

Abstract
Globoid cell leukodystrophy (GLD) is a demyelinating lysosomal storage disease due to the deficiency of the galactocerebrosidase (GALC) enzyme. The favorable outcome of hematopoietic stem and progenitor cell (HSPC)-based approaches in GLD and other similar diseases suggests HSPC gene therapy as a promising therapeutic option for patients. The path to clinical development of this strategy was hampered by a selective toxicity of the overexpressed GALC in the HSPC compartment. Here, we presented the optimization of a lentiviral vector (LV) in which miR-126 regulation was coupled to codon optimization of the human GALC cDNA to obtain a selective and enhanced enzymatic activity only upon transduced HSPCs differentiation. The safety of human GALC overexpression driven by this LV was extensively demonstrated in vitro and in vivo on human HSPCs from healthy donors. No perturbation in the content of proapoptotic sphingolipids, gene expression profile, and capability of engraftment and mutlilineage differentiation in chimeric mice was observed. The therapeutic potential of this LV was then assessed in a severe GLD murine model that benefited from transplantation of corrected HSPCs with longer survival and ameliorated phenotype as compared to untreated siblings. This construct has thus been selected as a candidate for clinical translation.
Introduction
Globoid cell leukodystrophy (GLD) or Krabbe disease is a rare, rapidly progressive, and severe demyelinating lysosomal storage disease (LSD) due to the deficiency of the galactocerebrosidase (GALC) enzyme. Since GALC is responsible for the hydrolysis of galactose from several glycosphingolipids involved in myelination, including galactosylceramide (GalCer) and its by-product galactosylsphingosine (psychosine, Psy), GALC dysfunction leads to the accumulation of substrates in the central (CNS) and peripheral nervous system (PNS). In particular, the accumulation of the cytotoxic sphingolipid Psy in myelinating cells and neurons leads to an overt primary and secondary demyelination.1–3 As a consequence, patients experience progressive and severe neurological symptoms like seizures, sensory loss, and rapid psychomotor deterioration.4 In the early infantile (EI) forms, clinical decline is rapid and death occurs a few years after the diagnosis. Many therapeutic approaches, alone and in combination,5 have been attempted in the most common murine model of the disease (the Twitcher mouse); however, the only one therapy thus far applied to GLD patients, on the basis of promising results in preclinical studies,6–10 is transplantation of hematopoietic stem cells (HSCT) derived from compatible healthy donors’ bone marrow (BM) or umbilical cord blood (CB). If applied very early in life, HSCT from unrelated CB can delay the onset of GLD and attenuate its clinical manifestations, even if long-term disability was reported in the majority of the transplanted children.11,12 Transplantation in patients presenting overt disease symptoms failed to substantially ameliorate their prognosis, and their disease progression was shown to be as rapid as in not-transplanted children.12 An autologous hematopoietic stem cell gene therapy approach could be of great relevance to overcome HSCT limited outcome since reducing transplant-related side effects, morbidity, and mortality could provide an added therapeutic benefit due to the potential for vector-mediated enzyme supra-physiological expression in hematopoietic stem and progenitor cells (HSPCs) and their progeny.13 Since newborn screening for GLD is not yet routinely available and the majority of patients are diagnosed after symptom onset (except when an older, already diagnosed affected sibling is present), this strategy could also spare precious time for search of a compatible donor.
We have already demonstrated the potential of autologous HSPC-gene therapy based on lentiviral vector (LV) transduction in the murine model of GLD.14 In this effort, we addressed the unexpected issue of the toxicity of LV-mediated murine GALC (mGALC) supra-physiological expression in murine HSPCs but not in their differentiated progeny15 by exploiting a murine GALC-encoding LV regulated by four target sequences for micro RNA 126 (miR-126), which is selectively expressed into immature hematopoietic cells and virtually absent in mature compartments.14 We proved the efficacy of this LV construct in protecting murine HSPCs from murine GALC overexpression-related toxicity. Moreover, the construct drove sustained enzyme expression up to therapeutic levels in the differentiated tissue progeny of the transduced HSPCs, as shown in the Trs mouse model of which we substantially improved survival and phenotype as compared not only to untreated affected siblings but also to affected mice transplanted with wild type HSPCs.14
We here investigated the best LV design for a proper clinical translation of these promising preclinical results. To this purpose, we employed a human GALC (hGALC) transgene of which we improved transcript stability and enzymatic expression by codon optimization and adjusted the number of miR-126 target sequences with the aim of balancing the need for repression of transgene expression in the human HSPC compartment with the requirement of sustained enzyme expression upon their differentiation. Moreover, we confirmed that the new construct developed for clinical translation retains therapeutic efficacy in a severe disease animal model.
Results
Optimization of an advanced LV construct for safe and efficient expression of the human GALC enzyme
In order to allow safe and efficient hGALC expression also in human HSPCs and their differentiated progeny, we confirmed the use of miR-126 posttranscriptional regulation, exploiting its capability to repress transgene expression in a differentiation stage-specific manner also in the human setting.14,16 We evaluated the regulatory potential of single versus multiple miR-126 target sequences at different stages of human HSPCs in vitro differentiation, namely, after 1 day and 14 days of liquid culture after second hit of transduction (t = 0). To this purpose, three regulated LVs with one, two, or four target sequences of miR-126 downstream to the hGALC cDNA were generated and tested on CB- and BM-derived HSPCs. Among the tested LV constructs, one miR-126 target (hereon referred to as hGALC_126T.LV) performed at best since it allowed a highly significant (P < 0.0001) repression of hGALC activity in both CB- and BM-HSPCs at +1 day of culture (Figure 1a) and a sustained hGALC expression upon in vitro cell differentiation (after 14 days of liquid culture) (Figure 1b), in the presence of comparable vector copy number (VCN) values with the other tested constructs (Supplementary Figure S1a). The slightly lower hGALC activity measured at +14 days in the cells transduced with hGALC_126T.LV as compared to the unregulated construct could be ascribed to presence of some residual miR-126 transcript in the in vitro culture progeny of our cells (data not shown). Thus, hGALC activity driven by hGALC_126T.LV could increase further in vivo in terminally differentiated cells that nearly completely lose miR-126 activity. The other tested constructs provided a stronger repression of enzyme activity in human HSPCs, persisting to some extent also upon cell differentiation, thus possibly affecting therapeutic enzyme delivery to affected tissues by the myeloid progeny of the transduced HSPCs. Importantly, the level of hGALC activity repression obtained by the use of hGALC_126T.LV in HSPCs was sufficient to restore the clonogenic potential of the transduced cells to what observed in control cells (Supplementary Figure S1b).
Figure 1.

Optimization of a LV for the expression of human GALC enzyme. (a) hGALC activity measured in BM- (n ≥ 4 donors) or CB- (n ≥ 7 donors) derived HSPCs upon transduction with hGALC.LV (no tags), hGALC_126T.LV (1x126T), hGALC_2x126T.LV (2x126T), hGALC_4x126T.LV (4x126T) or hIDUA.LV (ctrl) at +1 day (+1d) after transduction is reported. Values are expressed as percentage relative to the activity achieved upon hGALC.LV transduction (dotted line = 100%) (mean ± SEM). (b) hGALC activity measured in BM- (n ≥ 2 donors) or CB- (n ≥ 10 donors) derived HSPCs upon transduction with above-mentioned LVs at +14 days after transduction is reported. Values are expressed as fold to normal donor (ND) level (dotted line) (mean ± SEM). (c) hGALC activity values measured at +14 days in BM-HSPCs (n ≥ 5) or CB- (n ≥ 6) upon hGALC_126T.LV or hGALCC.O._126T.LV transduction. Values are normalized for VCN and expressed as fold to normal donor (ND) level (dotted line) (mean ± SEM). Statistical analysis was performed with one-way analysis of variance with Bonferroni posttest: *P < 0.05, **P < 0.01, *** P < 0.001, and ****P < 0.0001.
In order to further ameliorate the stability of hGALC long transcript, enhance hGALC expression in differentiated cells, and increase the therapeutic potential of the strategy, we generated a new LV carrying a codon optimized human Galc cDNA regulated by one miR-126 target. This PGK.hGALCC.O._1x126T.LV (from here on referred to as hGALCC.O._126T.LV) and a control vector were tested on CB- and BM-HSPCs with the protocol in use in the clinics allowing BM-derived HSPCs efficient transduction.13 This protocol resulted in very high transduction efficiency in CB-HSPCs in which >8.5 LV copies/genome were measured in the liquid culture progeny of the transduced cells with both tested vectors (Supplementary Figure S2). Even at this high VCN, the use of the regulated LV encoding codon-optimized hGALC resulted in a good repression of hGALC activity in undifferentiated cells (+1 day; Supplementary Figure S3). Importantly, upon differentiation, the cells transduced with hGALCC.O._126T.LV showed a sustained enzyme activity, which was significantly higher than the activity measured in the in vitro progeny of HSPCs transduced with the original hGALC_126T.LV with a noncodon optimized transgene (Figure 1c). Of note, hGALC activity measured in the progeny of hGALCC.O._126T.LV transduced BM-derived HSPCs was equal to 14-fold the basal normal donors’ level (Supplementary Figure S4), confirming the high therapeutic potential of this regulated LV.
hGALCC.O._126T.LV drives safe hGALC overexpression in human HSPCs
To determine whether hGALC expression driven by the regulated codon optimized hGALCC.O._126T.LV construct was well tolerated by human HSPCs, transduced cells were extensively characterized and analyzed in their functionality both in vitro and in vivo. Control vector, as anticipated above, was a similar LV encoding for the human Arylsulfatase A (ARSA) cDNA, being ARSA the lysosomal enzyme acting before GALC in the sulfatide catabolic pathway, under the control of the same hPGK promoter but without any miRNA regulation due to its safe expression profile13,17 (hARSA.LV).
BM- and CB-HSPCs transduced with hGALCC.O._126T.LV and control vectors were plated for colony-forming cell assay and Long-term Culture Initiating Cell assay (LTC-IC); a similar number of colony-forming cells were retrieved in all the tested conditions (Figure 2a and Supplementary Figure S5), with homogenous size, morphology, and differentiation pattern toward burst/colony-forming unit-erythroid (B/CFU-E) and colony-forming unit macrophage/granulocyte macrophage (CFU-M/GM) subsets (Figure 2b).
Figure 2.

Safe hGALC over-expression in human HSPCs. (a,b) HSPCs from BM- (n = 11 replicates derived from five donors for transduced cells and n = 5 donors for untransduced cells) and CB- (n = 6 donors) were transduced with the indicated LVs and plated for the clonogenic assay. The number (#) of (a) colonies and (b) differentiation toward B/CFU-E and CFU-M/GM subsets are reported (mean±SEM). (c,d) BM- (n = 9 replicates derived from three donors for transduced cells and n = 3 replicates derived from three donors for UT cells) and CB- (n = 6 donors) derived HSPCs transduced with indicated LVs were evaluated for (c) the occurrence of apoptosis and (d) maintenance of stemness markers at the end of the second hit of transduction, and after 1 day in culture. Apoptosis was assessed by cytofluorimetric analysis using AnnexinV/7AAD markers and identifying viable cells (AnnV-7AAD-), early apoptotic cells (AnnV+7AAD-), late apoptotic cells (AnnV+7AAD+), and necrotic cells (AnnV-7AAD+). Average values ± SEM are reported. On viable cells: ****P < 0.0001, ***P < 0.001, with unpaired t-test on hGALC- versus hARSA-expressing HSPCs. Percentages of CD34+ and CD38+ relative staining were assessed by cytofluorimetric analysis (mean ± SEM).
To exclude any latent early sign of toxicity induced by transduction and transgene expression in HSPCs, BM and CB cells were deeply analyzed for occurrence of apoptosis by AnnexinV/7AAD cytofluorimetric analysis performed soon after transduction and after 1 day in culture. The assessment showed comparable percentages of viable cells as well as early apoptotic events likely due to transduction in all tested conditions (Figure 2c). Moreover, hGALCC.O._126T.LV transduced human HSPCs showed similar surface marker expression as compared to control vector-transduced counterparts (Figure 2d).
We previously demonstrated that GALC-mediated toxicity and induction of apoptosis in murine HSPCs transduced with mGALC-expressing LV are associated with accumulation of Ceramide (in particular of C16 Ceramide, a specific well known proapoptotic stimulus18,19) and related proapoptotic sphingolipids. We thus measured these compounds in BM-HSPCs at +1 day and +14 days after transduction with the above-referenced vectors. The sphingolipid profile did not substantially differ between hGALCC.O._126T.LV and control vector transduced BM-HSPCs and their in vitro progeny, suggesting that regulated hGALC expression does not cause a perturbation of HSPCs sphingolipid content at both the tested time points (Figure 3a).
Figure 3.

Sphingolipid and gene expression profile of BM-derived HSPCs. (a) Dihydroceramide (total d18:0), ceramide (total d18:1), C16-ceramide (d18:1/16:0), globotriaosylceramide (Gb3), glucosyl/galactosyl ceramide (Glc/GalCer), lactosylceramide (LacCer), sphinganine (SPA), sphingosine (SPH) were measured at +1 day and +14 days in BM-derived HSPCs transduced as indicated. Sphingolipids concentration is expressed as pmol/μg protein content and values are reported on a logarithmic scale (+1 day: n = 3 donors; +14 days: n = 2 donors). (b) Unsupervised Affymetrix analysis of the gene expression profiles of BM-derived HSPCs transduced with hGALCC.O._126T.LV or hARSA.LV and control untransduced samples (UT) (n = 3 donors for each condition).
To further investigate whether LV-driven hGALC regulated expression could affect physiological processes of BM-HSPCs, the expression profiles of BM-derived HSPCs transduced with hGALCC.O._126T.LV and hARSA.LV were compared with those of untransduced (UT) cells at +1 day. Unsupervised hierarchical clustering by the dChip software showed a tendency to cluster in three main branches corresponding to transduction triplicates, indicating that overexpression of hGALC and hARSA was not associated with consistent changes in the gene expression profile of HSPCs (Figure 3b). A supervised clustering approach (fold change >2, P < 0.05) performed on hGALC and hARSA-transduced versus UT cells (Supplementary Figure S6a,b) and transduced versus UT cells (Supplementary Figure S6c) displayed overall 17 upregulated and 3 downregulated genes. Only 1 gene (Slc7a11) is in common among the three analyses (a, b, and c). None of the deregulated genes belong to the proto-oncogene database20 and none of them, with the exception of hARSA, belong to the sphingolipid metabolism pathway (KEGG hsa00600 pathway) indicating that the hGALC overexpression driven by the regulated LV has no impact on the sphingolipid enzymatic cascade.
Regulated hGALC overexpression does not impair repopulation and differentiation potential of BM-HSPCs
BM-HSPCs transduced with hGALCC.O._126T.LV and control vector were also analyzed in vivo for their repopulation and differentiation potential. Sublethally irradiated NSG mice were transplanted with BM-HSPCs transduced with hGALCC.O._126T.LV or with control vector. Mice were sacrificed 12 weeks after transplantation and hematopoietic organs were analyzed for human cell engraftment and differentiation. hGALC-transduced cells efficiently repopulated hematopoietic organs of all transplanted mice similar to control transduced cells (Figure 4a). Notably, multilineage differentiation of the grafted population in BM, liver, spleen, and thymus of the chimeric mice was observed (Figure 4b). No significant differences in the repopulation and differentiation capacities of hGALC- and control LV-transduced hHSPCs were noticed, thus enforcing the safety profile of regulated hGALC overexpression in these cells.
Figure 4.

Engraftment and differentiation of BM-derived HSPCs in tissues of transplanted NSG mice. BM-derived HSPCs (n = 3 donors) transduced with hGALCC.O._126T.LV or hARSA.LV were transplanted into sublethally irradiated NSG mice (n = 16 and 15 mice, respectively). (a) The percentage of human CD45+cells in BM, liver, spleen, and thymus of NSG mice was evaluated 12 weeks after transplantation. (b) Mice with >1% CD45+ cells in BM (hGALCC.O._126T.LV: n = 16; hARSA.LV: n = 15), liver (hGALCC.O._126T.LV: n = 16; hARSA.LV: n = 15) and spleen (hGALCC.O._126T.LV: n = 10; hARSA.LV: n = 9) and with >5% CD45+ cells in the thymus (hGALCC.O._126T.LV: n = 5; hARSA.LV: n = 2) were considered engrafted and on these CD45+ percentages a lineage marker evaluation was performed, including the following markers: CD34 (stem and progenitor cells), CD13 (myeloid cells), CD15 (granulocytes), CD19 (B lymphocytes), CD3 (pan-T lymphocytes), CD4 (T-helper lymphocytes), CD8 (cytotoxic T lymphocytes); percent marker-positive cells was calculated relative to CD45+ gated cells; in the thymus, the percent of CD3+ cells was calculated among CD45+ cells and then percent of CD4+ and CD8+ cells (indicated by arrow) was calculated relative to CD3+ gated cells.
Functionality of hGALCC.O._126T.LV in a severe GLD mouse model
In order to confirm that this hGALCC.O._126T.LV could be employed for future clinical translation, we tested it in a severe murine model of GLD for its capability to induce the production of a functional protein able to clear sphingolipid storages and contribute to phenotype amelioration. Lineage negative HSPCs isolated from Twitcher mice were transduced with hGALCC.O._126T.LV and hGALC activity was measured at above-normal values (1.6-fold the enzyme activity measured in the control LV-transduced wild-type cells) in their culture progeny (Figure 5a). An high VCN, comparable to controls, was also measured in the gene-corrected murine HSPCs (Figure 5b), suggesting the absence of the negative selection of highly transduced cells previously observed upon unregulated mGALC overexpression.14 Moreover, murine HSPCs transduced with the two vectors when plated for the colony-forming cell assay gave rise to a comparable number of colonies, indicating the absence of a functional impairment (Figure 5c) and supporting the tolerability of regulated hGALC expression driven by hGALCC.O._126T.LV also in murine HSPCs.
Figure 5.
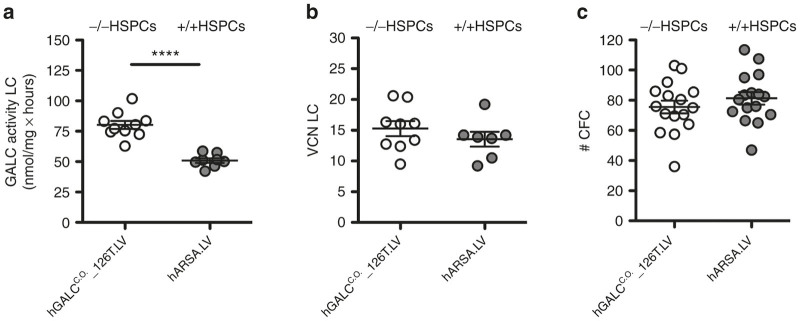
Safe hGALC over-expression in murine HSPCs. (a) HSPCs from BM of Twitcher mice (–/–) and wild-type (+/+) mice were transduced with hGALCC.O._126T.LV and hARSA.LV, respectively, and plated for liquid culture and clonogenic assay. (a) hGALC activity (n = 10 and 8 pools of donors respectively) and (b) VCN (n = 9 and 7 pools of donors, respectively) evaluated after 14 days of liquid culture, and the number (#) of colonies (n = 16 pools of donors each) (c) retrieved after 2 weeks of growth in semisolid medium are reported (mean ± SEM). Statistical analysis on hGALC activity values was performed with unpaired t-test; ****P < 0.0001.
hGALC-corrected Twitcher HSPCs were then transplanted into lethally irradiated p7 newborn Twitcher mice. Transplanted mice (-/- hGALC_126T) showed a statistically significant longer survival than untreated (UT) Twitcher mice (P = 0.0002); results were comparable to what observed in animals receiving wild-type control LV-transduced murine HSPCs (+/+ hARSA) (Figure 6a). This gain in survival was associated to a sustained engraftment of hGALCC.O._126T.LV-transduced cells in the BM of transplanted mice (Supplementary Table S1), and efficient hGALC enzyme delivery to the brain, spinal cord and liver, as observed in long-lived treated animals (Figure 6b). VCN measured in the BM significantly positively affected hGALC activity measured in brain and spinal cord and survival of gene therapy–treated mice (Figure 6c). These findings were supported by the evidence of transduced cells in these tissues (Table S1). Along with improved survival, gene therapy–treated mice also experienced reduced tremors and showed an amelioration of walking abilities (Figure 6d) as compared to untreated animals. To determine the effect of hGALC activity reconstitution on lysosomal storage in the nervous tissue, we visualized globoid cells containing undegraded sphingolipids by lectin immunofluorescence. Lectin+ storage was strongly increased in Twitcher brains as compared to wild-type counterparts, with a rostral to caudal gradient reflecting disease progression (Figure 6e). In the brain of long-lived gene therapy–treated mice, a significant reduction of the storage was observed in cortex, cerebellum, and pons (Figure 6e and Supplementary Figure S7). Reduction was also observed in the sciatic nerve, suggesting a gene therapy contribution also at the PNS level (Figure 6e). Astrogliosis was also measured by means of GFAP staining in treated and control mice. While a strong GFAP immunopositive signal was detected in Twitcher mice in both anterior and posterior brain areas, a significant reduction of the signal was observed in the cortex and pons of gene therapy–treated mice with a trend of decrease of GFAP staining also in the cerebellum (Figure 6f and Supplementary Figure S7). These results were comparable if not superior to those observed by transplanting control LV-transduced wild-type cells.
Figure 6.

Therapeutic efficacy of HSPC-based gene therapy with hGALCC.O._126T.LV in Twitcher mice. (a) Kaplan–Meier survival curves of Twitcher mice transplanted with either -/- HSPCs transduced by hGALCC.O._126T.LV (n = 9 transplanted mice) or +/+ HSPCs transduced by hARSA.LV (n = 12 transplanted mice) and untreated affected controls (UT) (n = 36 mice). Mice survival over day 40 (deaths up to this phase are expected as consequence of the conditioning regimen) are shown. UT mice survived on average 43 ± 3 days (maximal survival, 50 days); -/- hGALC_126T mice: 53 ± 9 days (maximal survival, 70 days); +/+ hARSA mice: 57 ± 9 days (maximal survival, 73 days). Log-rank (Mantel-Cox) test for curves comparison: -/- hGALC_126T versus UT P = 0.0002; hARSA versus UT P < 0.0001; -/- hGALC_126T versus +/+ hARSA P = ns. Comparison of overall survival was done by one-way analysis of variance with Bonferroni posttest: -/- hGALC_126T versus UT P < 0.0001; +/+ hARSA versus UT P < 0.0001; -/- hGALC_126T versus +/+ hARSA P = ns. (b) hGALC activity measured in the indicated tissues is reported for -/- hGALC_126T (n≥6) and +/+ hARSA (n ≥ 4) long-lived (≥47 days) mice. (c) Pearson’s positive correlations between VCN retrieved in the BM of -/- hGALC_126T mice and hGALC activity measured in brain (Pearson r = 0.91, P = 0.012, n = 6) and spinal cord (Pearson r=0.97, P = 0.001, n = 6), and correlation between VCN in the BM and survival of -/- hGALC_126T mice (Pearson r = 0.92, P = 0.01, n = 6) are reported. Lines represent linear regression of data (brain: r2 = 0.83; spinal cord: r2 = 0.95; survival: r2 = 0.84). (d) Walking patterns of a representative gene therapy–treated mouse and controls were recorded, as reported in methods. (e) Quantification of lectin+ area on tissue sections from cortex, cerebellum, pons, and sciatic nerve of untreated +/+ (n = 4), -/- (n = 4), -/- hGALC_126T (n = 6), and +/+ hARSA (n = 3). (f) Quantification of GFAP area on tissue sections from cortex, cerebellum and pons of untreated +/+ (n = 4), -/- (n = 4), -/- hGALC_126T (n = 6) and +/+ hARSA (n = 3). For (e and f), an average of ≥3 sections from each area were evaluated for each mouse. Statistical analysis for (e and f) was performed with one-way analysis of variance with Bonferroni posttest: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Discussion
There is a strong rationale for the development of a safe and effective gene therapy approach for GLD based on HSPCs and LVs, given the favorable preliminary results obtained with this treatment in a similar lysosomal storage disease, metachromatic leukodystrophy.13 The experimental work described here contributes to the clinical development of such approach by addressing key feasibility, efficacy, and safety issues related to the expression of the hGALC enzyme in hematopoietic cells.
Based on our previous data obtained in the disease murine model employing the murine Galc cDNA and murine HSPCs,14,15 we developed a LV construct containing the target sequences of miR-126 and the human Galc cDNA for clinical translation. This construct allowed preventing enzyme expression in early murine and human HSPCs that are plenty of miR-126, while drove sustained GALC activity in their differentiated progeny that is nearly deprived of miR-126. For clinical translation of this strategy we then constructed and tested different LVs in which the murine transgene was replaced by the human Galc cDNA and different levels of regulation were obtained by varying the number of miR-126 target sequences inserted downstream the transgene, based on the evidence that the miR-126 machinery is nearly fourfold more active in human versus murine cells.14 Interestingly, fewer miR-126 target sequences were enough to obtain a strong repression of human GALC expression in human HSPCs as compared to murine cells. These data are supported by the recent preclinical testing of a miR-126 regulated LV for the treatment of X-linked chronic granulomatous disease (X-CGD) in which two tandem repeats were used as best combination of miR-126T in human HSPCs, and more target sequences resulted in reduced transgene expression in differentiated cells.16 We further reduced the number of miR-126 target sequences to one in order to achieve the highest regulated enzymatic activity in differentiated cells and to avoid any possible risk of miR-126 de-targeting from its natural targets. Moreover, the low but detectable enzyme activity measured at +1 day after gene transfer in human HSPCs transduced with the regulated LV could also be beneficial to restore cellular functions that may be affected by complete human GALC deficiency in patients’ cells with null Galc mutations.15
In order to ameliorate the stability of hGALC long transcript and improve enzyme expression in differentiated cells, we took advantage of codon optimization. Recoding the transgene to optimize transcription and translation has already been demonstrated to improve LVs titers as well as protein production and the efficacy of therapies, i.e., for chronic granulomatous disease, hemophilia A and B, RAG1 and 2 SCID, and X-linked SCID.16,21–26 Upon codon optimization, while maintaining a high suppression of hGALC expression in human HSPCs even in the presence of only one miR-126T, we could obtain substantially increased enzyme expression, up to 14-folds over normal donors’ level, in the differentiated progeny of BM-derived HSPCs, improving the therapeutic potential of the optimized vector over a noncodon optimized one.
The codon optimized regulated LV was then tested for safety in highly transduced human HSPCs in order to assess whether the regulation provided by one miR-126 target sequence could prevent toxicity, apoptosis, and/or functional impairment. Importantly, transduced CB- and BM-HSPCs plated in semi-solid medium showed a normal clonogenic potential in terms of number and morphology of generated colonies and of differentiation toward B/CFU-E and CFU-M/GM lineages. This result is superimposable to what was obtained on CB-derived HSPCs upon transduction with the four target sequences-regulated LV expressing murine GALC.14 This preserved cell functionality was also corroborated by the lack of evidence of up-regulation of apoptotic markers in the transduced cells, as assessed by cytofluorimetric analysis, 1 day after transduction. Consistently, sphingolipid measurement in BM-derived HSPCs transduced with the hGALCC.O._126T.LV failed to demonstrate accumulation of proapoptotic species shortly after transduction and upon in vitro differentiation. Since sphingolipid metabolism is in constant flux with the balance between pro- and antiapoptotic metabolites determining cellular fate (concept known as “sphingolipid rheostat”27–30), these data suggest that the amount of sphingolipids produced by human GALC in human HSPCs transduced with the optimized construct could be efficiently managed by downstream enzymes able to bring them back to well tolerated levels, thus avoiding the induction of apoptosis. To further confirm these data and exclude any other perturbation at a molecular level, a gene expression analysis was performed on transduced BM-derived HSPCs. No significantly down- or upregulated transcripts were found shortly after transduction with hGALCC.O._126T.LV with respect to control LV. Moreover, the supervised analyses identified two main branches, untransduced and LV-transduced cells, independently from the gene carried by the vector. Among the transduced cells the hGALC and hARSA samples cluster according to donors, remarking that the hGALC- and hARSA-transduced cells show a comparable expression profile.
Once confirmed the safety and tolerability of hGALC expression driven by hGALCC.O._126T.LV in vitro, transplantation studies were performed to assess transduced human HSPCs functionality in vivo. Importantly, also in the challenging in vivo setting of human/mouse chimeras, BM-derived HSPCs transduced with the regulated LV demonstrated their full functionality by successful engraftment in NSG mice and multilineage differentiation as control LV-transduced cells.
Given the positive results obtained in human HSPCs, the hGALCC.O._126T.LV was tested in a severe murine model of GLD to prove its potential to produce a functional protein able and sufficient to clear pathologic storage and attenuate disease manifestations. The vector was used to safely transduce murine HSPCs that were then transplanted in lethally irradiated Twitcher neonates. The transplant resulted in efficient delivery of the corrective enzyme by the progeny of transplanted cells to neural and not-neural tissues, with positive correlation of enzyme activity with transduced cell engraftment and in a clear phenotype amelioration. A significant improvement in the lifespan of the treated mice, as compared to untreated Twitcher animals, was achieved as a proof-of-concept of enzyme functionality, and correlation of survival with transduced cell engraftment was shown. This prolonged survival, which is similar to what was obtained by transplanting HSPCs from wild-type donors, was associated to clearance of undegraded material and reduction of the astrogliosis in the brain of treated animals. Despite the level of enzyme expression measured in the culture progeny of murine HSPCs transduced with the codon optimized hGALC regulated LV is superimposable to what obtained with a murine GALC-encoding LV regulated by 4 miR-126 target sequences, the advantage of gene-corrected HSPCs over their wild-type counterpart previously observed in Trs mice using the regulated LV encoding murine Galc cDNA14 was not demonstrated. This difference could be due to the choice of a more challenging disease animal model (the Twitcher mouse, a total Galc knock out, as compared to Trs mice that have some residual enzyme activity) characterized by a more rapidly progressing disease. Moreover, hGALC codon optimized mRNA sequence could be translated with less efficiency in murine cells, being optimized on human tRNAs abundance, and the resulting human enzyme could lead to unfavorable interactions with murine activators and microenvironment. Of note, even if HSC gene therapy employing this or the previously tested LV14 allowed to significantly delay disease progression, this treatment alone, at least in the severe animal models of the disease, did not definitely prevent or halt disease progression, since mice still die with GLD symptoms. This is due to the complexity and rapid progression of the disease that will likely benefit from combinatorial approaches, as tested by others and us.31,32
In conclusion, we here demonstrated the safe and effective profile of our miR-126 regulated LV in driving high and functional human GALC expression in the progeny of both human and murine HSPCs, without impairing their viability and functionality. We expect that Krabbe HSPCs could benefit even more from this approach for the low but detectable enzyme activity measured soon after gene transfer that could restore cellular functions and for the sustained therapeutic activity achieved in the differentiated progeny that could promote high extent of surrounding cells cross-correction. Altogether, these findings provide a strong rationale for considering this LV the best candidate for the clinical translation of an ex vivo gene therapy approach for GLD.
Materials and Methods
Lentiviral vector production and titration
VSV-pseudotyped third-generation LVs were produced by cotransfection of transfer, packaging (pMD2.Lg/p.RRE and pRSV.Rev), and envelope constructs (pMD2.G) into 293T cells, as previously described.33 The following transfer constructs were used: pCCLsin.cPPT.hPGK.hARSA.Wpre; pCCLsin.cPPT.hPGK.hIDUA.Wpre. hGALC cDNA was originally provided by Dr Y. Eto and cloned in the LV alone (pCCLsin.cPPT.hPGK.hGALC.Wpre) or followed by one (pCCLsin.cPPT.hPGK.hGALC_1xmiR126T.Wpre), two (pCCLsin. cPPT.hPGK.hGALC_2xmiR126T.Wpre), or four (pCCLsin.cPPT.hPGK.hGALC_4xmiR126T.Wpre) miR-126 target sequences (miR-126T:GCATTATTACTCACGGTACGA). Codon optimized hGALC sequence (hGALCC.O) was synthesized (GeneArt Gene Synthesis; Life Technologies, Carlsbad, CA) and cloned in the LV with a downstream one miR-126 target sequence (pCCLsin.cPPT.hPGK.hGALCC.O_1xmiR126T.Wpre).
Mouse studies
FVB-Twitcher (FVB/Twi) mice were generated in our animal research facility by breeding heterozygous Twitcher (+/−) C57BL6 mice (Jackson Laboratory, Bor Harbor, ME) with wild-type (+/+) FVB mice. These mice referred to as Twitcher mice in this study have been previously described.31,34,35 Procedures were performed according to protocols approved by the Animal Care and Use Committee of the Fondazione San Raffaele (IACUC #425, 573) and communicated to the Ministry of Health and local authorities. Mice genotyping was performed on DNA from tail biopsies as described.36 NOD.Cg-PrkdcscidIl2rgtm1WjI/SzJ (NSG) mice were purchased by Jackson Laboratory.
Isolation, transduction, and transplantation of human CD34+ cells
Human bone marrow (BM)–derived HSPCs were purchased (2M-101C; Lonza, Verviers, Belgium) and prestimulated for 24 hours in CellGro medium (CellGenix, Freiburg, Germany) supplemented with hIL-3 (60 ng/μl), hTPO (100 ng/μl), hSCF (300 ng/μl), hFlt3-L (300 ng/μl) (all of them from Peprotech, Hamburg, Germany) and transduced with two hits of LV at MOI 100 as described.13
Human cord blood (CB)-derived HSPCs were either freshly purified from human cord blood after obtaining informed consent and on approval by the San Raffaele Hospital Bioethical Committee, or purchased from Lonza (2C-101). Cells were either prestimulated for 12 hours with the standard CB-HSPCs cytokine cocktail (hIL-6 (20 ng/μl), hTPO (20 ng/μl), hSCF (100 ng/μl), hFlt3-L (100 ng/μl)) in StemSpan serum-free medium (StemCell Technologies, Vancouver, British Columbia, Canada) and transduced with two hits of LV at MOI 100 (ref. 17) or prestimulated for 24 hours and transduced following BM-HSPCs protocol previously described. Untransplanted BM- and CB-derived HSPCs were kept in culture for 14 days in IMDM medium (Sigma Aldrich, St Louis, MO), 10% FBS (Euroclone, Pero, Italy), penicillin 100 U/ml and streptomycin 100 µg/ml, l-glutamine 2 mmol/l, and cytokines (hSCF (300 ng/μl), hIL3 (60 ng/μl), hIL6 (60 ng/μl)).
For NSG experiments, human BM-derived HSPCs were prestimulated and transduced with the indicated LVs. After transduction, 3 × 105 cells were washed and infused into the tail vein of sublethally irradiated (176 cGy - Rad Gil EN 60601- 1, Gilardoni, Mandello del Lario, Italy) 7–9-week-old female NSG mice. After 12 weeks, mice were sacrificed and BM, spleen, thymus, and liver were harvested and analyzed for lineages composition.
Isolation, transduction, and transplantation of murine hematopoietic cells
Young adult mice (4–8 weeks) were sacrificed with CO2, and BM was harvested by flushing femurs, tibias, and humerus. Murine HSPCs were purified (StemSep Mouse Hematopoietic Progenitor Cell Enrichment Kit; StemCell Technologies) and transduced in StemSpan serum-free medium supplemented with cytokines (mSCF (5 ng/ml), mFlt3L (10 ng/ml), mIL3 (10 ng/ml), mIL6 (20 ng/ml); all purchased from Peprotech) with LVs as previously described.37 Transduced cells (7.5 × 105 cells/mouse) were intravenously (jugular vein) injected into 7-day-old lethally irradiated (612 rad) -/-Twitcher mice. After 3 days, mice were injected intraperitoneally with 5 × 106 BM mononuclear cells from -/-Twitcher mice as supportive cells. Untransplanted murine HSPCs were kept in culture for 14 days in RPMI medium (Sigma), 10% FBS (Euroclone), penicillin 100 U/ml and streptomycin 100 µg/ml, l-glutamine 2 mmol/l, and cytokines (mSCF (5 ng/ml), mFlt3L (10 ng/ml), mIL3 (10 ng/ml), mIL6 (20 ng/ml); Peprotech).
Colony-forming cell assay
After transduction, HSPCs were washed, counted, and seeded at a density of 800 cells/ml (human) or 7,000 cells/ml (murine) in semi-solid medium (human MethoCult, H4434; murine Methocult, M3434; StemCell Technologies). After 14 days, colonies were scored and counted.
GALC activity
GALC activity was determined as previously described.38
Quantitative PCR
Genomic DNA was extracted from liquid culture samples with QIAamp DNA Blood Mini kit (Qiagen, Hilden, Germany), from liver (<100 mg) with QIAamp MIDI kit (Qiagen) and from brain and spinal cord tissues (<100 mg) by using TRI Reagent (Sigma). Quantitative PCR analysis was performed on at least 50 ng of total genomic DNA as described.17,39 The number of LV copies per cell (VCN) was calculated by the following equation: (ng LV/ng endogenous DNA) × (no. of LV integrations in the standard curve) × (sample ploidy/standard ploidy).
Immunofluorescence analysis
Mice were deeply anesthetized with 2,2,2-Tribromoethanol (Sigma) and intracardially perfused with 0.9% NaCl. Brains and sciatic nerve tissues were collected, equilibrated for 24 hours in 4% PFA followed by 48-hour incubation in 20 and 30% sucrose in D-PBS. After embedding in Tissue-Tek O.C.T. compound (Sakura, Torrance, CA) and deep-freezing on dry ice, tissues were cut in serial sagittal and transverse 14-μm-thick cryostat slices for brain and sciatic nerve, respectively. Sections mounted on SuperFrost slides (Thermo Scientific, Waltham, MA) were washed for 5 minutes in D-PBS and incubated with blocking solution (D-PBS, 10% goat serum, 0.3% Triton X-100) for 2 hours. The primary antibodies anti-GFAP (1:100, MAB3402; Millipore, Billerica, MA) and Lectins from Bandeiraea simplicifolia (Griffonia simplicifolia)-FITC (10 μg/ml; Sigma) were diluted in blocking solution. After incubation for 12 hours at 4 °C, sections were washed 3× with D-PBS and stained for 1 hour with secondary antibody goat antimouse IgG (1:500; Alexa Fluor 546; Molecular Probes, Eugene, OR) in D-PBS 10% goat serum and 0.1% Triton X-100. Nuclei were stained for 8 minutes at room temperature with ToProIII (1:1,000; Molecular Probes) in D-PBS. Sections were washed three times with D-PBS and mounted with FluorSave (Calbiochem, San Diego, CA). Samples were analyzed using a Zeiss Axioskop2 microscope using double laser confocal microscopy with Zeiss Plan-Neofluar objective lens (Zeiss, Arese, Italy). Images were acquired using a Radiance 2100 camera (Bio-Rad, Segrate, Italy) and LaserSharp 2000 acquisition software (Bio-Rad) and analyzed by Adobe Photoshop CS4 software (Adobe, San Jose, CA). For computer-aided image analysis, Image J software (NIH Image, Bethesda, MD) was used to quantify GFAP+ and lectin+ signal on images—size (pizel^2) 0–9.9999, circularity 0.00–1.0, measurements: pixel counts, total signal positive area (reported in graph), and average pixel size.
Flow cytometric analysis
Human cells from liquid culture or cells from tissues of transplanted NSG mice were resuspended in FACS blocking buffer (D-PBS, 1%BSA, 5%FBS and Fc Block (1:100; BD Pharmingen, San Diego, CA)) at 4 °C for 15 minutes, and then incubated with following antibodies: APC anti-human CD45 (Invitrogen, Carlsbad, CA) or APC-Cy7 anti-human CD45 (BD Pharmingen); RPE anti-human CD34 (Dako, Carpinteria, CA) or Pe-Cy7 anti-human CD34 (BD Biosciences); APC anti-human CD38 (BD Pharmingen), Pacific blue anti-human CD19 (Invitrogen), PE anti-human CD13 (BD Pharmingen), Amcyan anti-human CD3 (BD Biosciences, San Jose, CA), RPE anti-human CD4 (Invitrogen), Pacific blue anti-human CD8 (Invitrogen); FITC anti-human CD15 (BD Biosciences) at 4 °C for 20 minutes. Cells were then washed and resuspended in fixing solution (D-PBS, 1% paraformaldehyde, and 2% FBS) and analyzed by FACS LSRII (BD Biosciences). 5,000–10,000 events were scored. Results were analyzed by FlowJo 8.5.3 software (Tristar, Ashland, OR).
7AAD/annexin V staining
Annexin V–PE apoptosis detection kit (BD Pharmingen) was used to detect early apoptotic cells. Human HSPCs were washed with D-PBS and resuspended in Binding Buffer solution at the concentration of 106 cells/ml and labeled following manufacturer’s protocol. 7-Amino-actinomycin D (7AAD; BD Pharmingen) was also added to the cells to distinguish among live (7AAD−, AnnexinV−), early apoptotic (7AAD−, AnnexinV+), late apoptotic (7AAD+, AnnexinV+), and necrotic (7AAD+, AnnexinV−) cells. Samples were analyzed by flow cytometry with FACS LSRII (BD Biosciences) within 1 hour, and results were analyzed by the Flow-Jo 8.3 software (Tristar).
Lipidomic analyses
The lipid analyses of cell samples were performed by mass spectrometry according to the Standard Operating Procedures (SOP), the Lab Method Sheets (LMS), and data Processing Method Sheets (PMS) of Zora Biosciences Oy (Espoo, Finland).40 In brief, lipids were extracted from cell suspensions using a modified Folch lipid extraction, reconstituted in chloroform:methanol (1:2, v/v) and synthetic external standards are post-extract spiked to the extracts. Total protein concentration of cell samples was determined using the Micro BCA Protein Assay Kit (Thermo Scientific) according to the manufacturer’s instructions, and the absorbance of the samples is measured at 560 nm by Multiskan EX (Thermo Scientific). Lipids were analyzed on a hybrid triple quadrupole/linear ion trap mass spectrometer (5500 QTRAP; AB SCIEX, Foster City, CA) equipped with an ultra-high pressure liquid chromatography (UHPLC) system (CTC HTC PAL autosampler and Rheos Allegro pump (Flux Instruments, Basel, Switzerland)) using multiple reaction monitoring (MRM)–based method in negative ion mode according to the corresponding LMS. The mass spectrometry data files were processed using MultiQuant 2.0 (AB SCIEX) for producing a list of lipid names and peak areas. Lipids were normalized to their respective internal standard and sample volume. The concentrations of molecular lipids are presented as pmol/μg total protein.
Gene expression analyses
Affymetrix microarray analysis was used for transcriptional profiling. RNA was isolated from the BM-derived HSPCs by the RNeasy Plus Mini kit (Qiagen), reverse transcribed with a GeneChip 3′ IVT Express kit, and hybridized to Affymetrix HG-U133 Plus 2.0 GeneChip arrays (both from Affymetrix, Santa Clara, CA). Hybridization, staining, and scanning were carried out by standard Affymetrix protocols. Fluorescence signals were recorded by a GeneChip scanner 3000 (Affymetrix), and image analysis was performed with the GeneChip Operating Software (Affymetrix). All data analyses were performed in R, using Bioconductor libraries and R statistical packages. The robust multiarray average procedure was used to convert probe signals into expression values. Specifically, the intensity scores were adjusted for the background and normalized by quantile normalization. Log2 expression values were calculated using median polish summarization and the custom definition files for Human Gene U133 Plus 2.0 arrays, based on Entrez (18 185 Entrez genes, version 12; available at: http://brainarray.mbni.med.umich.edu/Brainarray/default.asp). Cluster analysis was carried out using probe sets reannotated with custom definition files, to ensure unequivocal probe-to-gene assignment. Differentially expressed genes were identified using DNA-Chip Analyzer software (dChip; available at: http://www.dchip.org) by selecting transcripts for which the difference in the level of expression between the two groups yielded a P value of 0.05 in a unsupervised and supervised analysis.
Walking abilities
Mice were colored at their upper paws with black Indian ink and lower paws with red Indian ink and were let walking in a corridor on a white sheet.
Statistical analysis
Analyses were made by one-way analysis of variance for repeated measurements using Bonferroni’s test for post hoc analysis. For pairwise comparisons, an unpaired Student’s t-test was used. All analyses were performed using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA).
Acknowledgments
We are kindly indebted to L. Sergi Sergi for the precious help in LV production. This work was supported by the Italian Telethon Foundation and the European Union (Leukotreat).
None of the authors declares conflict of interest.
References
- Jatana, M, Giri, S and Singh, AK (2002). Apoptotic positive cells in Krabbe brain and induction of apoptosis in rat C6 glial cells by psychosine. Neurosci Lett 330: 183–187. [DOI] [PubMed] [Google Scholar]
- Tanaka, K, Nagara, H, Kobayashi, T, Goto, I and Suzuki, K (1989). The twitcher mouse: attenuated processes of Schwann cells in unmyelinated fibers. Brain Res 503: 160–162. [DOI] [PubMed] [Google Scholar]
- Cantuti Castelvetri, LC, Givogri, MI, Zhu, H, Smith, B, Lopez-Rosas, A, Qiu, X et al. (2011). Axonopathy is a compounding factor in the pathogenesis of Krabbe disease. Acta Neuropathol 122: 35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg, B, Kollberg, H, Sourander, P and Akesson, HO (1969). Infantile globoid cell leucodystrophy (Krabbe’s disease). A clinical and genetic study of 32 Swedish cases 1953–1967. Neuropadiatrie 1: 74–88. [DOI] [PubMed] [Google Scholar]
- Li, Y and Sands, MS (2014). Experimental therapies in the murine model of globoid cell leukodystrophy. Pediatr Neurol 51: 600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogerbrugge, PM, Poorthuis, BJ, Romme, AE, van de Kamp, JJ, Wagemaker, G and van Bekkum, DW (1988). Effect of bone marrow transplantation on enzyme levels and clinical course in the neurologically affected twitcher mouse. J Clin Invest 81: 1790–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichioka, T, Kishimoto, Y, Brennan, S, Santos, GW and Yeager, AM (1987). Hematopoietic cell transplantation in murine globoid cell leukodystrophy (the twitcher mouse): effects on levels of galactosylceramidase, psychosine, and galactocerebrosides. Proc Natl Acad Sci USA 84: 4259–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo, A, Hoogerbrugge, PM, Suzuki, K, Poorthuis, BJ, Van Bekkum, DW and Suzuki, K (1988). Pathology of the peripheral nerve in the twitcher mouse following bone marrow transplantation. Brain Res 460: 178–183. [DOI] [PubMed] [Google Scholar]
- Suzuki, K, Hoogerbrugge, PM, Poorthuis, BJ, Bekkum, DW and Suzuki, K (1988). The twitcher mouse. Central nervous system pathology after bone marrow transplantation. Lab Invest 58: 302–309. [PubMed] [Google Scholar]
- Yeager, AM, Brennan, S, Tiffany, C, Moser, HW and Santos, GW (1984). Prolonged survival and remyelination after hematopoietic cell transplantation in the twitcher mouse. Science 225: 1052–1054. [DOI] [PubMed] [Google Scholar]
- Duffner, PK, Caviness, VS Jr, Erbe, RW, Patterson, MC, Schultz, KR, Wenger, DA et al. (2009). The long-term outcomes of presymptomatic infants transplanted for Krabbe disease: report of the workshop held on July 11 and 12, 2008, Holiday Valley, New York. Genet Med 11: 450–454. [DOI] [PubMed] [Google Scholar]
- Escolar, ML, Poe, MD, Provenzale, JM, Richards, KC, Allison, J, Wood, S et al. (2005). Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. N Engl J Med 352: 2069–2081. [DOI] [PubMed] [Google Scholar]
- Biffi, A, Montini, E, Lorioli, L, Cesani, M, Fumagalli, F, Plati, T et al. (2013). Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 341: 1233158. [DOI] [PubMed] [Google Scholar]
- Gentner, B, Visigalli, I, Hiramatsu, H, Lechman, E, Ungari, S, Giustacchini, A et al. (2010). Identification of hematopoietic stem cell-specific miRNAs enables gene therapy of globoid cell leukodystrophy. Sci Transl Med 2: 58ra84. [DOI] [PubMed] [Google Scholar]
- Visigalli, I, Ungari, S, Martino, S, Park, H, Cesani, M, Gentner, B et al. (2010). The galactocerebrosidase enzyme contributes to the maintenance of a functional hematopoietic stem cell niche. Blood 116: 1857–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiriaco, M, Farinelli, G, Capo, V, Zonari, E, Scaramuzza, S, Di Matteo, G et al. (2014). Dual-regulated lentiviral vector for gene therapy of X-linked chronic granulomatosis. Mol Ther 22: 1472–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capotondo, A, Cesani, M, Pepe, S, Fasano, S, Gregori, S, Tononi, L et al. (2007). Safety of arylsulfatase A overexpression for gene therapy of metachromatic leukodystrophy. Hum Gene Ther 18: 821–836. [DOI] [PubMed] [Google Scholar]
- Thomas, RL Jr, Matsko, CM, Lotze, MT and Amoscato, AA (1999). Mass spectrometric identification of increased C16 ceramide levels during apoptosis. J Biol Chem 274: 30580–30588. [DOI] [PubMed] [Google Scholar]
- Aflaki, E, Doddapattar, P, Radović, B, Povoden, S, Kolb, D, Vujić, N et al. (2012). C16 ceramide is crucial for triacylglycerol-induced apoptosis in macrophages. Cell Death Dis 3: e280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, T, Shen, H, Akagi, K, Morse, HC, Malley, JD, Naiman, DQ et al. (2002). New genes involved in cancer identified by retroviral tagging. Nat Genet 32: 166–174. [DOI] [PubMed] [Google Scholar]
- Moreno-Carranza, B, Gentsch, M, Stein, S, Schambach, A, Santilli, G, Rudolf, E et al. (2009). Transgene optimization significantly improves SIN vector titers, gp91phox expression and reconstitution of superoxide production in X-CGD cells. Gene Ther 16: 111–118. [DOI] [PubMed] [Google Scholar]
- Ward, NJ, Buckley, SM, Waddington, SN, Vandendriessche, T, Chuah, MK, Nathwani, AC et al. (2011). Codon optimization of human factor VIII cDNAs leads to high-level expression. Blood 117: 798–807. [DOI] [PubMed] [Google Scholar]
- Cantore, A, Nair, N, Della Valle, P, Di Matteo, M, Màtrai, J, Sanvito, F et al. (2012). Hyperfunctional coagulation factor IX improves the efficacy of gene therapy in hemophilic mice. Blood 120: 4517–4520. [DOI] [PubMed] [Google Scholar]
- van Til, NP, de Boer, H, Mashamba, N, Wabik, A, Huston, M, Visser, TP et al. (2012). Correction of murine Rag2 severe combined immunodeficiency by lentiviral gene therapy using a codon-optimized RAG2 therapeutic transgene. Mol Ther 20: 1968–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike-Overzet, K, Rodijk, M, Ng, YY, Baert, MR, Lagresle-Peyrou, C, Schambach, A et al. (2011). Correction of murine Rag1 deficiency by self-inactivating lentiviral vector-mediated gene transfer. Leukemia 25: 1471–1483. [DOI] [PubMed] [Google Scholar]
- Huston, MW, van Til, NP, Visser, TP, Arshad, S, Brugman, MH, Cattoglio, C et al. (2011). Correction of murine SCID-X1 by lentiviral gene therapy using a codon-optimized IL2RG gene and minimal pretransplant conditioning. Mol Ther 19: 1867–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuvillier, O, Pirianov, G, Kleuser, B, Vanek, PG, Coso, OA, Gutkind, S et al. (1996). Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 381: 800–803. [DOI] [PubMed] [Google Scholar]
- Bartke, N and Hannun, YA (2009). Bioactive sphingolipids: metabolism and function. J Lipid Res 50 (suppl.): S91–S96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hait, NC, Oskeritzian, CA, Paugh, SW, Milstien, S and Spiegel, S (2006). Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim Biophys Acta 1758: 2016–2026. [DOI] [PubMed] [Google Scholar]
- Hannun, YA and Obeid, LM (2008). Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 9: 139–150. [DOI] [PubMed] [Google Scholar]
- Ricca, A, Rufo, N, Ungari, S, Morena, F, Martino, S, Kulik, W et al. (2015). Combined gene/cell therapies provide long-term and pervasive rescue of multiple pathological symptoms in a murine model of globoid cell leukodystrophy. Hum Mol Genet 24: 3372–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins-Salsbury, JA, Shea, L, Jiang, X, Hunter, DA, Guzman, AM, Reddy, AS et al. (2015). Mechanism-based combination treatment dramatically increases therapeutic efficacy in murine globoid cell leukodystrophy. J Neurosci 35: 6495–6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follenzi, A and Naldini, L (2002). HIV-based vectors. Preparation and use. Methods Mol Med 69: 259–274. [PubMed] [Google Scholar]
- Lattanzi, A, Salvagno, C, Maderna, C, Benedicenti, F, Morena, F, Kulik, W et al. (2014). Therapeutic benefit of lentiviral-mediated neonatal intracerebral gene therapy in a mouse model of globoid cell leukodystrophy. Hum Mol Genet 23: 3250–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri, M, Ricca, A, di Girolamo, I, Alcala’-Franco, B, Cavazzin, C, Orlacchio, A et al. (2011). Neural stem cell gene therapy ameliorates pathology and function in a mouse model of globoid cell leukodystrophy. Stem Cells 29: 1559–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai, N, Inui, K, Tatsumi, N, Fukushima, H, Nishigaki, T, Taniike, M et al. (1996). Molecular cloning and expression of cDNA for murine galactocerebrosidase and mutation analysis of the twitcher mouse, a model of Krabbe’s disease. J Neurochem 66: 1118–1124. [DOI] [PubMed] [Google Scholar]
- Biffi, A, De Palma, M, Quattrini, A, Del Carro, U, Amadio, S, Visigalli, I et al. (2004). Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J Clin Invest 113: 1118–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino, S, Tiribuzi, R, Tortori, A, Conti, D, Visigalli, I, Lattanzi, A et al. (2009). Specific determination of beta-galactocerebrosidase activity via competitive inhibition of beta-galactosidase. Clin Chem 55: 541–548. [DOI] [PubMed] [Google Scholar]
- Biffi, A, Capotondo, A, Fasano, S, del Carro, U, Marchesini, S, Azuma, H et al. (2006). Gene therapy of metachromatic leukodystrophy reverses neurological damage and deficits in mice. J Clin Invest 116: 3070–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, HR, Sylvänne, T, Koistinen, KM, Tarasov, K, Kauhanen, D and Ekroos, K (2011). High throughput quantitative molecular lipidomics. Biochim Biophys Acta 1811: 925–934. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.