further improved, in part by optimizing the structures of the drug carriers. Here, HPMA copolymer based drug carriers are used as an example to discuss the possible strategies to augment the passive tumor accumulation.
Biocompatible long-circulating HPMA copolymer-based linear drug carriers
The molecular weights of the first-generation HPMA copolymer-based linear drug carriers used in research as well as in clinical trials were limited to below around 45 kDa, to ensure the safe renal excretion through glomerular filtration after intravenous administration (Seymour et al., 1987). Although these drug carriers have excellent safety profiles as shown in patients, they were quickly excreted with limited tumor accumulation and antitumor efficacy. On the other hand, in vivo research has demonstrated that a higher molecular weight of the polymer-drug conjugate contributes to the longer circulation retention time resulting in higher accumulation in the tumor tissue (Figure 8) (Seymour et al., 1995; Shiah et al., 2001a; Etrych et al., 2012). Using drug carriers with non-degradable polymer backbone, it is extremely hard to tune the molecular weight of the drug carriers to allow both safety and antitumor efficacy. With the advances in chemistry and living radical polymerization, the synthesis of backbone biodegradable multiblock linear HPMA copolymer drug carriers became possible; such carriers would achieve the enhanced tumor accumulation, augmented antitumor effects, and elimination of polymer carrier fragments from the body at the same time.
Figure 8.
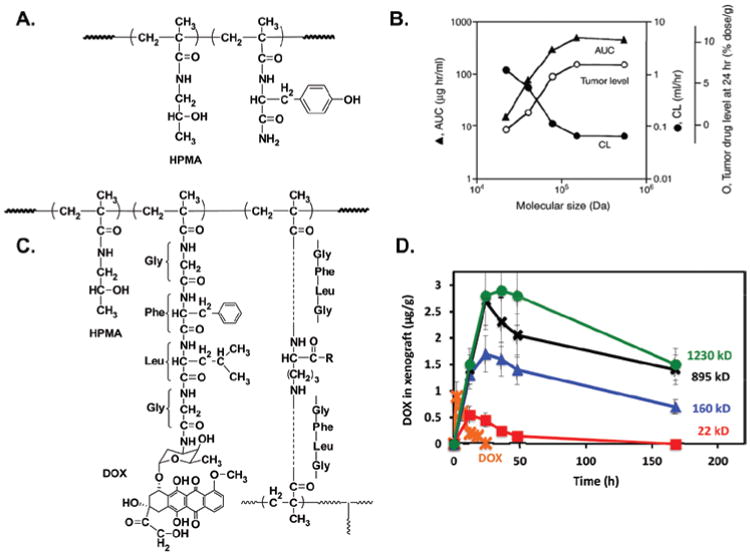
(A) The chemical structure of Tyr-HPMA-copolymer. (B) The relationship between the molecular size of i.v. administered 125I-labeled Tyr-HPMA-copolymers and the plasma concentration (AUC), tumor accumulation and renal clearance (CL) in sarcoma-180 tumor-bearing mice (Seymour et al., 1995; Fang et al., 2011). (C) The chemical structure of the branched HPMA copolymer-DOX conjugate. (D) Tumor accumulation of DOX in OVCAR-3 carcinoma bearing nu/nu mice after i.v. bolus of free DOX or HPMA copolymer-DOX conjugate with different molecular weights (Shiah et al., 2001a).
In linear biodegradable conjugates, multiple telechelic HPMA copolymer fragments with molecular weights below the renal threshold are linked together by spacers containing lysosomally degradable tetrapeptide GFLG sequences, thus leading to the intracellular biodegradation of the resultant high molecular weight (HMW) conjugate. The synthesis of this multiblock backbone degradable HPMA copolymer-drug conjugate usually consists of two main steps (Pan et al., 2011; Yang et al., 2011; Luo et al., 2011). First, the low molecular weight telechelic HPMA copolymer-drug fragments can be synthesized by RAFT (Reversible Addition-Fragmentation chain Transfer) copolymerization of HPMA, polymerizable drug-containing comonomers and possibly other polymerizable comonomers for imaging or targeting purposes. The RAFT copolymerization ensures precise control of the molecular weight and polydispersity of the copolymer fragments. In the second step, the polymer chains are extended by alkyne-azide or thiol-ene click reaction of the polymer fragments with the functionalized spacers, resulting in the conjugate with high molecular weight (100–300 kDa). Multiblock HPMA copolymer conjugates containing anti-cancer drugs DOX or gemcitabine were synthesized (Figure 9). The exposure of the conjugates to enzymes, model thiol proteinase papain or lysosomal cathepsin B, led to complete degradation of the backbone as well as efficient release of the drugs (Pan et al., 2011; Yang et al., 2011).
Figure 9.
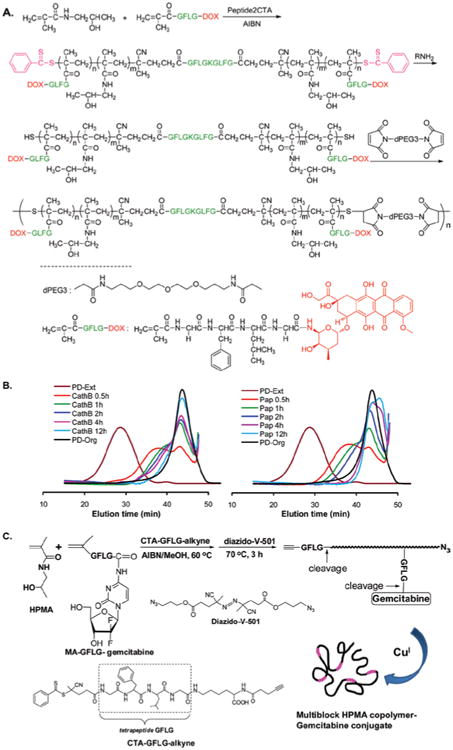
(A) The synthesis of multiblock HPMA copolymer-DOX conjugate. The HPMA copolymer segments containing DOX was synthesized by RAFT copolymerization of monomer HPMA and MA-GFLG-DOX in the presence of a bifunctional chain transfer agent containing GFLG sequences (Peptide2CTA). The chain was extended by the reaction of the telechelic α,ω-dithoil-HPMA copolymers with bismaleimide, yielding the multiblock biodegradable HPMA copolymer-DOX conjugate (Pan et al., 2011). (B) FPLC profiles showing the degradation of multibock HPMA copolymer-DOX conjugate by incubation with cathepsin B (left) and papain (right) at different time intervals. PD-Org: the initial telechelic HPMA copolymer-DOX conjugate; PD-Ext: the extended multiblock HPMA copolymer-DOX conjugate (Pan et al., 2011). C) The synthesis of multiblock HPMA copolymer-gemcitabine conjugate. The heterotelechelic HPMA copolymer-gemcitabine conjugate segments containing terminal alkyne and azide groups was firstly synthesized by RAFT copolymerization using the chain transfer agent containing GFLG sequence and alkyne group (CTA-GFLG-alkyne), followed by post-polymerization modification with diazido-V-501. The chain extension was achieved by Cu (I) catalyzed azide-alkyne click reaction (Yang et al., 2011). Adapted from Pan et al. (2011); Yang et al. (2011).
Biodegradable high molecular weight drug carriers with other architectures
The preparation of HMW HPMA copolymer based drug carriers can be achieved by other synthetic routes than those described above. Branched water-soluble HPMA copolymer-drug conjugates with molecular weights ranging from 100 to more than 1000 kDa for anti-cancer purposes have been synthesized in earlier studies. To allow degradation of the conjugates, lysosomally degradable tetrapeptide (GFLG) was incorporated into the crosslinking agent, N2,N5-bis(N-methacryloylglycylphenylalanylleucylglycyl)-ornithine. The final drug-containing conjugate was synthesized by one-step radical copolymerization of HPMA, the crosslinking agent, and the polymerizable comonomer containing anti-cancer drugs. The reaction was performed in the presence of a chain transfer agent, 3-mercaptopropionic acid (3-MPA), which lead to the controlled length of the primary polymer chains below the renal threshold. This method has been used to synthesize HMW branched HPMA copolymer-DOX conjugates (Dvořák et al., 1999; Shiah et al., 2001a). This approach permitted relatively reproducible synthesis of the biodegradable HMW polymers; however, with wider molecular weight distributions than the linear copolymers synthesized by living radical polymerization.
The grafted HMW HPMA copolymer-drug conjugate was also successfully synthesized, by both conventional free radical polymerization and RAFT polymerization (Etrych et al., 2008; Chytil et al., 2010). DOX, used as a model drug, was attached to the grafted polymeric drug carrier by hydrazone bonds, which can be cleaved under the mildly acidic intracellular pH. The grafted copolymer conjugate was synthesized by grafting the semitelechelic HPMA copolymers to the biodegradable spacer-containing side-chains of the primary HPMA copolymers, via several means, such as the reactions of N-hydroxysuccinimide (OSu), thiazolidine-2-thione (TT) or pyridyl disulfide (PDS) end groups in the semitelechelic copolymers with the respective functional groups, hydrazide, amino, and SH groups in the primary polymer precursor, respectively. The resultant grafted HMW HPMA copolymer-DOX conjugates can be degraded intracellularly upon the cleavage of the GFLG or disulfide spacers between the primary and grafted polymer chains, allowing renal clearance of the low molecular weight polymer fragments (Etrych et al., 2008). Recently, RAFT polymerization was employed to facilitate the synthesis of biodegradable HMW grafted HPMA copolymer-drug conjugate with well-defined structure and narrow polydispersity (Chytil et al., 2010). Briefly, HPMA copolymers terminated with thiocarbonylthio groups were firstly synthesized by RAFT copolymerization; then the thicarbonylthio groups were removed and the thiol-terminated HPMA copolymers were modified to introduce different functional groups. The resultant semitelechelic polymer precursors can be grafted to the side chains of another type of HPMA copolymer chains to yield the final grafted conjugate.
Studies investigating the in vivo behaviors of both the branched and grafted HPMA copolymer-drug conjugates showed significantly prolonged blood circulation and enhanced tumor accumulation of the HMW conjugates than the low molecular weight ones in tumor-bearing mice models, with concomitant significantly higher antitumor activity in vivo (Shiah et al., 2001a; Etrych et al., 2008, 2012).
Strategies to improve tumor tissue penetration
After extravasation, nanomedicines and small molecule drugs still need to travel sufficiently deep to reach as many cancer cells as possible to elicit the tumor cell killing effect. But barriers, including the increased IFP, high cellular density, and dense ECM further impede the efficient diffusion of anti-cancer therapeutics (Minchinton & Tannock, 2006). Earlier studies of doxorubicin intratumoral distribution have shown the heterogeneous drug accumulation, with more doxorubicin around the tumor vessels but little or no doxorubicin in tumor hypoxia regions (Primeau et al., 2005). Nanomedicines usually have much larger size than small molecule anti-cancer drugs and it is more difficult to penetrate the tumor tissue (Lammers et al., 2012). So paradoxically, the high molecular weight nanomedicines can enhance the EPR effect by extravasation, but at the risk of reducing tumor tissue penetration compared to the low molecular weight counterpart. Therefore, it would be important to tailor the size of the nanomedicine so that it possesses both long circulation time and tumor penetration properties.
Several additional strategies have been explored to improve the tissue penetration of nanomedicines, most of which were designed to overcome the aforementioned anatomical and physiological barriers, by pretreatment or co-administration of various tumor microenvironment-modifying agents.
The choice of the “tumor penetration improving agents” largely depends on the dominant factors that inhibit the drug transport in different tumor types. Enzymatic degradation of different ECM components has sensitized certain tumors to standard chemotherapies and nanomedicines. For example, studies showed that pretreatments by collagenase could increase the penetration of antibodies and viral nanoparticles in metastatic ovarian carcinoma model and human melanoma xenografts, respectively (McKee et al., 2006; Choi et al., 2006). Early studies also demonstrated the chemotherapy sensitizing effect of hyaluronidase in certain tumor cell lines (St Croix et al., 1998). The proposed mechanism is that hyaluronidase can degrade hyaluronan (HA) and loosen the ECM, thus reducing the adhesion of the anti-cancer drugs to ECM and increasing the drug exposure to cancer cells.
Recently, the role of HA in remodeling tumor microenvironment and drug resistance has been more clearly demonstrated. Moreover, a PEGylated formulation of hyaluronidase has been developed with promising chemo-sensitizing effects by improving tumor penetration (Thompson et al., 2010; Provenzano et al., 2012). Certain cancers are enriched with HA in the ECM, with pancreatic ductal adenocarcinoma (PDA) as a perfect example. PDA is well known for its lethality, extremely hypovascular microenvironment and thus severe resistance to chemotherapeutics such as gemcitabine. Studies have shown that the aberrantly high accumulation of HA contributes to the dense ECM and the increased tumor IFP, subsequently compressing the tumor vasculature and largely impeding the delivery of therapeutics (Provenzano et al., 2012). Based on the pathological role of HA, strategies to break down HA have the potential to facilitate the penetration of drugs or nanoparticles. rHuPH20 is a FDA-approved recombinant human hyaluronidase, only used locally due to its rapid inactivation in the body. In contrast, the PEGylated rHuPH20, PEGPH20, possesses significantly increased circulation half-life and makes the systemic intravenous administration possible. Systemic administration of PEGPH20 to mice bearing PDA depleted HA, decreased the IFP rapidly to the level as in normal pancreas within 24 h of treatment and subsequently reopened the tumor vessel lumens widely. This remodeling is considered irreversible since these changes are still present even long time after the stop of treatment. More beneficial effects were observed following the combination therapy of PEGPH20 with the standard chemotherapeutic gemcitabine. The tumor microenvironment was further remodeled extensively with deaths of stromal cells and secondary reduction of collagen deposition, leading to soft and highly vascularized tumor tissue. The combination therapy significantly increased the response rate to gemcitabine, improved the overall survival and reduced the metastatic burden (Figure 10) (Provenzano et al., 2012).
Figure 10.
The tumor remodeling and anti-tumor effects of Gemcitabine + PEGPH20 (PEGylated recombinant human hyaluronidase PH20) combination therapy on KrasLSL-G12D/+;Trp53LSL-R172H/+;Cre (KPC) mice. (A) Tumor IFP at survival endpoint decreased significantly after treatment with gemcitabine + PEGPH20 (GP; n = 9) than with gemcitabine (G; n = 9) only. *p < 0.0001. (B and C) Tumor was hypovascular following gemcitabine treatment (B), while tumor vasculatures were increased following gemcitabine + PEGPH20 combination therapy (C). D) Tumor volume changes following Gemcitabine and Gemcitabine + PEGPH20 combination treatments after one cycle (*p = 0.009). (E) Kaplan-Meier survival curves of mice in different treatment groups: control (n = 16), PEGPH20 (n = 15), Gem (n = 16), and Gem + PEGPH20 (n = 14). *p = 0.004 demonstrated the significant difference in median overall survival of Gem (55.5 days) and Gemcitabine + PEGPH20 (91.5 days) treated mice. (F) Metastatic burden in Gemcitabine + sPEGPH20 treated mice was significantly decreased compared with Gem treatment alone (G) (*p = 0.014). Adapted with permission from Provenzano et al. (2012).
For certain cancers such as some types of pancreatic cancer (PDA), breast and prostate cancers that promote stromal desmoplasia, methods inhibiting stromal cells and fibrosis such as administration of low dose transforming growth factor-β improve the penetration of therapeutics and enhance their anti-cancer effect (Olive et al., 2009; Kano et al., 2007).
Another distinct strategy to improve the tumor tissue penetration is to actively drive the therapeutics deeply into the tumor tissue by conjugating certain targeting moieties with tissue penetrating functions to nanomedicines or free drugs. These peptides usually contain a key sequence motif R/KXXR/K at the C terminal, which allows specific binding to an overexpressed cell surface receptor neuropilin-1 on tumor cells (Ruoslahti, 2012; Sugahara et al., 2009). The binding is thought to induce rapid endocytic/exocytic bulk transport pathway that allows the transport of nanomedicines across multiple cell layers deep into the tumor tissue. More interestingly, even the co-administration of nanoparticles or free drugs with unconjugated peptides would lead to enhanced penetration of the therapeutics into the core of the tumor tissues (Sugahara et al., 2010). This is probably due to the activation of the bulk transport system that takes along other molecules with the peptides together. An example of this type of tissue-penetrating peptides includes iRGD (CRGDKGPD), in which the internal R/KXXR/K motif is exposed to C terminus after protease cleavage of the whole sequence upon binding to the tumor vasculature.
Issues regarding to the interaction of the drug payload with the therapeutic targets
Drug payload and potency
An important goal of a drug delivery system is to deliver optimal amount of the bioactive therapeutic agents to tumor cells while minimizing the drug amount in normal organs. The amount of the anti-cancer drug reaching its cellular/subcellular target is determined by the delivery efficiencies in various processes including the access of nanomedicines to the tumor tissue, transport of nanomedicines to tumor cells within tumor microenvironment, uptake by tumor cells as well as the release of the drug payload at the site of action. These issues are discussed respectively in the context of this review. Besides these aspects, the capacity of drug loading into the nanocarrier also limits the dose delivered to tumor (Taurin et al., 2012). Thus, it is essential to develop drug conjugation chemistries that allow sufficient incorporation of drug into the drug carrier and separation of the conjugate from unbound drug. In parallel, precise characterization of the drug amount in terms of the weight % of all drug carrier components is necessary to facilitate the accurate in vitro and in vivo efficacy and safety evaluations of nanomedicines (Duncan & Gaspar, 2011). To be noted, the potency of the therapeutic agent to be delivered affects the safety and efficacy of the resulting drug delivery system. Due to the physiological properties of nanocarriers, it is impossible to absolutely avoid the accumulation of drug in normal organs, though it is possible to reduce certain nonspecific toxicities of the delivered drugs. Certain nanomedicine formulations show strong anti-cancer efficacy, but severe toxicity induced by the potent anti-cancer drug in other organs can prevent their clinical uses. For example, the once marketed anti-cancer formulation Mylotarg (conjugate of anti-CD33 antibody with calicheamicin) was withdrawn from clinical use due to its severe side effect found in high incidence during the post-marketing surveillance (Hütter & Schlenk, 2011). On the other hand, the drug needs to be potent enough so that the dose required achieving therapeutic effect is within the practical range for formulation and administration. Thus, the choice of appropriate anti-cancer drug plays an important role for the development of successful nanomedicines. An example of a successful antibody-drug conjugate is the recently approved anti-CD30 antibody-auristatin conjugate for the treatment of Hodgkin's lymphoma and anaplastic large cell lymphoma (Rothe et al., 2012).
Cellular uptake and subcellular trafficking
As early as 1906, the concept of “magic bullet” was proposed by Paul Ehrlich and the importance of biorecognition for drug efficacy was emphasized (Ehrlich, 1906). Numerous drug delivery systems employing active targeting moieties have been tested in vitro and in vivo, showing more effective binding and cellular uptake than the corresponding non-targeted systems. For example, studies evaluating HPMA copolymer-anti-PSMA antibody conjugate on PSMA overexpressing prostate cancer cells showed that the antibody after conjugation had comparably strong binding affinity with that of the free antibody in vitro (Liu et al., 2009). The subsequent endocytosis of the actively targeted conjugate into prostate cancer cells was faster than the control conjugate with nonspecific IgG. Further characterizations demonstrated multiple endocytotic pathways involved including clathrin-mediated endocytosis, clathrin-/caveolae-independent endocytosis and macropinocytosis, eventually directing the endosomal and lysosomal trafficking of drug containing conjugate. In another in vitro cancer model, HPMA copolymer-anti-cancer drug-OV-TL16 antibody conjugate showed enhanced binding and internalization into ovarian carcinoma cells, with concomitant higher cytotoxicity than the non-targeted conjugate (Omelyanenko et al., 1998b). Furthermore, in vivo evaluations of HPMA copolymer-DOX-OV-TL16 antibody and HPMA copolymer-Mce6-OV-TL16 antibody conjugate combination therapy did show significantly enhanced tumor accumulation and therapeutic efficacy of the targeted conjugates than non-targeted conjugates (Shiah et al., 2001b).
The polymer conjugate platform with versatile synthetic possibilities, as well as the use of smaller sized targeting moieties such as antibody fragments, natural ligands and peptides enables the incorporation of multiple targeting ligands into the same nanocarrier. This multivalent effect can further enhance the biorecognition, resulting in increased therapeutic efficacy (David et al., 2001).
Whereas the advantageous cellular interactions of actively targeted nanocarriers have been confirmed in various studies, the effect of active targeting in overall tumor accumulation is controversial across different studies. Many studies did not reveal significantly higher tumor accumulation following targeted nanomedicines than non-targeted ones (Kirpotin et al., 2006; Bartlett et al., 2007; Gabizon et al., 2003; Kwon et al., 2012). In a study of folate receptor (FR)-targeted liposome, the co-administration of free folate could not affect the extent of tumor accumulation of the targeted liposomes, suggesting the negligible effect of active targeting on tumor accumulation (Gabizon et al., 2003). In contrast to this, enhanced tumor accumulations of targeted nanomedicines have been observed in other studies (Seymour et al., 2002; ElBayoumi & Torchilin, 2009). These results are most likely due to the effective cell entry and retention of the nanocarriers, which in turn reduces the potential clearance of the nanocarriers already entering the tumor microenvironment. However, the mechanism of active targeting indicates its potential limitations, that is, before reaching cancer cells, various barriers such as dense stromal cells and ECM between tumor vessels and cancer cells may prevent the interaction of cancer cells with the conjugate, rendering the active targeting less effective.
Intracellular vs. extracellular drug release
The ideal anti-cancer nanomedicine constructs satisfying both enhanced efficacy and safety should be stable in the circulation and selectively release the therapeutic payload at the tumor site. The controlled release of therapeutics is one of the important advantages of macromolecular therapeutics over traditional free drug treatment. Both intracellular and extracellular drug release strategies have been designed based on the specific biological properties of cancer cells and tumor microenvironment. No matter which strategy is chosen, the foremost criterium for an efficient macromolecular therapeutics is to achieve favorable drug release kinetics.
For intracellular drug release, the specific lysosomal enzymes and the reductive or mildly acidic intracellular environment have been employed to design the biodegradable linkers. For example, a tetrapeptide biodegradable spacer GFLG, commonly used in many HPMA copolymer-anti-cancer drug conjugates, can be cleaved fast by the lysosome specific enzyme cathepsin B (Rejmanová et al., 1983). This oligopeptide sequence is perfectly stable in the circulation, due to the absence of relevant enzymes in blood. However, the cleavage efficiency of GFLG can be heterogeneous depending on the expression levels of cathepsin B in different individuals. Spacers independent of intracellular enzyme activities include the hydrazone bond responsive to the low pH and the reductive disulfide bond (Ulbrich & Šubr, 2010). Data showed that the hydrazone linker is not completely stable in the circulation at neutral pH, but releasing the payload very fast at the intracellular pH (Ulbrich et al., 2003; Ulbrich & Šubr, 2010).
For extracellular drug release, spacers sensitive to the acidic pH or the specific enzymes present in tumor microenvironment such as MMPs can be employed. Besides tumor specific spacers, other linkers cleavable in specific tissues can be used for the treatment of cancers in certain organs (Gao et al., 2006, 2007, 2008, 2009). For example, colon-specific delivery can take advantage of the specific microbial enzyme activities such as azo-reductase activity in the colon. The linker composed of an aromatic azo bond and a 1,6-elimination spacer was designed and incorporated into the side chains of the HPMA copolymer-9-aminocamptothecin (9-AC) conjugate (Gao et al., 2006). The fast and efficient release of the unmodified drug from the linker led to prolonged mice survival from colon cancer (Gao et al., 2007, 2008, 2009).
The choice of different linkers should be based on the physiochemical properties of cancer intra- and extracellular environment, as well as the types of cancer. The nanomedicines for extracellular drug release probably allow deeper penetration of free drug payload in the tumor than the nanosized drug containing carriers. On the other hand, nanomedicines for intracellular release can take advantage of the distinct internalization pathways, potentially overcoming or preventing the multidrug resistance of the cancer cells to free drugs.
Conclusions and outlook
Considerable milestones have been achieved in the field of anti-cancer nanomedicines, including the development of biocompatible materials, the advanced synthetic or other chemical approaches for the design of nanocarriers, the discovery of the targetability of nanomedicines, the improved understanding of the relationship between cancer biology and nanomedicine design, the introduction of nanomedicines into clinical evaluations and applications. There is no doubt that nanomedicine development for cancer treatments has strong scientific foundations. However, anti-cancer nanomedicine development is still far from perfection, due to both the potential limitations in material science and the understandings of cancer biology. In particular, the influences of the complex pathophysiological features of cancer on the behavior of nanomedicines have been underestimated. In this review, the complexities of cancer cell biology, microenvironment and disease progression, as well as several possible strategies in nanomedicine design to address some aspects of these complexities were discussed.
For better nanomedicine designs, further considerations are needed. First, the various functional components of nanomedicines should be incorporated comprehensively, to make the best of advantages and avoid the disadvantages of the nanoformulations. Second, the structure and the components of nanocarriers should be well controlled. Third, tumor type or stage, even patient subpopulation-specific designs are needed considering the intertumoral heterogeneity.
Acknowledgments
Declaration of interest: Related research in authors' laboratory was supported in part by NIH Grants CA 51578, GM 69847, CA 132831, and GM 95606 and by the University of Utah Research Foundation.
References
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alitalo K, Tammela T, Petrova TV. Lymphangiogenesis in development and human disease. Nature. 2005;438:946–953. doi: 10.1038/nature04480. [DOI] [PubMed] [Google Scholar]
- Allen TM. Ligand-targeted therapeutics in anticancer therapy. Nat Rev Cancer. 2002;2:750–763. doi: 10.1038/nrc903. [DOI] [PubMed] [Google Scholar]
- Arap W, Pasqualini R, Ruoslahti E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science. 1998;279:377–380. doi: 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- Atkinson RL, Zhang M, Diagaradjane P, Peddibhotla S, Contreras A, Hilsenbeck SG, Woodward WA, Krishnan S, Chang JC, Rosen JM. Thermal enhancement with optically activated gold nanoshells sensitizes breast cancer stem cells to radiation therapy. Sci Transl Med. 2010;2:55ra79. doi: 10.1126/scitranslmed.3001447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacac M, Stamenkovic I. Metastatic cancer cell. Annu Rev Pathol. 2008;3:221–247. doi: 10.1146/annurev.pathmechdis.3.121806.151523. [DOI] [PubMed] [Google Scholar]
- Banzato A, Bobisse S, Rondina M, Renier D, Bettella F, Esposito G, Quintieri L, Meléndez-Alafort L, Mazzi U, Zanovello P, Rosato A. A paclitaxel-hyaluronan bioconjugate targeting ovarian cancer affords a potent in vivo therapeutic activity. Clin Cancer Res. 2008;14:3598–3606. doi: 10.1158/1078-0432.CCR-07-2019. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Bar EE, Chaudhry A, Lin A, Fan X, Schreck K, Matsui W, Piccirillo S, Vescovi AL, DiMeco F, Olivi A, Eberhart CG. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells. 2007;25:2524–2533. doi: 10.1634/stemcells.2007-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME. Impact of tumor-specific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging. Proc Natl Acad Sci USA. 2007;104:15549–15554. doi: 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco E, Hsiao A, Ruiz-Esparza GU, Landry MG, Meric-Bernstam F, Ferrari M. Molecular-targeted nanotherapies in cancer: enabling treatment specificity. Mol Oncol. 2011;5:492–503. doi: 10.1016/j.molonc.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- Bourseau-Guilmain E, Béjaud J, Griveau A, Lautram N, Hindré F, Weyland M, Benoit JP, Garcion E. Development and characterization of immuno-nanocarriers targeting the cancer stem cell marker AC133. Int J Pharm. 2012;423:93–101. doi: 10.1016/j.ijpharm.2011.06.001. [DOI] [PubMed] [Google Scholar]
- Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat Rev Cancer. 2005;5:744–749. doi: 10.1038/nrc1694. [DOI] [PubMed] [Google Scholar]
- Broxterman HJ, Gotink KJ, Verheul HM. Understanding the causes of multidrug resistance in cancer: a comparison of doxorubicin and sunitinib. Drug Resist Updat. 2009;12:114–126. doi: 10.1016/j.drup.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Bruce WR, Van Der Gaag H. A quantitative assay for the number of murine lymphoma cells capable of proliferation in vivo. Nature. 1963;199:79–80. doi: 10.1038/199079a0. [DOI] [PubMed] [Google Scholar]
- Campbell RB, Fukumura D, Brown EB, Mazzola LM, Izumi Y, Jain RK, Torchilin VP, Munn LL. Cationic charge determines the distribution of liposomes between the vascular and extravascular compartments of tumors. Cancer Res. 2002;62:6831–6836. [PubMed] [Google Scholar]
- Canal F, Sanchis J, Vicent MJ. Polymer–drug conjugates as nano-sized medicines. Curr Opin Biotechnol. 2011;22:894–900. doi: 10.1016/j.copbio.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- Chandna P, Khandare JJ, Ber E, Rodriguez-Rodriguez L, Minko T. Multifunctional tumor-targeted polymer-peptide-drug delivery system for treatment of primary and metastatic cancers. Pharm Res. 2010;27:2296–2306. doi: 10.1007/s11095-010-0235-2. [DOI] [PubMed] [Google Scholar]
- Cho K, Wang X, Nie S, Chen ZG, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14:1310–1316. doi: 10.1158/1078-0432.CCR-07-1441. [DOI] [PubMed] [Google Scholar]
- Choi KY, Chung H, Min KH, Yoon HY, Kim K, Park JH, Kwon IC, Jeong SY. Self-assembled hyaluronic acid nanoparticles for active tumor targeting. Biomaterials. 2010;31:106–114. doi: 10.1016/j.biomaterials.2009.09.030. [DOI] [PubMed] [Google Scholar]
- Choi J, Credit K, Henderson K, Deverkadra R, He Z, Wiig H, Vanpelt H, Flessner MF. Intraperitoneal immunotherapy for metastatic ovarian carcinoma: Resistance of intratumoral collagen to antibody penetration. Clin Cancer Res. 2006;12:1906–1912. doi: 10.1158/1078-0432.CCR-05-2141. [DOI] [PubMed] [Google Scholar]
- Chung YL, Jian JJ, Cheng SH, Tsai SY, Chuang VP, Soong T, Lin YM, Horng CF. Sublethal irradiation induces vascular endothelial growth factor and promotes growth of hepatoma cells: implications for radiotherapy of hepatocellular carcinoma. Clin Cancer Res. 2006;12:2706–2715. doi: 10.1158/1078-0432.CCR-05-2721. [DOI] [PubMed] [Google Scholar]
- Chytil P, Etrych T, Kříž J, Šubr V, Ulbrich K. N-(2-Hydroxypropyl) methacrylamide-based polymer conjugates with pH-controlled activation of doxorubicin for cell-specific or passive tumour targeting. Synthesis by RAFT polymerisation and physicochemical characterisation. Eur J Pharm Sci. 2010;41:473–482. doi: 10.1016/j.ejps.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17:313–319. doi: 10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]
- Collins AT, Maitland NJ. Prostate cancer: regeneration of interest in the prostate. Nat Rev Urol. 2009;6:184–186. doi: 10.1038/nrurol.2009.48. [DOI] [PubMed] [Google Scholar]
- Collins I, Workman P. New approaches to molecular cancer therapeutics. Nat Chem Biol. 2006;2:689–700. doi: 10.1038/nchembio840. [DOI] [PubMed] [Google Scholar]
- Colombo M, Corsi F, Foschi D, Mazzantini E, Mazzucchelli S, Morasso C, Occhipinti E, Polito L, Prosperi D, Ronchi S, Verderio P. HER2 targeting as a two-sided strategy for breast cancer diagnosis and treatment: Outlook and recent implications in nanomedical approaches. Pharmacol Res. 2010;62:150–165. doi: 10.1016/j.phrs.2010.01.013. [DOI] [PubMed] [Google Scholar]
- David A, Kopečková P, Rubinstein A, Kopeček J. Enhanced biorecognition and internalization of HPMA copolymers containing multiple or multivalent carbohydrate side-chains by human hepatocarcinoma cells. Bioconjug Chem. 2001;12:890–899. doi: 10.1021/bc010026v. [DOI] [PubMed] [Google Scholar]
- Davies C de L, Lundstrøm LM, Frengen J, Eikenes L, Burland ØS, Kaalhus O, Hjelstuen MH, Brekken C. Radiation improves the distribution and uptake of liposomal doxorubicin (caelyx) in human osteosarcoma xenografts. Cancer Res. 2004;64:547–553. doi: 10.1158/0008-5472.can-03-0576. [DOI] [PubMed] [Google Scholar]
- Davis ME. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Mol Pharm. 2009;6:659–668. doi: 10.1021/mp900015y. [DOI] [PubMed] [Google Scholar]
- Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- Dick JE. Stem cell concepts renew cancer research. Blood. 2008;112:4793–4807. doi: 10.1182/blood-2008-08-077941. [DOI] [PubMed] [Google Scholar]
- Dick JE. Looking ahead in cancer stem cell research. Nat Biotechnol. 2009;27:44–46. doi: 10.1038/nbt0109-44. [DOI] [PubMed] [Google Scholar]
- Duncan R, Gaspar R. Nanomedicine(s) under the microscope. Mol Pharm. 2011;8:2101–2141. doi: 10.1021/mp200394t. [DOI] [PubMed] [Google Scholar]
- Dvorák M, Kopečková P, Kopeček J. High-molecular weight HPMA copolymer-adriamycin conjugates. J Control Release. 1999;60:321–332. doi: 10.1016/s0168-3659(99)00087-5. [DOI] [PubMed] [Google Scholar]
- Edlund M, Sung SY, Chung LW. Modulation of prostate cancer growth in bone microenvironments. J Cell Biochem. 2004;91:686–705. doi: 10.1002/jcb.10702. [DOI] [PubMed] [Google Scholar]
- Ehrlich P. Studies in Immunity. New York: Plenum Press; 1906. [Google Scholar]
- ElBayoumi TA, Torchilin VP. Tumor-targeted nanomedicines: enhanced antitumor efficacy in vivo of doxorubicin-loaded, long-circulating liposomes modified with cancer-specific monoclonal antibody. Clin Cancer Res. 2009;15:1973–1980. doi: 10.1158/1078-0432.CCR-08-2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etrych T, Chytil P, Mrkvan T, Šírová M, Říhová B, Ulbrich K. Conjugates of doxorubicin with graft HPMA copolymers for passive tumor targeting. J Control Release. 2008;132:184–192. doi: 10.1016/j.jconrel.2008.04.017. [DOI] [PubMed] [Google Scholar]
- Etrych T, Šubr V, Strohalm J, Šírová M, Říhová B, Ulbrich K. HPMA copolymer-doxorubicin conjugates: The effects of molecular weight and architecture on biodistribution and in vivo activity. J Control Release. 2012 doi: 10.1016/j.jconrel.2012.06.029. [DOI] [PubMed] [Google Scholar]
- Fang J, Nakamura H, Maeda H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63:136–151. doi: 10.1016/j.addr.2010.04.009. [DOI] [PubMed] [Google Scholar]
- Fang J, Sawa T, Maeda H. Factors and mechanism of “EPR” effect and the enhanced antitumor effects of macromolecular drugs including SMANCS. Adv Exp Med Biol. 2003;519:29–49. doi: 10.1007/0-306-47932-X_2. [DOI] [PubMed] [Google Scholar]
- Feld R, Rubinstein LV, Weisenberger TH. Sites of recurrence in resected stage I non-small-cell lung cancer: a guide for future studies. J Clin Oncol. 1984;2:1352–1358. doi: 10.1200/JCO.1984.2.12.1352. [DOI] [PubMed] [Google Scholar]
- Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147:992–1009. doi: 10.1016/j.cell.2011.11.016. [DOI] [PubMed] [Google Scholar]
- Furth J, Kahn M. The transmission of leukemia of mice with a single cell. Am J Cancer. 1937;31:276–282. [Google Scholar]
- Gabizon A, Horowitz AT, Goren D, Tzemach D, Shmeeda H, Zalipsky S. In vivo fate of folate-targeted polyethylene-glycol liposomes in tumor-bearing mice. Clin Cancer Res. 2003;9:6551–6559. [PubMed] [Google Scholar]
- Gao J, Graves S, Koch U, Liu S, Jankovic V, Buonamici S, El Andaloussi A, Nimer SD, Kee BL, Taichman R, Radtke F, Aifantis I. Hedgehog signaling is dispensable for adult hematopoietic stem cell function. Cell Stem Cell. 2009;4:548–558. doi: 10.1016/j.stem.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Sun J, Li H, Liu W, Zhang Y, Li B, Qian W, Wang H, Chen J, Guo Y. Lyophilized HER2-specific PEGylated immunoliposomes for active siRNA gene silencing. Biomaterials. 2010;31:2655–2664. doi: 10.1016/j.biomaterials.2009.11.112. [DOI] [PubMed] [Google Scholar]
- Gao SQ, Lu ZR, Petri B, Kopečková P, Kopeček J. Colon-specific 9-aminocamptothecin-HPMA copolymer conjugates containing a 1,6-elimination spacer. J Control Release. 2006;110:323–331. doi: 10.1016/j.jconrel.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Gao SQ, Lu ZR, Kopečková P, Kopeček J. Biodistribution and pharmacokinetics of colon-specific HPMA copolymer–9-aminocamptothecin conjugate in mice. J Control Release. 2007;117:179–185. doi: 10.1016/j.jconrel.2006.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao SQ, Sun Y, Kopečková P, Peterson CM, Kopeček J. Pharmacokinetic modeling of absorption behavior of 9-aminocamptothecin (9-AC) released from colon-specific HPMA copolymer-9-AC conjugate in rats. Pharm Res. 2008;25:218–226. doi: 10.1007/s11095-007-9465-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao SQ, Sun Y, Kopečková P, Peterson CM, Kopeček J. Antitumor efficacy of colon-specific HPMA copolymer/9-aminocamptothecin conjugates in mice bearing human-colon carcinoma xenografts. Macromol Biosci. 2009;9:1135–1142. doi: 10.1002/mabi.200900147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill PS, Wernz J, Scadden DT, Cohen P, Mukwaya GM, von Roenn JH, Jacobs M, Kempin S, Silverberg I, Gonzales G, Rarick MU, Myers AM, Shepherd F, Sawka C, Pike MC, Ross ME. Randomized phase III trial of liposomal daunorubicin versus doxorubicin, bleomycin, and vincristine in AIDS-related Kaposi's sarcoma. J Clin Oncol. 1996;14:2353–2364. doi: 10.1200/JCO.1996.14.8.2353. [DOI] [PubMed] [Google Scholar]
- Goda K, Fenyvesi F, Bacsó Z, Nagy H, Márián T, Megyeri A, Krasznai Z, Juhász I, Vecsernyés M, Szabó G., Jr Complete inhibition of P-glycoprotein by simultaneous treatment with a distinct class of modulators and the UIC2 monoclonal antibody. J Pharmacol Exp Ther. 2007;320:81–88. doi: 10.1124/jpet.106.110155. [DOI] [PubMed] [Google Scholar]
- Gordon AN, Fleagle JT, Guthrie D, Parkin DE, Gore ME, Lacave AJ. Recurrent epithelial ovarian carcinoma: a randomized phase III study of pegylated liposomal doxorubicin versus topotecan. J Clin Oncol. 2001;19:3312–3322. doi: 10.1200/JCO.2001.19.14.3312. [DOI] [PubMed] [Google Scholar]
- Gradishar WJ, Tjulandin S, Davidson N, Shaw H, Desai N, Bhar P, Hawkins M, O'Shaughnessy J. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J Clin Oncol. 2005;23:7794–7803. doi: 10.1200/JCO.2005.04.937. [DOI] [PubMed] [Google Scholar]
- Greco F, Vicent MJ, Gee S, Jones AT, Gee J, Nicholson RI, Duncan R. Investigating the mechanism of enhanced cytotoxicity of HPMA copolymer-Dox-AGM in breast cancer cells. J Control Release. 2007;117:28–39. doi: 10.1016/j.jconrel.2006.10.012. [DOI] [PubMed] [Google Scholar]
- Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–659. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, Lander ES. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146:633–644. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Harisinghani MG, Barentsz J, Hahn PF, Deserno WM, Tabatabaei S, van de Kaa CH, de la Rosette J, Weissleder R. Noninvasive detection of clinically occult lymph-node metastases in prostate cancer. N Engl J Med. 2003;348:2491–2499. doi: 10.1056/NEJMoa022749. [DOI] [PubMed] [Google Scholar]
- Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, Jain RK, McDonald DM. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156:1363–1380. doi: 10.1016/S0002-9440(10)65006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidel JD, Davis ME. Clinical developments in nanotechnology for cancer therapy. Pharm Res. 2011;28:187–199. doi: 10.1007/s11095-010-0178-7. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure - an obstacle in cancer therapy. Nat Rev Cancer. 2004;4:806–813. doi: 10.1038/nrc1456. [DOI] [PubMed] [Google Scholar]
- Hess KR, Varadhachary GR, Taylor SH, Wei W, Raber MN, Lenzi R, Abbruzzese JL. Metastatic patterns in adenocarcinoma. Cancer. 2006;106:1624–1633. doi: 10.1002/cncr.21778. [DOI] [PubMed] [Google Scholar]
- Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, Goodell MA, Brenner MK. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci USA. 2004;101:14228–14233. doi: 10.1073/pnas.0400067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann I, Stover EH, Cullen DE, Mao J, Morgan KJ, Lee BH, Kharas MG, Miller PG, Cornejo MG, Okabe R, Armstrong SA, Ghilardi N, Gould S, de Sauvage FJ, McMahon AP, Gilliland DG. Hedgehog signaling is dispensable for adult murine hematopoietic stem cell function and hematopoiesis. Cell Stem Cell. 2009;4:559–567. doi: 10.1016/j.stem.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongrapipat J, Kopečková P, Liu J, Prakongpan S, Kopeček J. Combination chemotherapy and photodynamic therapy with fab' fragment targeted HPMA copolymer conjugates in human ovarian carcinoma cells. Mol Pharm. 2008a;5:696–709. doi: 10.1021/mp800006e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongrapipat J, Kopečková P, Prakongpan S, Kopeček J. Enhanced antitumor activity of combinations of free and HPMA copolymer-bound drugs. Int J Pharm. 2008b;351:259–270. doi: 10.1016/j.ijpharm.2007.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horning SJ, Younes A, Jain V, Kroll S, Lucas J, Podoloff D, Goris M. Efficacy and safety of tositumomab and iodine-131 tositumomab (Bexxar) in B-cell lymphoma, progressive after rituximab. J Clin Oncol. 2005;23:712–719. doi: 10.1200/JCO.2005.07.040. [DOI] [PubMed] [Google Scholar]
- Hsu MJ, Juliano RL. Interactions of liposomes with the reticuloendothelial system. II: Nonspecific and receptor-mediated uptake of liposomes by mouse peritoneal macrophages. Biochim Biophys Acta. 1982;720:411–419. doi: 10.1016/0167-4889(82)90120-3. [DOI] [PubMed] [Google Scholar]
- Hütter ML, Schlenk RF. Gemtuzumab ozogamicin in non-acute promyelocytic acute myeloid leukemia. Expert Opin Biol Ther. 2011;11:1369–1380. doi: 10.1517/14712598.2011.604630. [DOI] [PubMed] [Google Scholar]
- Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci USA. 2011;108:1397–1402. doi: 10.1073/pnas.1018898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain RK. Transport of molecules across tumor vasculature. Cancer Metastasis Rev. 1987;6:559–593. doi: 10.1007/BF00047468. [DOI] [PubMed] [Google Scholar]
- Jain RK. Determinants of tumor blood flow: a review. Cancer Res. 1988;48:2641–2658. [PubMed] [Google Scholar]
- Jain RK. Delivery of molecular and cellular medicine to solid tumors. J Control Release. 1998;53:49–67. doi: 10.1016/s0168-3659(97)00237-x. [DOI] [PubMed] [Google Scholar]
- Jain RK, Munn LL, Fukumura D. Dissecting tumour pathophysiology using intravital microscopy. Nat Rev Cancer. 2002;2:266–276. doi: 10.1038/nrc778. [DOI] [PubMed] [Google Scholar]
- Jain RK, Tong RT, Munn LL. Effect of vascular normalization by antiangiogenic therapy on interstitial hypertension, peritumor edema, and lymphatic metastasis: insights from a mathematical model. Cancer Res. 2007;67:2729–2735. doi: 10.1158/0008-5472.CAN-06-4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7:653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- Johnson RN, Kopečková P, Kopeček J. Synthesis and evaluation of multivalent branched HPMA copolymer-Fab' conjugates targeted to the B-cell antigen CD20. Bioconjug Chem. 2009;20:129–137. doi: 10.1021/bc800351m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juweid M, Neumann R, Paik C, Perez-Bacete MJ, Sato J, van Osdol W, Weinstein JN. Micropharmacology of monoclonal antibodies in solid tumors: direct experimental evidence for a binding site barrier. Cancer Res. 1992;52:5144–5153. [PubMed] [Google Scholar]
- Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C, Guise TA, Massagué J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- Kano MR, Bae Y, Iwata C, Morishita Y, Yashiro M, Oka M, Fujii T, Komuro A, Kiyono K, Kaminishi M, Hirakawa K, Ouchi Y, Nishiyama N, Kataoka K, Miyazono K. Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF-beta signaling. Proc Natl Acad Sci USA. 2007;104:3460–3465. doi: 10.1073/pnas.0611660104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, Isaacs JT, Berman DM, Beachy PA. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431:707–712. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen UB, Marks JD, Benz CC, Park JW. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006;66:6732–6740. doi: 10.1158/0008-5472.CAN-05-4199. [DOI] [PubMed] [Google Scholar]
- Klein CA. Parallel progression of primary tumours and metastases. Nat Rev Cancer. 2009;9:302–312. doi: 10.1038/nrc2627. [DOI] [PubMed] [Google Scholar]
- Kopeček J, Kopečková P. HPMA copolymers: origins, early developments, present, and future. Adv Drug Deliv Rev. 2010;62:122–149. doi: 10.1016/j.addr.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouros-Mehr H, Bechis SK, Slorach EM, Littlepage LE, Egeblad M, Ewald AJ, Pai SY, Ho IC, Werb Z. GATA-3 links tumor differentiation and dissemination in a luminal breast cancer model. Cancer Cell. 2008;13:141–152. doi: 10.1016/j.ccr.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon IK, Lee SC, Han B, Park K. Analysis on the current status of targeted drug delivery to tumors. J Control Release. 2012 doi: 10.1016/j.jconrel.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laakkonen P, Akerman ME, Biliran H, Yang M, Ferrer F, Karpanen T, Hoffman RM, Ruoslahti E. Antitumor activity of a homing peptide that targets tumor lymphatics and tumor cells. Proc Natl Acad Sci USA. 2004;101:9381–9386. doi: 10.1073/pnas.0403317101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammers T. Improving the efficacy of combined modality anticancer therapy using HPMA copolymer-based nanomedicine formulations. Adv Drug Deliv Rev. 2010;62:203–230. doi: 10.1016/j.addr.2009.11.028. [DOI] [PubMed] [Google Scholar]
- Lammers T, Peschke P, Kühnlein R, Šubr V, Ulbrich K, Debus J, Huber P, Hennink W, Storm G. Effect of radiotherapy and hyperthermia on the tumor accumulation of HPMA copolymer-based drug delivery systems. J Control Release. 2007;117:333–341. doi: 10.1016/j.jconrel.2006.10.032. [DOI] [PubMed] [Google Scholar]
- Lammers T, Hennink WE, Storm G. Tumour-targeted nanomedicines: principles and practice. Br J Cancer. 2008;99:392–397. doi: 10.1038/sj.bjc.6604483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammers T, Šubr V, Ulbrich K, Peschke P, Huber PE, Hennink WE, Storm G. Simultaneous delivery of doxorubicin and gemcitabine to tumors in vivo using prototypic polymeric drug carriers. Biomaterials. 2009;30:3466–3475. doi: 10.1016/j.biomaterials.2009.02.040. [DOI] [PubMed] [Google Scholar]
- Lammers T, Kiessling F, Hennink WE, Storm G. Drug targeting to tumors: principles, pitfalls and (pre-) clinical progress. J Control Release. 2012;161:175–187. doi: 10.1016/j.jconrel.2011.09.063. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Galoforo SS, Berns CM, Erdos G, Gupta AK, Ways DK, Corry PM. Effect of ionizing radiation on AP-1 binding activity and basic fibroblast growth factor gene expression in drug-sensitive human breast carcinoma MCF-7 and multidrug-resistant MCF-7/ADR cells. J Biol Chem. 1995;270:28790–28796. doi: 10.1074/jbc.270.48.28790. [DOI] [PubMed] [Google Scholar]
- Leu AJ, Berk DA, Lymboussaki A, Alitalo K, Jain RK. Absence of functional lymphatics within a murine sarcoma: a molecular and functional evaluation. Cancer Res. 2000;60:4324–4327. [PubMed] [Google Scholar]
- Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–1309. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- Leunig M, Yuan F, Menger MD, Boucher Y, Goetz AE, Messmer K, Jain RK. Angiogenesis, microvascular architecture, microhemodynamics, and interstitial fluid pressure during early growth of human adenocarcinoma LS174T in SCID mice. Cancer Res. 1992;52:6553–6560. [PubMed] [Google Scholar]
- Li C, Ke S, Wu QP, Tansey W, Hunter N, Buchmiller LM, Milas L, Charnsangavej C, Wallace S. Tumor irradiation enhances the tumor-specific distribution of poly(L-glutamic acid)-conjugated paclitaxel and its antitumor efficacy. Clin Cancer Res. 2000;6:2829–2834. [PubMed] [Google Scholar]
- Li SD, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm. 2008;5:496–504. doi: 10.1021/mp800049w. [DOI] [PubMed] [Google Scholar]
- Lim KJ, Bisht S, Bar EE, Maitra A, Eberhart CG. A polymeric nanoparticle formulation of curcumin inhibits growth, clonogenicity and stem-like fraction in malignant brain tumors. Cancer Biol Ther. 2011;11:464–473. doi: 10.4161/cbt.11.5.14410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Kopečková P, Bühler P, Wolf P, Pan H, Bauer H, Elsässer-Beile U, Kopeček J. Biorecognition and subcellular trafficking of HPMA copolymer-anti-PSMA antibody conjugates by prostate cancer cells. Mol Pharm. 2009;6:959–970. doi: 10.1021/mp8002682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu ZR, Kopečková P, Kopeček J. Polymerizable Fab' antibody fragments for targeting of anticancer drugs. Nat Biotechnol. 1999;17:1101–1104. doi: 10.1038/15085. [DOI] [PubMed] [Google Scholar]
- Luo K, Yang J, Kopečková P, Kopeček J. Biodegradable Multiblock Poly[N-(2-hydroxypropyl)methacrylamide] via Reversible Addition-Fragmentation Chain Transfer Polymerization and Click Chemistry. Macromolecules. 2011;44:2481–2488. doi: 10.1021/ma102574e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maduro JH, Pras E, Willemse PH, de Vries EG. Acute and longterm toxicity following radiotherapy alone or in combination with chemotherapy for locally advanced cervical cancer. Cancer Treat Rev. 2003;29:471–488. doi: 10.1016/s0305-7372(03)00117-8. [DOI] [PubMed] [Google Scholar]
- Maeda H, Matsumura Y, Kato H. Purification and identification of [hydroxyprolyl3]bradykinin in ascitic fluid from a patient with gastric cancer. J Biol Chem. 1988;263:16051–16054. [PubMed] [Google Scholar]
- Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- Malugin A, Kopečková P, Kopeček J. HPMA copolymer-bound doxorubicin induces apoptosis in ovarian carcinoma cells by the disruption of mitochondrial function. Mol Pharm. 2006;3:351–361. doi: 10.1021/mp050065e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamaeva V, Rosenholm JM, Bate-Eya LT, Bergman L, Peuhu E, Duchanoy A, Fortelius LE, Landor S, Toivola DM, Lindén M, Sahlgren C. Mesoporous silica nanoparticles as drug delivery systems for targeted inhibition of Notch signaling in cancer. Mol Ther. 2011;19:1538–1546. doi: 10.1038/mt.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- Matsumura Y, Kataoka K. Preclinical and clinical studies of anticancer agent-incorporating polymer micelles. Cancer Sci. 2009;100:572–579. doi: 10.1111/j.1349-7006.2009.01103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee TD, Grandi P, Mok W, Alexandrakis G, Insin N, Zimmer JP, Bawendi MG, Boucher Y, Breakefield XO, Jain RK. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res. 2006;66:2509–2513. doi: 10.1158/0008-5472.CAN-05-2242. [DOI] [PubMed] [Google Scholar]
- Milano J, McKay J, Dagenais C, Foster-Brown L, Pognan F, Gadient R, Jacobs RT, Zacco A, Greenberg B, Ciaccio PJ. Modulation of notch processing by gamma-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol Sci. 2004;82:341–358. doi: 10.1093/toxsci/kfh254. [DOI] [PubMed] [Google Scholar]
- Miller K, Eldar-Boock A, Polyak D, Segal E, Benayoun L, Shaked Y, Satchi-Fainaro R. Antiangiogenic antitumor activity of HPMA copolymer-paclitaxel-alendronate conjugate on breast cancer bone metastasis mouse model. Mol Pharm. 2011;8:1052–1062. doi: 10.1021/mp200083n. [DOI] [PubMed] [Google Scholar]
- Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583–592. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- Minko T, Kopečková P, Kopeček J. Chronic exposure to HPMA copolymer-bound adriamycin does not induce multidrug resistance in a human ovarian carcinoma cell line. J Control Release. 1999;59:133–148. doi: 10.1016/s0168-3659(98)00186-2. [DOI] [PubMed] [Google Scholar]
- Minko T, Kopečková P, Kopeček J. Efficacy of the chemotherapeutic action of HPMA copolymer-bound doxorubicin in a solid tumor model of ovarian carcinoma. Int J Cancer. 2000;86:108–117. doi: 10.1002/(sici)1097-0215(20000401)86:1<108::aid-ijc17>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Minko T, Kopečková P, Kopeček J. Preliminary evaluation of caspases-dependent apoptosis signaling pathways of free and HPMA copolymer-bound doxorubicin in human ovarian carcinoma cells. J Control Release. 2001;71:227–237. doi: 10.1016/s0168-3659(01)00220-6. [DOI] [PubMed] [Google Scholar]
- Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2:584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- Nadali F, Pourfathollah AA, Alimoghaddam K, Nikougoftar M, Rostami S, Dizaji A, Azizi E, Zomorodipour A, Ghavamzadeh A. Multidrug resistance inhibition by antisense oligonucleotide against MDR1/mRNA in P-glycoprotein expressing leukemic cells. Hematology. 2007;12:393–401. doi: 10.1080/10245330701283991. [DOI] [PubMed] [Google Scholar]
- Nahrendorf M, Keliher E, Marinelli B, Leuschner F, Robbins CS, Gerszten RE, Pittet MJ, Swirski FK, Weissleder R. Detection of macrophages in aortic aneurysms by nanoparticle positron emission tomography-computed tomography. Arterioscler Thromb Vasc Biol. 2011;31:750–757. doi: 10.1161/ATVBAHA.110.221499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri D, Bicknell R. Tumour vascular targeting. Nat Rev Cancer. 2005;5:436–446. doi: 10.1038/nrc1627. [DOI] [PubMed] [Google Scholar]
- Nguyen DX, Bos PD, Massagué J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- Nguyen QT, Olson ES, Aguilera TA, Jiang T, Scadeng M, Ellies LG, Tsien RY. Surgery with molecular fluorescence imaging using activatable cell-penetrating peptides decreases residual cancer and improves survival. Proc Natl Acad Sci USA. 2010;107:4317–4322. doi: 10.1073/pnas.0910261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama N, Nori A, Malugin A, Kasuya Y, Kopečková P, Kopeček J. Free and N-(2-hydroxypropyl)methacrylamide copolymer-bound geldanamycin derivative induce different stress responses in A2780 human ovarian carcinoma cells. Cancer Res. 2003;63:7876–7882. [PubMed] [Google Scholar]
- Noguchi Y, Wu J, Duncan R, Strohalm J, Ulbrich K, Akaike T, Maeda H. Early phase tumor accumulation of macromolecules: a great difference in clearance rate between tumor and normal tissues. Jpn J Cancer Res. 1998;89:307–314. doi: 10.1111/j.1349-7006.1998.tb00563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northfelt DW, Dezube BJ, Thommes JA, Miller BJ, Fischl MA, Friedman-Kien A, Kaplan LD, Du Mond C, Mamelok RD, Henry DH. Pegylated-liposomal doxorubicin versus doxorubicin, bleomycin, and vincristine in the treatment of AIDS-related Kaposi's sarcoma: results of a randomized phase III clinical trial. J Clin Oncol. 1998;16:2445–2451. doi: 10.1200/JCO.1998.16.7.2445. [DOI] [PubMed] [Google Scholar]
- Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- Nowotnik DP, Cvitkovic E. ProLindac (AP5346): a review of the development of an HPMA DACH platinum Polymer Therapeutic. Adv Drug Deliv Rev. 2009;61:1214–1219. doi: 10.1016/j.addr.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Nugent LJ, Jain RK. Extravascular diffusion in normal and neoplastic tissues. Cancer Res. 1984;44:238–244. [PubMed] [Google Scholar]
- O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- O'Brien ME, Wigler N, Inbar M, Rosso R, Grischke E, Santoro A, Catane R, Kieback DG, Tomczak P, Ackland SP, Orlandi F, Mellars L, Alland L, Tendler C CAELYX Breast Cancer Study Group. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann Oncol. 2004;15:440–449. doi: 10.1093/annonc/mdh097. [DOI] [PubMed] [Google Scholar]
- O'Brien ME, Socinski MA, Popovich AY, Bondarenko IN, Tomova A, Bilynsky BT, Hotko YS, Ganul VL, Kostinsky IY, Eisenfeld AJ, Sandalic L, Oldham FB, Bandstra B, Sandler AB, Singer JW. Randomized phase III trial comparing single-agent paclitaxel Poliglumex (CT-2103, PPX) with single-agent gemcitabine or vinorelbine for the treatment of PS 2 patients with chemotherapy-naïve advanced non-small cell lung cancer. J Thorac Oncol. 2008;3:728–734. doi: 10.1097/JTO.0b013e31817c6b68. [DOI] [PubMed] [Google Scholar]
- Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, Denicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, Reichelt S, Howat WJ, Chang A, Dhara M, Wang L, Rückert F, Grützmann R, Pilarsky C, Izeradjene K, Hingorani SR, Huang P, Davies SE, Plunkett W, Egorin M, Hruban RH, Whitebread N, McGovern K, Adams J, Iacobuzio-Donahue C, Griffiths J, Tuveson DA. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson ES, Jiang T, Aguilera TA, Nguyen QT, Ellies LG, Scadeng M, Tsien RY. Activatable cell penetrating peptides linked to nanoparticles as dual probes for in vivo fluorescence and MR imaging of proteases. Proc Natl Acad Sci USA. 2010;107:4311–4316. doi: 10.1073/pnas.0910283107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omelyanenko V, Kopečková P, Gentry C, Kopeček J. Targetable HPMA copolymer-adriamycin conjugates. Recognition, internalization, and subcellular fate. J Control Release. 1998a;53:25–37. doi: 10.1016/s0168-3659(97)00235-6. [DOI] [PubMed] [Google Scholar]
- Omelyanenko V, Gentry C, Kopečková P, Kopeček J. HPMA copolymer-anticancer drug-OV-TL16 antibody conjugates. II. Processing in epithelial ovarian carcinoma cells in vitro. Int J Cancer. 1998b;75:600–608. doi: 10.1002/(sici)1097-0215(19980209)75:4<600::aid-ijc18>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Oravecz-Wilson KI, Philips ST, Yilmaz OH, Ames HM, Li L, Crawford BD, Gauvin AM, Lucas PC, Sitwala K, Downing JR, Morrison SJ, Ross TS. Persistence of leukemia-initiating cells in a conditional knockin model of an imatinib-responsive myeloproliferative disorder. Cancer Cell. 2009;16:137–148. doi: 10.1016/j.ccr.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padera TP, Kadambi A, di Tomaso E, Carreira CM, Brown EB, Boucher Y, Choi NC, Mathisen D, Wain J, Mark EJ, Munn LL, Jain RK. Lymphatic metastasis in the absence of functional intratumor lymphatics. Science. 2002;296:1883–1886. doi: 10.1126/science.1071420. [DOI] [PubMed] [Google Scholar]
- Padera TP, Stoll BR, Tooredman JB, Capen D, di Tomaso E, Jain RK. Pathology: cancer cells compress intratumour vessels. Nature. 2004;427:695. doi: 10.1038/427695a. [DOI] [PubMed] [Google Scholar]
- Pan H, Sima M, Kopečková P, Wu K, Gao S, Liu J, Wang D, Miller SC, Kopeček J. Biodistribution and pharmacokinetic studies of bone-targeting N-(2-hydroxypropyl)methacrylamide copolymer-alendronate conjugates. Mol Pharm. 2008;5:548–558. doi: 10.1021/mp800003u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H, Yang J, Kopečková P, Kopeček J. Backbone degradable multiblock N-(2-hydroxypropyl)methacrylamide copolymer conjugates via reversible addition-fragmentation chain transfer polymerization and thiol-ene coupling reaction. Biomacromolecules. 2011;12:247–252. doi: 10.1021/bm101254e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegram MD, Konecny G, Slamon DJ. The molecular and cellular biology of HER2/neu gene amplification/overexpression and the clinical development of herceptin (trastuzumab) therapy for breast cancer. Cancer Treat Res. 2000;103:57–75. doi: 10.1007/978-1-4757-3147-7_4. [DOI] [PubMed] [Google Scholar]
- Peschke P, Klein V, Wolber G, Friedrich E, Hahn EW. Morphometric analysis of bromodeoxyuridine distribution and cell density in the rat Dunning prostate tumor R3327-AT1 following treatment with radiation and/or hyperthermia. Histol Histopathol. 1999;14:461–469. doi: 10.14670/HH-14.461. [DOI] [PubMed] [Google Scholar]
- Pierce GB, Wallace C. Differentiation of malignant to benign cells. Cancer Res. 1971;31:127–134. [PubMed] [Google Scholar]
- Pluen A, Netti PA, Jain RK, Berk DA. Diffusion of macromolecules in agarose gels: comparison of linear and globular configurations. Biophys J. 1999;77:542–552. doi: 10.1016/S0006-3495(99)76911-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primeau AJ, Rendon A, Hedley D, Lilge L, Tannock IF. The distribution of the anticancer drug Doxorubicin in relation to blood vessels in solid tumors. Clin Cancer Res. 2005;11:8782–8788. doi: 10.1158/1078-0432.CCR-05-1664. [DOI] [PubMed] [Google Scholar]
- Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz A, Bucana C, Fogler WE, Poste G, Fidler IJ. Biochemical, morphological, and ultrastructural studies on the uptake of liposomes by murine macrophages. Cancer Res. 1981;41:487–494. [PubMed] [Google Scholar]
- Rejmanová P, Pohl J, Baudyš M, Kostka V, Kopeček J. Polymers containing enzymatically degradable bonds. 8. Degradation of oligopeptide sequences in N-(2-hydroxypropyl)methacrylamide copolymers by bovine spleen cathepsin B. Makromol Chem. 1983;184:2009–2020. [Google Scholar]
- Ren Y, Wang Y, Zhang Y, Wei D. Overcoming multidrug resistance in human carcinoma cells by an antisense oligodeoxynucleotide-doxorubicin conjugate in vitro and in vivo. Mol Pharm. 2008;5:579–587. doi: 10.1021/mp800001j. [DOI] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, Maira G, Parati EA, Stassi G, Larocca LM, De Maria R. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468:824–828. doi: 10.1038/nature09557. [DOI] [PubMed] [Google Scholar]
- Říhová B, Kopečková P, Strohalm J, Rossmann P, Vĕtvička V, Kopeček J. Antibody-directed affinity therapy applied to the immune system: in vivo effectiveness and limited toxicity of daunomycin conjugated to HPMA copolymers and targeting antibody. Clin Immunol Immunopathol. 1988;46:100–114. doi: 10.1016/0090-1229(88)90010-4. [DOI] [PubMed] [Google Scholar]
- Říhová B, Strohalm J, Hoste K, Jelínková M, Hovorka O, Kováf M, Plocová D, Šírová M, Št'astný M, Schacht E, Ulbrich K. Immunoprotective therapy with targeted anticancer drugs. Macromolecular Symposia. 2001;172:21–28. [Google Scholar]
- Roberts WG, Palade GE. Neovasculature induced by vascular endothelial growth factor is fenestrated. Cancer Res. 1997;57:765–772. [PubMed] [Google Scholar]
- Rosen JM, Jordan CT. The increasing complexity of the cancer stem cell paradigm. Science. 2009;324:1670–1673. doi: 10.1126/science.1171837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothe A, Sasse S, Goergen H, Eichenauer DA, Lohri A, Jäger U, Bangard C, Böll B, von Bergwelt Baildon M, Theurich S, Borchmann P, Engert A. Brentuximab vedotin for relapsed or refractory CD30+ hematologic malignancies: the German Hodgkin Study Group experience. Blood. 2012;120:1470–1472. doi: 10.1182/blood-2012-05-430918. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E. Peptides as targeting elements and tissue penetration devices for nanoparticles. Adv Mater. 2012;24:3747–3756. doi: 10.1002/adma.201200454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Kittler O, Ragg T, Daskalakis A, Granzow M, Ahr A, Blankenstein TJ, Kaufmann M, Diebold J, Arnholdt H, Muller P, Bischoff J, Harich D, Schlimok G, Riethmuller G, Eils R, Klein CA. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci USA. 2003;100:7737–7742. doi: 10.1073/pnas.1331931100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder A, Heller DA, Winslow MM, Dahlman JE, Pratt GW, Langer R, Jacks T, Anderson DG. Treating metastatic cancer with nanotechnology. Nat Rev Cancer. 2012;12:39–50. doi: 10.1038/nrc3180. [DOI] [PubMed] [Google Scholar]
- Segal E, Pan H, Ofek P, Udagawa T, Kopečková P, Kopeček J, Satchi-Fainaro R. Targeting angiogenesis-dependent calcified neoplasms using combined polymer therapeutics. PLoS ONE. 2009;4:e5233. doi: 10.1371/journal.pone.0005233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour LW, Duncan R, Strohalm J, Kopeček J. Effect of molecular weight (Mw) of N-(2-hydroxypropyl)methacrylamide copolymers on body distribution and rate of excretion after subcutaneous, intraperitoneal, and intravenous administration to rats. J Biomed Mater Res. 1987;21:1341–1358. doi: 10.1002/jbm.820211106. [DOI] [PubMed] [Google Scholar]
- Seymour LW, Miyamoto Y, Maeda H, Brereton M, Strohalm J, Ulbrich K, Duncan R. Influence of molecular weight on passive tumour accumulation of a soluble macromolecular drug carrier. Eur J Cancer. 1995;31A:766–770. doi: 10.1016/0959-8049(94)00514-6. [DOI] [PubMed] [Google Scholar]
- Seymour LW, Ferry DR, Anderson D, Hesslewood S, Julyan PJ, Poyner R, Doran J, Young AM, Burtles S, Kerr DJ Cancer Research Campaign Phase I/II Clinical Trials committee. Hepatic drug targeting: phase I evaluation of polymer-bound doxorubicin. J Clin Oncol. 2002;20:1668–1676. doi: 10.1200/JCO.2002.20.6.1668. [DOI] [PubMed] [Google Scholar]
- Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138:822–829. doi: 10.1016/j.cell.2009.08.017. [DOI] [PubMed] [Google Scholar]
- Shah NP, Skaggs BJ, Branford S, Hughes TP, Nicoll JM, Paquette RL, Sawyers CL. Sequential ABL kinase inhibitor therapy selects for compound drug-resistant BCR-ABL mutations with altered oncogenic potency. J Clin Invest. 2007;117:2562–2569. doi: 10.1172/JCI30890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, Wong KK, Brandstetter K, Wittner B, Ramaswamy S, Classon M, Settleman J. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharkey RM, Goldenberg DM. Targeted therapy of cancer: new prospects for antibodies and immunoconjugates. CA Cancer J Clin. 2006;56:226–243. doi: 10.3322/canjclin.56.4.226. [DOI] [PubMed] [Google Scholar]
- Shiah JJ, Sun Y, Peterson CM, Kopeček J. Biodistribution of free and N-(2-hydroxypropyl)methacrylamide copolymer-bound mesochlorin e6 and adriamycin in nude mice bearing human ovarian carcinoma OVCAR-3 xenografts. J Control Release. 1999;61:145–157. doi: 10.1016/s0168-3659(99)00113-3. [DOI] [PubMed] [Google Scholar]
- Shiah JG, Sun Y, Peterson CM, Straight RC, Kopeček J. Antitumor activity of N-(2-hydroxypropyl)methacrylamide copolymer-mesochlorine e6 and adriamycin conjugates in combination treatments. Clin Cancer Res. 2000;6:1008–1015. [PubMed] [Google Scholar]
- Shiah JG, Dvořák M, Kopečková P, Sun Y, Peterson CM, Kopeček J. Biodistribution and antitumour efficacy of long-circulating N-(2-hydroxypropyl)methacrylamide copolymer-doxorubicin conjugates in nude mice. Eur J Cancer. 2001a;37:131–139. doi: 10.1016/s0959-8049(00)00374-9. [DOI] [PubMed] [Google Scholar]
- Shiah JG, Sun Y, Kopečková P, Peterson CM, Straight RC, Kopeček J. Combination chemotherapy and photodynamic therapy of targetable N-(2-hydroxypropyl)methacrylamide copolymer-doxorubicin/mesochlorin e(6)-OV-TL 16 antibody immunoconjugates. J Control Release. 2001b;74:249–253. doi: 10.1016/s0168-3659(01)00325-x. [DOI] [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Sorbera LA, Graul A, Silvestre J, Castañer J. Oncolytic, antiarthritic, matrix metalloproteinase inhibitor. Drugs Fut. 1999;24:16. [Google Scholar]
- Spano D, Zollo M. Tumor microenvironment: a main actor in the metastasis process. Clin Exp Metastasis. 2012;29:381–395. doi: 10.1007/s10585-012-9457-5. [DOI] [PubMed] [Google Scholar]
- St Croix B, Man S, Kerbel RS. Reversal of intrinsic and acquired forms of drug resistance by hyaluronidase treatment of solid tumors. Cancer Lett. 1998;131:35–44. doi: 10.1016/s0304-3835(98)00199-2. [DOI] [PubMed] [Google Scholar]
- Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Girard OM, Hanahan D, Mattrey RF, Ruoslahti E. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 2009;16:510–520. doi: 10.1016/j.ccr.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Greenwald DR, Ruoslahti E. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science. 2010;328:1031–1035. doi: 10.1126/science.1183057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- Tammela T, Alitalo K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell. 2010;140:460–476. doi: 10.1016/j.cell.2010.01.045. [DOI] [PubMed] [Google Scholar]
- Tang C, Ang BT, Pervaiz S. Cancer stem cell: target for anti-cancer therapy. FASEB J. 2007;21:3777–3785. doi: 10.1096/fj.07-8560rev. [DOI] [PubMed] [Google Scholar]
- Tardi PG, Gallagher RC, Johnstone S, Harasym N, Webb M, Bally MB, Mayer LD. Coencapsulation of irinotecan and floxuridine into low cholesterol-containing liposomes that coordinate drug release in vivo. Biochim Biophys Acta. 2007;1768:678–687. doi: 10.1016/j.bbamem.2006.11.014. [DOI] [PubMed] [Google Scholar]
- Tardi P, Johnstone S, Harasym N, Xie S, Harasym T, Zisman N, Harvie P, Bermudes D, Mayer L. In vivo maintenance of synergistic cytarabine:daunorubicin ratios greatly enhances therapeutic efficacy. Leuk Res. 2009;33:129–139. doi: 10.1016/j.leukres.2008.06.028. [DOI] [PubMed] [Google Scholar]
- Tassa C, Shaw SY, Weissleder R. Dextran-coated iron oxide nanoparticles: a versatile platform for targeted molecular imaging, molecular diagnostics, and therapy. Acc Chem Res. 2011;44:842–852. doi: 10.1021/ar200084x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taurin S, Nehoff H, Greish K. Anticancer nanomedicine and tumor vascular permeability; Where is the missing link? J Control Release. 2012 doi: 10.1016/j.jconrel.2012.07.013. [DOI] [PubMed] [Google Scholar]
- Teng Y, Wang X, Wang Y, Ma D. Wnt/beta-catenin signaling regulates cancer stem cells in lung cancer A549 cells. Biochem Biophys Res Commun. 2010;392:373–379. doi: 10.1016/j.bbrc.2010.01.028. [DOI] [PubMed] [Google Scholar]
- Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Fernández-del Castillo C, Yajnik V, Antoniu B, McMahon M, Warshaw AL, Hebrok M. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851–856. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CB, Shepard HM, O'Connor PM, Kadhim S, Jiang P, Osgood RJ, Bookbinder LH, Li X, Sugarman BJ, Connor RJ, Nadjsombati S, Frost GI. Enzymatic depletion of tumor hyaluronan induces antitumor responses in preclinical animal models. Mol Cancer Ther. 2010;9:3052–3064. doi: 10.1158/1535-7163.MCT-10-0470. [DOI] [PubMed] [Google Scholar]
- Tsuruo T, Iida H, Tsukagoshi S, Sakurai Y. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res. 1981;41:1967–1972. [PubMed] [Google Scholar]
- Ulbrich K, Etrych T, Chytil P, Jelínková M, Říhová B. HPMA copolymers with pH-controlled release of doxorubicin: in vitro cytotoxicity and in vivo antitumor activity. J Control Release. 2003;87:33–47. doi: 10.1016/s0168-3659(02)00348-6. [DOI] [PubMed] [Google Scholar]
- Ulbrich K, Šubr V. Structural and chemical aspects of HPMA copolymers as drug carriers. Adv Drug Deliv Rev. 2010;62:150–166. doi: 10.1016/j.addr.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Veiseh O, Kievit FM, Ellenbogen RG, Zhang M. Cancer cell invasion: treatment and monitoring opportunities in nanomedicine. Adv Drug Deliv Rev. 2011;63:582–596. doi: 10.1016/j.addr.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergote IB, Micha JP, Pippitt CH, Jr, Rao GG, Spitz DL, Reed N, Dark GG, Garcia A, Maslyar DJ, Rustin GJ. Phase II study of NKTR-102 in women with platinum-resistant/refractory ovarian cancer. J Clin Oncol. 2010;28(15suppl) abstract5013. [Google Scholar]
- Vicent MJ, Greco F, Nicholson RI, Paul A, Griffiths PC, Duncan R. Polymer therapeutics designed for a combination therapy of hormone-dependent cancer. Angew Chem Int Ed Engl. 2005;44:4061–4066. doi: 10.1002/anie.200462960. [DOI] [PubMed] [Google Scholar]
- Villares GJ, Zigler M, Wang H, Melnikova VO, Wu H, Friedman R, Leslie MC, Vivas-Mejia PE, Lopez-Berestein G, Sood AK, Bar-Eli M. Targeting melanoma growth and metastasis with systemic delivery of liposome-incorporated protease-activated receptor-1 small interfering RNA. Cancer Res. 2008;68:9078–9086. doi: 10.1158/0008-5472.CAN-08-2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradov S, Wei X. Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine (Lond) 2012;7:597–615. doi: 10.2217/nnm.12.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner V, Dullaart A, Bock AK, Zweck A. The emerging nanomedicine landscape. Nat Biotechnol. 2006;24:1211–1217. doi: 10.1038/nbt1006-1211. [DOI] [PubMed] [Google Scholar]
- Wang J, Lou P, Lesniewski R, Henkin J. Paclitaxel at ultra low concentrations inhibits angiogenesis without affecting cellular microtubule assembly. Anticancer Drugs. 2003;14:13–19. doi: 10.1097/00001813-200301000-00003. [DOI] [PubMed] [Google Scholar]
- Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, Fligelman B, Leversha M, Brennan C, Tabar V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010;468:829–833. doi: 10.1038/nature09624. [DOI] [PubMed] [Google Scholar]
- Wang Y, Oliver G. Current views on the function of the lymphatic vasculature in health and disease. Genes Dev. 2010;24:2115–2126. doi: 10.1101/gad.1955910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, Ali S, Abbruzzese JL, Gallick GE, Sarkar FH. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009;69:2400–2407. doi: 10.1158/0008-5472.CAN-08-4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZA, Shen MM. Revisiting the concept of cancer stem cells in prostate cancer. Oncogene. 2011;30:1261–1271. doi: 10.1038/onc.2010.530. [DOI] [PubMed] [Google Scholar]
- Wiseman GA, White CA, Witzig TE, Gordon LI, Emmanouilides C, Raubitschek A, Janakiraman N, Gutheil J, Schilder RJ, Spies S, Silverman DH, Grillo-López AJ. Radioimmunotherapy of relapsed non-Hodgkin's lymphoma with zevalin, a 90Y-labeled anti-CD20 monoclonal antibody. Clin Cancer Res. 1999;5:3281s–3286s. [PubMed] [Google Scholar]
- Wolf K, Mazo I, Leung H, Engelke K, von Andrian UH, Deryugina EI, Strongin AY, Bröcker EB, Friedl P. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J Cell Biol. 2003;160:267–277. doi: 10.1083/jcb.200209006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K, Wu YI, Liu Y, Geiger J, Tam E, Overall C, Stack MS, Friedl P. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat Cell Biol. 2007;9:893–904. doi: 10.1038/ncb1616. [DOI] [PubMed] [Google Scholar]
- Wu J, Akaike T, Maeda H. Modulation of enhanced vascular permeability in tumors by a bradykinin antagonist, a cyclooxygenase inhibitor, and a nitric oxide scavenger. Cancer Res. 1998;58:159–165. [PubMed] [Google Scholar]
- Wu K, Liu J, Johnson RN, Yang J, Kopeček J. Drug-free macromolecular therapeutics: induction of apoptosis by coiled-coil-mediated cross-linking of antigens on the cell surface. Angew Chem Int Ed Engl. 2010;49:1451–1455. doi: 10.1002/anie.200906232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K, Yang J, Liu J, Kopeček J. Coiled-coil based drug-free macromolecular therapeutics: In vivo efficacy. J Control Release. 2012;157:126–131. doi: 10.1016/j.jconrel.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Wang F, Wen Z, Zhan C, Feng L, Liu Y, Wei X, Xie C, Lu W. LyP-1-conjugated PEGylated liposomes: A carrier system for targeted therapy of lymphatic metastatic tumor. J Control Release. 2012;157:118–125. doi: 10.1016/j.jconrel.2011.07.034. [DOI] [PubMed] [Google Scholar]
- Yan Z, Zhan C, Wen Z, Feng L, Wang F, Liu Y, Yang X, Dong Q, Liu M, Lu W. LyP-1-conjugated doxorubicin-loaded liposomes suppress lymphatic metastasis by inhibiting lymph node metastases and destroying tumor lymphatics. Nanotechnology. 2011;22:415103. doi: 10.1088/0957-4484/22/41/415103. [DOI] [PubMed] [Google Scholar]
- Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Yang J, Luo K, Pan H, Kopečková P, Kopeček J. Synthesis of Biodegradable Multiblock Copolymers by Click Coupling of RAFT-Generated HeterotelechelicPolyHPMA Conjugates. React Funct Polym. 2011;71:294–302. doi: 10.1016/j.reactfunctpolym.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, Morrison SJ. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, Jain RK. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55:3752–3756. [PubMed] [Google Scholar]
- Yuan F, Salehi HA, Boucher Y, Vasthare US, Tuma RF, Jain RK. Vascular permeability and microcirculation of gliomas and mammary carcinomas transplanted in rat and mouse cranial windows. Cancer Res. 1994;54:4564–4568. [PubMed] [Google Scholar]
- Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, Huang Y, Hu X, Su F, Lieberman J, Song E. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–1123. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, Haug JS, Rupp D, Porter-Westpfahl KS, Wiedemann LM, Wu H, Li L. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Yang J, Kopeček J. Selective inhibitory effect of HPMA copolymer-cyclopamine conjugate on prostate cancer stem cells. Biomaterials. 2012;33:1863–1872. doi: 10.1016/j.biomaterials.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]