

Abstract
Dimethoxycurcumin (DMC) is an analog of curcumin with superior efficacy in various disease models. Currently, drug delivery system research on DMC is very limited, and it has become a huge challenge to realize further developments and clinical applications. In the present study, a kind of amphiphilic block copolymer, N-t-butoxycarbonyl-phenylalanine terminated monomethoxyl poly (ethylene glycol)-b-poly (ε-caprolactone), or mPEG-PCL-Phe(Boc), was prepared from monomethoxyl poly (ethylene glycol)-b-poly (ε-caprolactone) (mPEG-PCL) with its hydroxyl terminal chemically converted into N-t-butoxycarbonyl-phenylalanine (Boc-Phe). This copolymer was determined to have a fairly low critical micelle concentration (2.56×10−3 mg/mL) and passive targeting potential to tumor tissue, and thus was applied to develop a polymeric micellar formulation of DMC for the first time. The DMC-loaded micelles prepared by thin-film hydration method had typical shell–core structure, with an average particle size of 17.9±0.4 nm and a polydispersity index of 0.045±0.011. The drug loading capacity and entrapment efficiency were 9.94%±0.15% and 97.22%±0.18%, respectively, indicating a high-affinity interaction between DMC and the copolymer. At a concentration of 2 mg/mL, the reconstituted micelle solution could be maintained for at least 10 days at room temperature, and displayed a low initial burst release followed by a sustained release in vitro. Pharmacokinetic study in rats revealed that in vivo drug exposure of DMC was significantly increased and prolonged by intravenously administering DMC-loaded micelles when compared with the same dose of free DMC dissolved in dimethyl sulfoxide. Furthermore, in vivo distribution results from tumor-bearing nude mice demonstrated that this micellar formulation significantly changed the biodistribution profile of DMC and increased drug accumulation in tumors. Therefore, the polymeric micellar formulation of DMC, based on the amphiphilic block copolymer, mPEG-PCL-Phe(Boc), could provide a desirable method for delivering DMC, especially for applications in cancer therapy.
Keywords: dimethoxycurcumin, polymeric micelles, characterization, pharmacokinetic profile, biodistribution, tumor targeting
Introduction
Natural products have played a very important role in drug discovery and development throughout history.1 Curcumin is a natural polyphenol derived from turmeric, which is a member of the ginger family and has been used as a food coloring, additive, and medicine in Asian countries for centuries.2,3 Numerous studies have demonstrated that curcumin possesses a broad spectrum of bioactivities, especially the ability to prevent the development of various types of cancer cells while sparing normal cells.4–7 However, the clinical application of curcumin has been severely hindered, mainly due to its undesirable physicochemical and pharmacokinetic properties, such as low water solubility, metabolic instability, and poor bioavailability.8–11 During the past few decades, great attention has been paid to structure modification to overcome these problems, and various novel structural analogs of curcumin have been designed for screening.12–14
Dimethoxycurcumin (DMC; Figure 1) is a kind of lipophilic analog of curcumin with all the phenolic hydroxyl groups methylated, and can be easily obtained by synthetic or semi-synthetic methods.15,16 A variety of experimental studies based on cells and animals have showed that such structural modifications brought about great improvement of DMC in chemical and metabolic stability, as well as anti-inflammatory and anti-tumor activities. Recently, this compound has been identified as a novel androgen-receptor antagonist, and therefore has aroused tremendous research interest.16,17 However, drug delivery system research on DMC is very limited, and it has become a huge challenge for to realize further developments and clinical applications. Some recent studies have been carried out to overcome the poor water solubility of DMC, such as liposomal formulations incorporating DMC into charged liposomes, and covalent conjugates with water soluble poly (amidoamine) (PAMAM) dendrimers (generations 3.5 and 4).18,19 Nevertheless defects in all these methods are obvious, and mainly include complicated preparation processes, thermodynamic instability, low drug loading capacity, and low entrapment efficiency.
Figure 1.
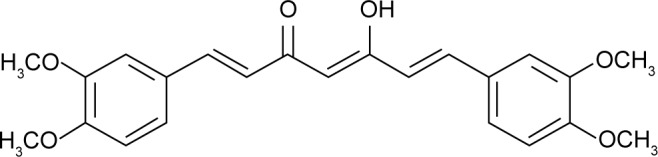
The structure of dimethoxycurcumin (DMC).
Nanotechnology is becoming more and more attractive in drug delivery and target therapy. Synthetic polymeric nanocarriers (generally amphiphilic block copolymers) are a promising tool for nanomedicine therapeutics because of their chemical versatility and good biocompatibility.20 The three most widely studied block copolymer classes are characterized by their hydrophobic blocks, and are poly (propylene oxide), poly (L-amino acid)s, and poly (ester)s. Through the balance between entropy and enthalpy, polymeric micelles (PMs) in aqueous solution are auto-assembly formed into nanoscopic core/shell structures with the hydrophilic “head” regions in contact with surrounding solvent, while sequestering the hydrophobic single-tail regions in the micelle center.21,22 Reasonable design of the hydrophilic shell could also reduce drug uptake by the reticuloendothelial system (RES) and mononuclear macrophage system (MPS), thus prolonging the blood circulation time.23 With the advantages of solubilization of poorly soluble molecules, sustained release, and size advantages, and protection of encapsulated substances from degradation and metabolism, PMs used for drug delivery have shown the abilities to attenuate toxicities, to enhance delivery to desired biological sites, and to improve the therapeutic efficacy of active pharmaceutical ingredients.24–28 Moreover, the process of preparing stable drug-loaded micelles is usually simple and controllable, and easy to industrialize. So far, formulation techniques with polymeric copolymers have be successfully applied to some of the most challenging molecules in the pharmaceutical industry, such as monomethoxyl poly (ethylene glycol)–polylactide copolymer (mPEG-PDLLA) for paclitaxel (Nanoxel), poly (ethylene glycol)–poly (acrylic acid) (PEG-PAA) for doxorubicin (NK-911), and PEGylated glutaminase (PEG-PGA) for SN-38 (NK-012).21,25 To the best of our knowledge, there have been no reports about PM formulation of DMC up to now.
In the present study, PMs were developed for drug delivery of DMC for the first time. In order to improve affinity to drugs and thus acquire high drug loading capacity and entrapment efficiency, monomethoxyl poly (ethylene glycol)-b-poly (ε-caprolactone) (mPEG-PCL) was used as a carrier after its hydroxyl terminal was chemically converted into N-t-butoxycarbonyl-phenylalanine (Boc-Phe). Then, the copolymer mPEG-PCL-Phe(Boc) was obtained, and DMC was incorporated to form drug-loaded micelles by conventional thin-film hydration method. Both the copolymer and drug-loaded PMs were characterized. Pharmacokinetic and biodistribution properties of this polymeric micellar formulation of DMC were further investigated in rats and tumor-bearing nude mice using the same dose of free DMC dissolved in dimethyl sulfoxide (DMSO) as reference. The findings demonstrated that DMC-loaded mPEG-PCL-Phe(Boc) micelles could significantly improve the pharmacokinetic profile, biodistribution, and tumor accumulation of DMC, and therefore could be a desirable drug delivery system of DMC, especially for applications in cancer therapy.
Materials and methods
Chemicals
Both DMC and bis-demethoxycurcumin, used as an internal standard (IS) for bioanalysis, were prepared by our laboratory and were tested for purity, which was >98.0%, using high-performance liquid chromatography (HPLC). Stannous octoate (Sn(Oct)2); ε-caprolactone (ε-CL); and monomethoxyl poly (ethylene glycol) (mPEG) with a number-average molecular weight (MN) of 2,000 were obtained from Sigma-Aldrich Co (St Louis, MO, USA). Pivaloyl chloride and 4-pyrrolidinopyridine were purchased from Wako Co (Osaka, Japan). N-t-butoxycarbonyl-phenylalanine (Boc-Phe) was provided by GL Biochem Co, Ltd (Shanghai, People’s Republic of China), and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DiR) was supplied by AAT Bioquest, Inc (Sunnyvale, CA, USA), respectively. Acetonitrile and methanol were both of HPLC grade and were purchased from Oceanpak Alexative Chemical (Gothenburg, Sweden). All other chemicals were reagent grade and were used as received. Purified water used throughout the study was provided by Wahaha Group Co, Ltd, (Hangzhou, People’s Republic of China).
Cells and animals
A549 human lung cells were purchased from the Cell Bank of the Chinese Academy of Science (Shanghai, People’s Republic of China). The cells were maintained at 37°C in a humidified incubator containing 5% CO2, were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) cell culture medium supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich Co), and were split every 2 days. All experiments were performed on cells in the logarithmic phase of growth. No ethics statement was required from the institutional review board for the use of this cell line.
Male Sprague Dawley rats weighing 200–250 g were purchased from the Laboratory Animal Services Center of Luye Pharma Group Ltd (Yantai, People’s Republic of China). Female Balb/c nude mice (about 6 weeks of age) were supplied by Beijing Weitong Lihua Laboratory Animal Co, Ltd (Beijing, People’s Republic of China). All animals used were exposed to a 12-hour light/dark cycle in a specific pathogen-free environment and received food and water ad libitum throughout the studies. Animals were allowed to acclimate for at least 1 week in their respective animal facilities before experimentation. All procedures involving animals and their care gained ethics approval from the appropriate Institutional Animal Care and Use Committee before initiation.
Synthesis of mPEG-PCL-Phe(Boc)
Synthesis of the amphiphilic block copolymer mPEG-PCL-Phe(Boc) involved preparation of mPEG-PCL and following modification of terminal hydroxyl groups with Boc-Phe. By ring-open polymerization of ε-CL, mPEG-PCL copolymer was synthesized as reported previously.29,30 Briefly, anhydrous mPEG (10 g), ε-CL (12 g), and Sn(Oct)2 (12 mg) were added into a dried round-bottomed flask under vacuum and stirred at 130°C for 24 hours. Then, the product was dissolved in dichloromethane, followed by precipitation in chilled ether. We filtered the solvent by vacuum filtration through a Buchner funnel containing a qualitative filter paper (3 μm pore size; Fushun Civil Filter Paper Factory, Fushun, People’s Republic of China), the precipitate mPEG-PCL was collected, and the precipitate was then dried in vacuo at room temperature for terminal modification.
In a round-bottomed flask, Boc-Phe (6.65 g, 25 mmol) and triethylamine (3.5 mL, 25 mmol) were dissolved in 50 mL ethyl acetate, and were chilled to −10°C. Then, pivaloyl chloride (3.05 mL, 25 mmol) was added, and the mixture was stirred at room temperature for 2 hours. After evaporating the solvent, mPEG-PCL (10 g), triethylamine (3.5 mL, 25 mmol), and 4-pyrrolidinopyridine (370.5 mg, 2.5 mmol) were added successively, and then were dissolved in chilled dichloromethane (50 mL). The mixture was stirred at 0°C for 2 hours followed by a 24-hour stirring regime at room temperature. After evaporating the solvent, the residue was re-crystallized from ethanol three times to obtain the title copolymer mPEG-PCL-Phe(Boc).
Characterization of mPEG-PCL-Phe(Boc)
Proton nuclear magnetic resonance (1H-NMR) spectroscopy was conducted for chemical structure analysis with an Avance™ 400 spectrometer (Bruker Optik GmbH, Ettlingen, Germany) operating at 400 MHz and 25°C. The copolymer was dissolved in deuterated chloroform (CDCl3) at a concentration of 20 mg/mL. Chemical shifts (δ) were given in parts per million (ppm) using tetra-methylsilane (TMS) as an internal reference.
Relative molecular weights (MN, MW for number-average and weight-average, respectively) and molecular weight distribution (polydispersity index [PDI], MW/MN) were determined by a gel permeation chromatography (GPC) system equipped with a Waters 1515 isocratic HPLC pump, a Waters 2414 refractive index detector, and a set of columns (Waters Styragel® HR3, HR4, and HR5, 7.8 mm ×300 mm). GPC measurements were carried out at 25°C using tetrahydrofuran as an eluent at a flow rate of 1 mL/min. Monodisperse polystyrene standard samples were used for calibration.
The critical micelle concentration (CMC) value was determined by fluorescence spectroscopy (RF-5301; Shimadzu Co, Kyoto, Japan) using pyrene as a fluorescence probe, as described previously.31,32 In brief, 100 μL of pyrene solution (0.1 mg/mL in benzene) was aliquoted into a series of 10 mL test tubes, and the solvent benzene was evaporated under nitrogen stream at room temperature for 5 hours. A series of aqueous polymeric solutions (concentrations ranging from 10 ng/mL to 1 mg/mL) were added separately to obtain the final concentration of pyrene of 1 μg/mL, and then were sonicated for 20 minutes at room temperature, and finally were incubated at 45°C for 4 hours. After keeping all the samples at 25°C overnight, they were subjected to the assay of fluorescence intensity at an excitation wavelength of 343 nm, using a scanning speed of 250 nm/min, and an excitation and emission slit of 5.0 nm and 2.5 nm, respectively. The curve of fluorescence intensity ratio (I384/I373) versus logarithmic concentration (logC) (C in mg/mL) was plotted to calculate the CMC.
In vivo optical imaging was performed to investigate the tissue-targeting potential of mPEG-PCL-Phe(Boc) using A549 tumor-bearing nude mice as the model animal. DiR was used as a probe after encapsulating into PMs according to methods reported previously.33,34 Briefly, female Balb/c nude mice were implanted with 1×107 A549 human lung cancer cells by subcutaneous injection in the right breast fat pad. When tumors were about 200 mm3 in size, mice were administered a single intravenous dose (0.25 mg/kg) of DiR solution in ethanol or its micellar solution diluted in 0.9% sterile saline, respectively. At various time points after dosing (0.167, 1, 2, 4, 6, and 8 hours), the mice were anesthetized (5% chloral hydrate), and fluorescence imaging pictures were individually recorded using an in vivo imaging system (Fx Pro; Onex Corporation, Toronto, ON, Canada) at an excitation wavelength of 720 nm and an emission wavelength of 790 nm. At 8 hours post-injection, all the mice were sacrificed by cervical dislocation; tumors and some major organs (heart, liver, spleen, lung, kidney, ovary, and uterus) were harvested immediately, twice washed with ice-cold phosphate-buffered saline (PBS), and were then subjected to fluorescence imaging analysis under the same conditions as described in this section.
Preparation of PMs
A DMC-loaded PM (DMC-PM) system was prepared using a thin-film hydration method.35,36 Briefly, DMC (30 mg) and mPEG-PCL-Phe(Boc) (270 mg) were dissolved in 5.0 mL of absolute ethanol. After 5 minutes of stirring, the organic solvent was completely evaporated on a rotary evaporator under reduced pressure at 40°C to obtain a transparent, thin film. Then, the resulting film was dissolved by the addition of purified water at 50°C to obtain a transparent DMC-incorporated micellar solution. The solution was filtered through a 0.22 μm filter, followed by lyophilization in ampoule bottles. Blank micelles were also prepared in the same way, but without the addition of DMC. All the freeze-dried micelles were stored at 4°C until use.
Characterization of PMs
Morphological characterization and particle sizing
The morphology of the DMC-PM system was examined by transmission electron microscopy (TEM). The prepared micellar solution was sonicated for 0.5 hours to allow micelles to distribute uniformly. One drop of the micellar solution (1 mg/mL) was deposited on a carbon-coated copper grid and was stained with 1.0% (w/w) phosphotungstic acid (PTA), was air-dried, and was then observed at 100 kV accelerating voltage using a JEM-1400 TEM (JEOL, Tokyo, Japan). The mean diameter and particle size distribution were measured by dynamic light scattering (DLS) method (Delsa™ Nano C particle analyzer; Beckman Coulter Inc, Brea, CA, USA) at 25°C with a scattering angle of 90°. For this analysis, DMC-PM solution (1 mg/mL) was also sonicated for 0.5 hours to distribute PMs uniformly. The PDI was calculated to evaluate the size distribution. All measurements were performed in triplicate, and the results were expressed as means ± standard deviations (SD).
Drug loading and entrapment efficiency
Drug loading (DL) and entrapment efficiency (EE) of the DMC-PM system were determined by the HPLC-ultraviolet (HPLC-UV) method. Briefly, accurately measured 1 mL aqueous micellar solution containing about 1 mg DMC/mL was centrifuged at 8,000 rpm for 10 minutes to distinguish between encapsulated and free DMC. Free DMC was separated from the micelles under high speed centrifugal force and formed into precipitates due to its poor water solubility. Then, the supernatant (encapsulated DMC) was diluted to 100-fold with methanol to disrupt the micelles. All of the samples were injected into an Agilent 1100 HPLC system (Agilent Technologies, Santa Clara, CA, USA) equipped with a G1312A binary pump, G1322A online degasser, G1313A autosampler, a G1314A variable wavelength detector (VWD), and a G1316A thermostatted column compartment. Measurements were carried out on a Luna column (4.6×250 mm, C18; Torrance Technology, Torrance, CA, USA) at 30°C, and the detection wavelength was set at 420 nm. The mobile phase was composed of acetonitrile and 0.2% aqueous acetic acid (55/45, v/v), and it was delivered at a flow rate of 1 mL/min. The injection volume was 20 μL, and the run time was 15 minutes. The linear range for the concentration of DMC was found to be 1–50 μg/mL (y=0.6755x, R2=0.99). The DL and EE were calculated according to Equations 1 and 2, all of the results were the mean of three test runs, and results were expressed as the mean ± SD:
| (1) |
| (2) |
where the amount of DMC in micelles refers to the quantity of DMC that was encapsulated into the micelles; the amount of micelles refers to the total amount of DMC and copolymer, and the initial amount of DMC means the theoretical amount of DMC used for the preparation of micelles.
Differential scanning calorimetry
The thermodynamic characteristics of free DMC, blank mPEG-PCL-Phe(Boc) PMs, and the DMC-PM system were detected by differential scanning calorimetry (DSC) (DMA Q800; TA Instruments, New Castle, DE, USA). Samples were loaded into aluminum pans with a perforated lid for scanning. The temperature was raised from 20°C–200°C under a dynamic nitrogen atmosphere at a heating rate of 10°C/min and with a temperature modulation of 1°C/30 seconds. The DSC curves were recorded.
In vitro stability
Lyophilized DMC-PMs in ampoule bottles was dissolved in purified water (2 mg DMC/mL), and the particle size and concentration of DMC in the solutions were immediately detected by DLS and HPLC, respectively. Then, the micelle solutions were stored at room temperature and in 4°C conditions. When DMC was released from the micelles, precipitation occurred, and turbidity was observed due to low solubility, and the content of DMC in micelles decreased. In addition, particle size of micelles also changed when the solutions were instable. Solutions were monitored periodically for any changes in appearance, particle size, and drug content by visual observation, DLS, and HPLC, respectively. Chemical stability was evaluated by measuring the degradation of DMC. All of the samples were centrifuged (8,000 rpm ×10 minutes) to remove the precipitate, and the particle size and concentration of DMC in the solutions were determined. The concentration and stability of DMC in the supernatant were determined by HPLC methods, as described in the Drug loading and entrapment efficiency section. All measurements were conducted in triplicate, and the results were expressed as the mean ± SD.
In vitro drug release
Using PBS solution (pH 7.4) containing 0.5% Tween-80 as a release medium, the in vitro drug release assay of DMC-PMs was carried out by using dialysis tubing, as previously reported.37,38 In brief, 2 mL of PBS solution containing DMC-PMs with a DMC concentration of 2 mg/mL was sealed in a dialysis bag (Spectra/Por® molecular weight cut-off [MWCO]=6,000–8,000 Da), was immersed into 40 mL of release medium, and then was shaken gently at 70 rpm using a digital water bath oscillator (model HZS-HA; Harbin Dongming Co, Harbin, Heilongjiang, People’s Republic of China). An aliquot (1 mL) of the medium out of the dialysis membrane was withdrawn at various time intervals, and the same volume of fresh release medium was added to maintain a sink condition.39 The amount of released DMC in the incubation medium was quantified by HPLC as described in Drug loading and entrapment efficiency section. The release of DMC from micelles was calculated according to methods described by Zheng et al.40 The results were the average of triplicate measurements and were expressed as mean ± SD.
Pharmacokinetic study in rats
After fasting overnight with water ad libitum, 12 male Sprague Dawley rats were randomly divided into two groups (n=6/group) and were administered a single intravenous dose (10 mg DMC/kg of body weight) of DMC-PMs diluted, as appropriate, in 0.9% saline or free DMC dissolved in DMSO (sb-DMC), respectively. At various time points after dosing, serial bleeds were collected from the jugular vein cannulas and were separated by centrifuging (8,000 rpm ×10 minutes) to obtain plasma for analysis of DMC plasma concentration.
Biodistribution study in tumor-bearing nude mice
Female Balb/c nude mice were inoculated subcutaneously with A549 human lung cancer cells to obtain a tumor-bearing model animal, according to the method described in the Characterization of mPEG-PCL-Phe(Boc) section. When tumors were about 200 mm3 in size, the mice were random-ized (n=4/group) such that the mean and median tumor sizes were similar between groups. Mice were then administered a single intravenous dose (50 mg DMC/kg of body weight) of DMC-PMs diluted, as appropriate, in 0.9% saline or sb-DMC in DMSO. At 0.25, 0.5, 1, 2, 4, and 8 hours after drug administration, four mice were sacrificed. Samples of tumor, heart, liver, spleen, lung, kidneys, and brain were harvested immediately, thoroughly washed with ice-cold 0.9% saline, and were then stored at −0°C until analyzed for DMC.
Analysis of DMC in plasma and tissue
Plasma and tissue concentrations of DMC were determined by reverse-phase HPLC-UV. Briefly, 100 μL of plasma sample was mixed with 10 μL of bis-demethoxycurcumin (IS, 100 μg/mL), 50 μL of hydrochloric acid (HCl, 1 mol/L), and 3 mL of ethyl acetate. The mixtures were vortexed, ultrasonic extracted, and were then centrifuged at 8,000 rpm for 10 minutes. After that, the organic layer was transferred to a clean Eppendorf tube and was evaporated to dryness under pressure by using a gas-blowing concentrator (model HGC-24A; Tianjin Hengao Science and Technology Development Co., Ltd., Tianjin, People’s Republic of China) at 40°C. The extraction residue was reconstituted in 100 μL of mobile phase, and 20 μL aliquots were injected into the HPLC system. All tissues were homogenized with 5% glucose solution in appropriate concentration (heart, spleen, lung and brain 1:25; kidney 1:6; liver and tumor 1:2) using a homogenizer (model F8; Fluko, Shanghai, People’s Republic of China); then, 1 mL aliquots of tissue homogenate were extracted for HPLC analysis.
The HPLC analysis using an Agilent 1100 system was carried out on a Luna column (4.6×250 mm, C18; Torrance Technology) under the same conditions as described in the Drug loading and entrapment efficiency section. According to the US Food and Drug Administration (FDA) guidelines for the validation of bioanalytical methods, the method was fully validated in terms of selectivity, linearity, accuracy, precision, stability, and recovery.41 The results are summarized in Table 1, indicating that this assay could be applied to the quantification of DMC in plasma and tissues.
Table 1.
The method used to validate results of HPLC quantification of dimethoxycurcumin (DMC) in plasma and tissue samples
| Samples | Linearity (0.05–50 μg/mL) | Precision (%)
|
Accuracy (%) | Recovery (%) | |
|---|---|---|---|---|---|
| Intra-day | Inter-day | ||||
| Plasma | y=0.5020x, R2=0.99 | 4.89 | 6.24 | 100.6 | 88.75 |
| Heart | y=0.3407x, R2=0.99 | 6.84 | 7.13 | 101.4 | 90.32 |
| Liver | y=0.6916x, R2=0.99 | 5.48 | 7.31 | 98.78 | 88.62 |
| Spleen | y=0.2601x, R2=0.99 | 6.91 | 7.22 | 99.56 | 90.72 |
| Lung | y=0.2614x, R2=0.99 | 5.33 | 6.71 | 100.4 | 87.56 |
| Kidney | y=0.7298x, R2=0.99 | 5.92 | 6.88 | 99.78 | 79.45 |
| Brain | y=0.2829x, R2=0.99 | 6.13 | 7.41 | 100.5 | 80.73 |
| Tumor | y=0.4827x, R2=0.99 | 5.96 | 7.26 | 101.3 | 93.41 |
Notes: The concentrations of DMC in plasma and tissue samples were determined by reverse-phase HPLC with UV detection. Plasma or tissue samples were mixed with 10 μL of bis-demethoxycurcumin (100 μg/mL), 50 μL of HCl (1 mol/L) and 3 mL of ethyl acetate. The mixtures were vortexed, ultrasonic extracted, and were then centrifuged at 8,000 rpm for 10 minutes. After that, the organic layer was transferred to a clean Eppendorf tube and was evaporated to dryness under pressure by using a gas-blowing concentrator at 40°C. The extraction residue was reconstituted in 100 μL of mobile phase, and 20 μL aliquots were injected into the HPLC system. According to the US FDA guidelines for the validation of bioanalytical methods, the method was fully validated in terms of selectivity, linearity, accuracy, precision, stability, and recovery.41
Abbreviations: HPLC, high-performance liquid chromatography; UV, ultraviolet; US FDA, US Food and Drug Administration.
Statistical analyses
The plasma/tissue concentration–time data were analyzed with a non-compartmental model by DAS 2.1 software (Mathematical Pharmacology Professional Committee of China, Shanghai, People’s Republic of China) to obtain the pharmacokinetic parameters. The area under the curve (AUC) was calculated by the linear trapezoidal rule; other pharmacokinetic parameters were calculated by standard methods.
Statistics were analyzed by SPSS software for Windows (version 17.0; SPSS, Chicago, IL, USA). One-way analysis of variance (ANOVA) and two-tailed Student’s t-tests were performed for statistical evaluation. A P-value <0.05 was considered statistically significant.
Results and discussion
Synthesis and characterization of mPEG-PCL-Phe(Boc)
In the current research, we synthesized mPEG-PCL-Phe(Boc) diblock copolymer for the encapsulation of DMC to enhance its solubility and stability. The synthetic scheme is briefly summarized in Figure 2. The first step was the ring-opening polymerization of mPEG-PCL using Sn(Oct)2 as a catalyst and mPEG as a macroinitiator. In the second step, the mixed anhydride reacted with hydroxyl-terminated mPEG-PCL to be capped by Boc-Phe. The yield of the mPEG-PCL-Phe(Boc) was about 95%, the synthetic diblock copolymer was shown to be biocompatible and safe, and the materials used are all acceptable in medical practice.
Figure 2.

Synthesis of mPEG-PCL-Phe(Boc) block copolymer.
Abbreviations: mPEG, monomethoxyl poly (ethylene glycol); PCL, b-poly (ε-caprolactone); Phe, phenylalanine; Boc, N-t-butoxycarbonyl; mPEG-PCL-Phe(Boc), N-t-butoxycarbonyl-phenylalanine terminated monomethoxyl poly (ethylene glycol)-b-poly (ε-caprolactone).
As illustrated in Figure 3, 1H-NMR spectra were used to analyze the chemical structures of the copolymers. The 1H-NMR spectrum of mPEG-PCL copolymer is shown in Figure 3B. The presence of methene (CH2) protons in mPEG was observed at 3.67 ppm (wavelength peak a), and the chemical shifts of methene (CH2) protons in PCL were found at 1.40 ppm (peak b), 1.66 ppm (peak c), 2.31 ppm (peak d), and 4.08 ppm (peak e). Compared to mPEG-PCL, new peaks at 1.44 ppm (peak Boc), 3.22 ppm (peak f), and 7.15 ppm (peak g) appeared due to the grafting reaction with Boc-Phe, while the other peaks of mPEG and PCL remained constant (Figure 3A), indicating that the desired copolymer was formed and that no other reaction had occurred.
Figure 3.

1H-NMR spectra of mPEG-PCL-Phe(Boc) (A), and mPEG-PCL (B).
Abbreviations: 1H-NMR, proton nuclear magnetic resonance; mPEG, monomethoxyl poly (ethylene glycol); PCL, b-poly (ε-caprolactone); Phe, phenylalanine; Boc, N-t-butoxycarbonyl; mPEG-PCL-Phe(Boc), N-t-butoxycarbonyl-phenylalanine terminated monomethoxyl poly (ethylene glycol)-b-poly (ε-caprolactone).
The molecular weight (MN and MW) results from GPC showed that the end-capping treatment led to a slight increase compared to mPEG-PCL, while the PDI value was almost the same (Table 2). The MNs from the GPC results using a polystyrene calibration curve were corrected by a factor of 0.57, which has been reported previously.42 Corrected GPC results showed that the molecular weight of the mPEG-PCL copolymer was 3,654 (2,000 for mPEG and 1,654 for PCL). After the grafting reaction, the molecular weight of mPEG-PCL-Phe(Boc) increased to 3,826 (2,000 for mPEG and 1,826 for hydrophobic part), and the increased value corresponded to the molecular weight of Boc-Phe. The PDI results from GPC showed that there was no obvious change before and after the end-capping treatment, and the low PDI values indicated a narrow molecular weight range for the copolymers.
Table 2.
Molecular weight characterization of mPEG-PCL and mPEG-PCL-Phe(Boc) by GPC
| Copolymer | MNa | MWa | PDIa | MNb |
|---|---|---|---|---|
| mPEG-PCL | 6,411 | 6,868 | 1.071 | 3,654 |
| mPEG-PCL-Phe(Boc) | 6,712 | 7,143 | 1.064 | 3,826 |
Notes: Relative molecular weights (MN, MW) and molecular weight distribution (PDI, MW/MN) were determined by a GPC system. The number-average molecular weights (MNs) from the GPC results were derived using a polystyrene calibration curve and were corrected by a factor of 0.57.
Obtained from GPC based on the polystyrene calibration.
Corrected GPC results, MNa ×0.57.
Abbreviations: mPEG-PCL, monomethoxyl poly (ethylene glycol)-b-poly (ε-caprolactone); mPEG-PCL-Phe(Boc), N-t-butoxycarbonyl-phenylalanine terminated monomethoxyl poly (ethylene glycol)-b-poly (ε-caprolactone); GPC, gel permeation chromatography; MN, number-average molecular weight; MW, weight-average molecular weight; PDI, polydispersity index.
The CMC value of mPEG-PCL-Phe(Boc) copolymer was detected by using the fluorescence spectroscopy method and by employing pyrene as the probe. The fluorescence intensity could not be detected at the concentration below CMC value, whereas higher polymer concentration wrapped pyrene to produce a strong fluorescence intensity. The fluorescence intensity was collected at wavelengths across the emission spectrum (Figure 4A). The fluorescence intensity ratio of I384/I373 versus logC was plotted in Figure 4B, where the cross-point of the two tangents corresponded to the CMC value. From these results, we calculated the CMC of the copolymer was 2.56×10−3 mg/mL, which is fairly low compared to low molecular weight surfactants.43 The fraction ratio of hydrophobic part and hydrophilic part of the copolymer was similar in the micelles, which contributed to the low CMC value.44 Low CMC value ensured the preservation of micelle stability and structural integrity under high dilution conditions that could be encountered in vivo. Low CMC value could also reduce drug degradation, thereby reducing metabolism to improve bioavailability in vivo.45,46
Figure 4.
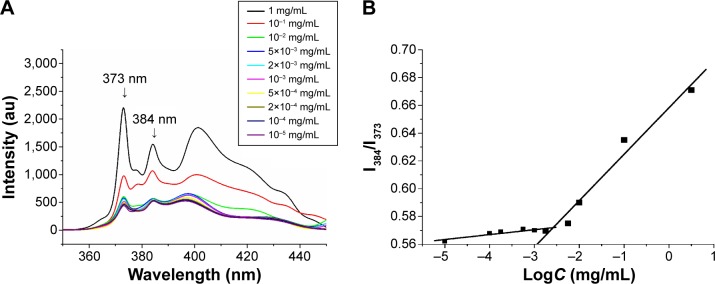
The fluorescence emission spectra of pyrene as a function of copolymer concentration in water (mg/mL) (A). The fluorescence intensity ratio of I384/I373 from pyrene emission spectra versus the log of the concentration (logC; mg/mL), a measurement used to determine the CMC for mPEG-PCL-Phe(Boc) (B).
Abbreviations: CMC, critical micelle concentration; mPEG, monomethoxyl poly (ethylene glycol); PCL, b-poly (ε-caprolactone); Phe, phenylalanine; Boc, N-t-butoxycarbonyl; mPEG-PCL-Phe(Boc), N-t-butoxycarbonyl-phenylalanine terminated monomethoxyl poly (ethylene glycol)-b-poly (ε-caprolactone).
The in vivo fluorescence images of tumor-bearing nude mice after receiving DiR solution and DiR-loaded micelles are displayed in Figure 5A. Compared to the DiR solution, the DiR-loaded micelles quickly penetrated the whole body within 10 minutes; the highest signal of free DiR solution appeared at 4 hours after intravenous injection, indicating circulation of the micelles in the blood stream.47 Micelle formulations penetrated most mouse tissues; free DiR signal was mostly focused in the liver tissue region, and there was no signal observed in the tumor region in the free DiR group throughout the entire monitoring period. Although the whole body fluorescence of the two formulations progressively decreased over time, the free DiR signal decreased more rapidly. Accordingly, it was thought that mPEG-PCL-Phe(Boc) micelles might facilitate the delivery of antitumor drugs into the tumor, with long-term retention in the tumor, which would enhance the antitumor effect. The excised tissues fluorescence images at 8 hours are illustrated in Figure 5B. Strong fluorescence signals were detected in the liver, lung, and tumor tissues of the mice injected with DiR-loaded micelles, suggesting a selective accumulation of the amphiphilic copolymer at the target sites. Free DiR was mainly observed in liver, spleen, and lung tissues, with a higher fluorescence intensity compared to the micelle formulation at 8 hours, while there was no DiR signal observed in the tumor tissue of the DiR solution (Figure 5B). As the distribution of free DiR was limited, the signal was a little higher in metabolic regions (liver, spleen, and lung tissues), and more DiR accumulated in organs related to RES, namely the liver and spleen. Due to the increased water solubility after encapsulation, DiR-loaded micelles could rapidly deliver the model drug into blood circulation, and display wide distribution and controlled release characteristics. The difference of fluorescence signals intensity in tumor between the two groups further indicated an obvious passive drug targeting effect to tumor of the micelles. Thus the present amphiphilic copolymer mPEG-PCL-Phe(Boc) has the potential to endow some hydrophobic active pharmaceutical ingredients with satisfactory pharmacokinetic profiles, mainly including long circulation, wide distribution, and tumor targeting.
Figure 5.

The time-dependent in vivo fluorescence images of DiR solution and DiR-loaded micelles in A549 tumor-bearing female nude mice after tail vein injection (A). The location of tumors is illustrated by black arrows. In vitro fluorescence images of excised organs and tumor tissues of nude mice at 8 h after injection of DiR solution and DiR-loaded micelle formulations (B).
Abbreviations: h, hours; min, minutes; DiR, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide.
Preparation and characterization of DMC-loaded PMs
PMs were easily prepared to wrap DMC by thin-film hydration method. After evaporating the dissolved organic solvent, DMC was encapsulated by the hydrophobic part of the copolymer by using a thermodynamic stability feature in water. The practical preparation process was simple, controllable, and it would be easy to scale up preparation to realize both lab and industrial production of PMs.48
Morphological characterization and particle sizing
As the TEM image shows in Figure 6A, morphology of DMC-PMs was found to be spherical or nearly spherical, with an obviously dark outer shell and a light inner core. The dark layers were formed by the reaction of the hydrophilic shell and PTA. The shell–core structure could contribute to the long circulation in blood because of the hydrophilic barriers and consequent low drug leakage.49 The distribution of micelles was homogeneous, with no adhesion and no reunion phenomenon. The particle size and size distribution results detected by DLS are shown in Figure 6B. As shown, the average particle size of DMC-PMs was 17.9±0.4 nm, and the PDI was 0.045±0.011, which means a narrow particle size distribution. Such small particle size and narrow size distributions are suitable for many drug delivery systems (such as intravenous injection and oral administration), and allow passive accumulation in tumors through the enhanced permeability and retention (EPR) effect; such characteristics also curtail arrest by RES and MPS.
Figure 6.
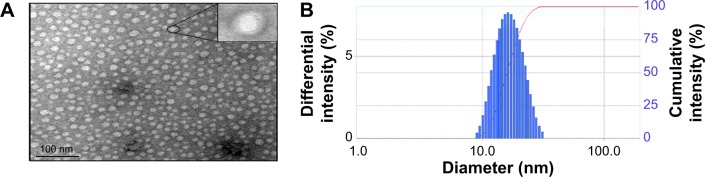
TEM image (×100,000) (A), and the particle size distribution (B) of DMC-PM.
Note: The inset of (A) clearly illustrated a typical shell-core structure of the micelle particles.
Abbreviations: TEM, transmission electron microscopy; DMC-PM, dimethoxycurcumin-loaded polymeric micelle.
Drug loading and entrapment efficiency
DL and EE results for DMC-PMs were 9.94%±0.15% and 97.22%±0.18%, respectively. The final DL and EE were neared to the theoretical loading, which indicated low drug wastage. High DL and EE were mostly due to hydrophobic interaction between DMC and the hydrophobic segment of the amphiphilic block polymer. These results further proved that the amphiphilic block polymer increased the solubility of DMC and decreased the usage amount of copolymers. Thus, increased solubility of DMC could ensure the degree by which a human body can use the drug and may ensure a better clinical therapeutic effect. Low amount of copolymers is safe to all biological organisms, although the copolymers themselves have a good biocompatibility.
Differential scanning calorimetry
The thermograms of free DMC, blank mPEG-PCL-Phe(Boc) micelles, and DMC-PMs were detected by DSC. As illustrated in Figure 7, the melting point of sb-DMC was about 130°C, and the melting point of blank mPEG-PCL-Phe(Boc) micelles was about 43°C in the DSC curve. There was only one melting point in the curve for DMC-PMs, and the intrinsic melting transition peak of DMC vanished in DMC-PMs, which indicated that DMC had been thoroughly encapsulated by the copolymers, rather than adhering to the surface of them. This result corresponded to the high DLC and EE values exhibited by the PMs. The solubility of DMC was greatly increased by encapsulating into the PM core, which could meet the needs for clinical trials and further evaluation.
Figure 7.
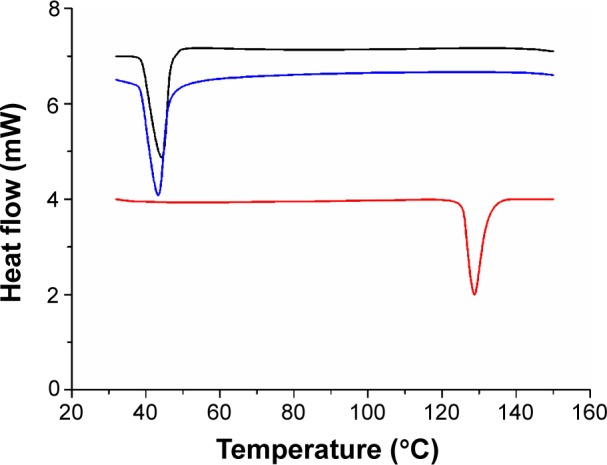
The DSC curves of free DMC (in red), blank mPEG-PCL-Phe(Boc) micelles (in black), and DMC-PMs (in blue).
Abbreviations: DSC, differential scanning calorimetry; mPEG, monomethoxyl poly (ethylene glycol); PCL, b-poly (ε-caprolactone); Phe, phenylalanine; Boc, N-t-butoxycarbonyl; DMC-PMs, dimethoxycurcumin-loaded polymeric micelles; mPEG-PCL-Phe(Boc), N-t-butoxycarbonyl-phenylalanine terminated monomethoxyl poly (ethylene glycol)-b-poly (ε-caprolactone).
In vitro stability
Reconstituted DMC-PMs (2 mg/mL) were clarified and showed a high concentration of DMC (Figure 8). By observing turbidity of the reconstituted micellar solutions both at room temperature and at 4°C, micellar solutions could be kept clarified for 10 days at room temperature and 1 month at 4°C. In addition, after storing the micelle solutions at room temperature and at 4°C for more than 10 days, free DMC in its precipitated state remained stable, and no other impurity occurred, which was tested by HPLC. In room temperature, the mean particle size of the micelles increased from 17.6 nm to 34.7 nm slowly (Figure 9A). However, during the period of storage at 4°C, the mean particle size ranged from 17.4 nm to 19.5 nm and increased only slightly at a steady pace (Figure 9A). The drug content of the micellar solutions decreased slightly during the first day, and then stayed almost stable at both temperatures (Figure 9B). It is well known that the interaction between hydrophobic drugs and the micelle core environment is a very important factor in micelle stability.50 After reconstituting the lyophilized micelles in aqueous solutions in the present study, the hydrophobic segment of the micelles was strong enough to hold DMC within micelle cores for up to 10 days both at room temperature and at 4°C in vitro. The micelles’ excellent physicochemical stability in the external environment may indicate a long lifetime in vivo.
Figure 8.

The appearances of DMC-PMs before lyophilization (A), lyophilized powder (B), and reconstituted solution (C).
Abbreviation: DMC-PMs, dimethoxycurcumin-loaded polymeric micelles.
Figure 9.
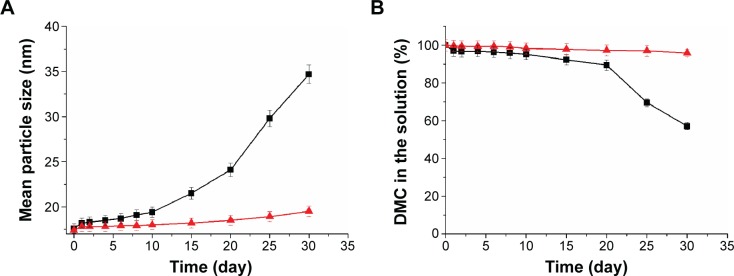
Mean particle size (A) and dimethoxycurcumin (DMC) content (B) of DMC-PMs stored at room temperature (■) and at 4°C (▲).
Note: Each point represents mean ± standard deviation (n=3).
Abbreviation: DMC-PMs, dimethoxycurcumin-loaded polymeric micelles.
In vitro drug release
The behavior of DMC released from micelles under in vitro conditions was determined by using the dialysis tubing method.37,38 The cumulative release of DMC versus time is plotted in Figure 10. The results showed that DMC-PMs displayed a sustained release behavior for up to 10 days. The drug’s rapid release during the first 24 hours was about 21.8%, but slow release was maintained in the following 9 days. The initial burst release could be explained by drug desorption from the copolymers because of the osmotic pressure difference inside and outside the copolymer.24,46,51 In clinical applications, this initial burst release may allow the drug to arrive quickly in organs and tumor sites to protect organs or to fight against cancer cells. In our present study, the degradation of DMC-PMs was slowed down by the decrease of osmotic pressure in the in vitro release medium, the drug release was thus retarded. The sustained release of DMC-PMs was likely due to the stronger interaction between DMC and the mPEG-PCL-Phe(Boc) copolymer. It is possible that DMC-PMs in vivo also display sustained release property similar to that in vitro. However, a series of follow-up animal experiments on pharmacokinetics and biodistribution may be needed to further demonstrate the actual situation in vivo for this polymeric micellar formulation of DMC.
Figure 10.
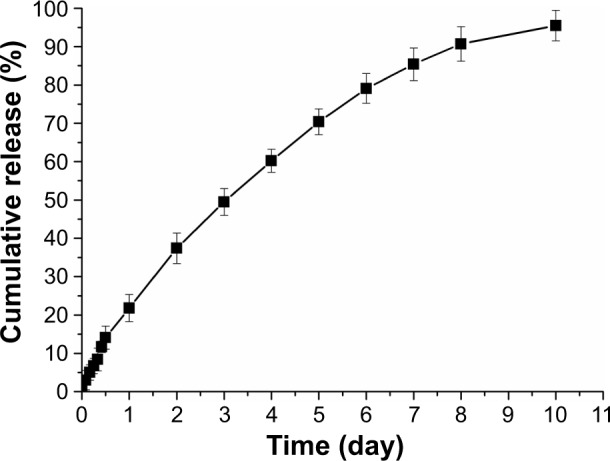
In vitro release profile of DMC-PMs.
Note: Each point represents mean ± standard deviation (n=3).
Abbreviation: DMC-PMs, dimethoxycurcumin-loaded polymeric micelles.
Pharmacokinetic study in rats
The pharmacokinetic characteristics of sb-DMC and DMC-PMs in rats are shown in Figure 11. Plasma DMC levels in the sb-DMC control group rapidly declined to below the lowest limit of quantification (LLOQ) (40 ng/mL) after 10 hours. Different results were observed in the DMC-PM group. Plasma DMC levels declined obviously in the first 0.33 hours, while a slow decrease was observed subsequently. In addition, plasma concentrations in the DMC-PM group were obviously higher than those in the control group at each time point. Even at 24 hours after dosing, plasma concentrations of DMC in the DMC-PM group were still above the LLOQ. Plasma pharmacokinetic parameters of rats including the maximum concentration (Cmax), plasma half-life (t1/2), area under the curve (AUC0–∞), mean residence time (MRT0–∞), clearance (Cl), and volume of distribution (V) were calculated, as listed in Table 3 and using a non-compartmental analysis model. As shown in Table 3, Cmax of DMC-PMs was significantly higher than that of the sb-DMC solution (P<0.05; Table 3), which was proportional to the amount of physically entrapped DMC present. DMC-PM group’s mean t1/2 value was much longer than that of the sb-DMC group (P<0.05; Table 3), which suggested that entrapping DMC into micelles could prolong in vivo residence time, thereby achieving a long-term therapeutic effect. The micelle group’s mean AUC0–∞ value was 3-fold higher than that of sb-DMC group, which indicated a higher DMC exposure yielded by the DMC-PM formulation, and an increase in the DMC systemic bioavailability in vivo. The Cl value of DMC-PMs was smaller than that of sb-DMC, but the MRT0–∞ of micelles was 3-fold that of the pure drug, which indicated that entrapping DMC into micelles could delay drug elimination, and that the time DMC stayed in blood was longer with DMC-PMs. In addition, the DMC-loaded micelle group’s V value was 1-fold higher than that of the sb-DMC group, which meant a much higher drug distribution in the tissue. The DMC-PM formulation changed the pharmacokinetic behavior of DMC in rats, which could obviously prolong drug circulation and increase exposure to DMC in vivo. DMC-PMs showed some sustained drug release characteristics and showed an increased possibility of extensive drug distribution in rat tissues.
Figure 11.
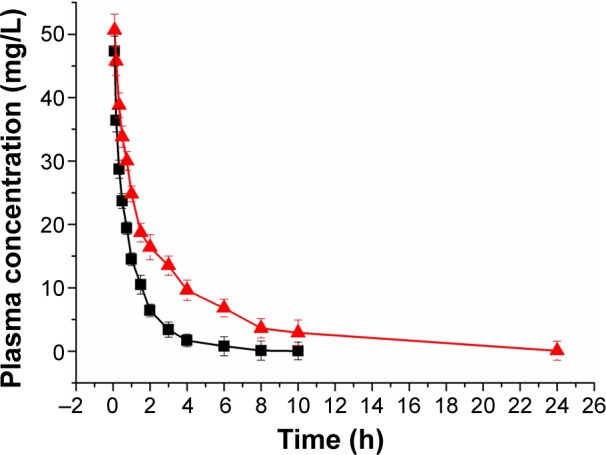
Plasma concentration–time curves after intravenous administration of DMC-PMs (▲) and sb-DMC (■) in Sprague Dawley rats at a single dosage of 10 mg/kg.
Note: Each point represents mean ± standard deviation (n=6).
Abbreviations: h, hours; DMC-PMs, dimethoxycurcumin-loaded polymeric micelles; sb-DMC, free DMC dissolved in dimethyl sulfoxide.
Table 3.
Pharmacokinetic parameters of dimethoxycurcumin (DMC) in plasma from rats intravenously administering sb-DMC or DMC-PMs at a dose of 10 mg/kg
| Parameter | sb-DMC | DMC-PMs |
|---|---|---|
| Cmax (mg·L−1) | 47.32±2.37 | 50.64±2.53* |
| t1/2 (h) | 0.92±0.03 | 2.81±0.34* |
| AUC0–∞ (mg·h·L−1) | 48.49±1.85 | 132.74±8.17* |
| MRT0–∞ (h) | 1.33±0.05 | 4.05±0.49* |
| V (L·kg−1) | 0.27±0.02 | 0.31±0.04 |
| Cl (L·h−1·kg−1) | 0.21±0.01 | 0.08±0.01* |
Notes: Statistical analysis was performed using a one-way ANOVA followed by a Student’s t-test. Results are presented as mean ± standard deviation, n=6.
P<0.05 versus sb-DMC.
Abbreviations: h, hours; sb-DMC, free dimethoxycurcumin (DMC) dissolved in dimethyl sulfoxide; DMC-PMs, dimethoxycurcumin loaded polymeric micelles; Cmax, the maximum concentration; t1/2, plasma half-life; AUC0–∞, area under the curve; MRT0–∞, mean residence time; Cl, clearance; V, volume of distribution; ANOVA, analysis of variance.
Biodistribution study in tumor-bearing nude mice
Biodistribution studies were performed in tumor-bearing nude mice to investigate the amount of drug that reached vital organs and tumors. A549 human lung cancer cells were implanted into the right breast fat pad of nude mice; the content of DMC in the main organs (heart, liver, spleen, lung, kidneys, and brain) and tumors at 0.25, 0.5, 1, 2, 4, and 8 hours is shown in Tables 4 and 5, and the pharmacokinetic parameters of tested tissues are shown in Table 6. The experimental results showed that DMC in both formulations was rapidly distributed to most tissues within 15 minutes; in addition, the present study revealed more drug localization in most organs and tumors with the micelle formulation. The two formulations at the same intravenous dosage of DMC showed similar tissue distribution trends in tumor-bearing nude mice. However, the animals administered with DMC-PMs had much higher DMC content in tumor, heart and liver at each sample collection time point when compared with those administered with sb-DMC. This result indicated that the formulation of DMC-PMs has significantly improved tissue distribution characteristics of DMC. The Cmax and AUC0–∞ values yielded by DMC-PMs were significantly higher than those of sb-DMC in most tissues, except in brain tissue. The higher drug level and drug exposure yielded by DMC-PMs in different tissues may be explained on the basis of the nanoparticle-like nature of micelles, but in brain, micelles were stable enough to maintain a hydrophilic shell that kept the drug outside of the blood–brain barrier. The Cl values of DMC-PMs were smaller than those of sb-DMC in most tissues, except in brain, which indicated that entrapping DMC into micelles could delay drug elimination. The mean t1/2 value of the DMC-PM group was much longer than that of the sb-DMC group for heart, liver, kidney, and tumor tissues, but in the remaining tissues; in addition, the mean t1/2 value of the micelle group was lower than that of sb-DMC group, with no significant difference, which suggested that DMC content was decreasing rapidly in some tissues due its metabolism effects.52 The mean MRT0–∞ value for the micelle group was much higher than that of the sb-DMC group in tumor and heart tissues, but in other tissues, the mean MRT0–∞ value for the micelle group was a little lower than that of the sb-DMC group; these results suggested that entrapping DMC into micelles could prolong drug in vivo residence time in heart and in tumor tissues, thereby achieving an increase in DMC systemic bioavailability and a long-term therapeutic effect. Taken together, these results suggested that DMC content with micelles was higher in most tissues, especially in heart, liver, and tumor tissues, and that DMC elimination rate was significantly slower with the micelle formulation. Because the concentration of DMC in heart and liver tissues was very high in the present study, DMC-PMs could be used in future to cure heart or liver diseases, or to deliver otherwise organ-damaging drugs, to protect the heart and liver from harm. In vivo fluorescence images (Figure 5) indicated that the mPEG-PCL-Phe(Boc) copolymer itself had tumor-targeting ability. In the current biodistribution study, compared to free DMC formulation, DMC-loaded micelles were superior in tumor-targeting ability based on EPR effect. In summary, we believe that DMC-loaded micelles could be used as a tumor-targeting drug delivery system.
Table 4.
Tissue distribution of dimethoxycurcumin (DMC) in nude mice intravenously administered DMC-PMs at a dose of 50 mg/kg
| Time (h) | Heart (μg/g) | Liver (μg/g) | Spleen (μg/g) | Lung (μg/g) | Kidney (μg/g) | Brain (μg/g) | Tumor (μg/g) |
|---|---|---|---|---|---|---|---|
| 0.25 | 5.47±0.30 | 3.96±0.17 | 0.63±0.11 | 0.40±0.02 | 1.21±0.05 | 0.26±0.04 | 1.04±0.12 |
| 0.5 | 2.40±0.11 | 1.20±0.07 | 0.55±0.11 | 1.20±0.08 | 0.55±0.04 | 0.18±0.02 | 0.60±0.09 |
| 1 | 1.21±0.16 | 0.50±0.04 | 0.46±0.10 | 1.54±0.06 | 0.17±0.02 | 0.08±0.01 | 0.24±0.03 |
| 2 | 0.60±0.15 | 0.12±0.02 | 0.30±0.01 | 0.80±0.07 | 0.09±0.02 | 0.06±0.01 | 0.19±0.02 |
| 4 | 0.40±0.01 | 0.07±0.01 | 0.18±0.01 | 0.34±0.03 | 0.04±0.01 | 0.05±0.01 | 0.09±0.01 |
| 8 | 0.18±0.02 | 0.03±0.01 | 0.07±0.02 | 0.16±0.02 | 0.01±0.01 | 0.04±0.02 | 0.05±0.02 |
Note: Results are presented as mean ± standard deviation, n=4.
Abbreviations: h, hours; DMC-PMs, dimethoxycurcumin-loaded polymeric micelles.
Table 5.
Tissue distribution of dimethoxycurcumin (DMC) in nude mice intravenously administering sb-DMC at a dose of 50 mg/kg
| Time (h) | Heart (μg/g) | Liver (μg/g) | Spleen (μg/g) | Lung (μg/g) | Kidney (μg/g) | Brain (μg/g) | Tumor (μg/g) |
|---|---|---|---|---|---|---|---|
| 0.25 | 1.00±0.14 | 1.25±0.19 | 0.47±0.10 | 1.36±0.09 | 0.88±0.04 | 0.36±0.06 | 0.48±0.05 |
| 0.5 | 0.50±0.10 | 0.70±0.13 | 0.44±0.09 | 0.80±0.09 | 0.40±0.04 | 0.25±0.03 | 0.20±0.03 |
| 1 | 0.18±0.05 | 0.47±0.12 | 0.38±0.02 | 0.47±0.07 | 0.21±0.03 | 0.12±0.03 | 0.13±0.02 |
| 2 | 0.07±0.01 | 0.20±0.10 | 0.26±0.01 | 0.26±0.03 | 0.18±0.03 | 0.08±0.01 | 0.08±0.01 |
| 4 | 0.04±0.02 | 0.12±0.01 | 0.16±0.01 | 0.18±0.02 | 0.05±0.01 | 0.06±0.02 | 0.05±0.01 |
| 8 | 0.01±0.01 | 0.05±0.02 | 0.07±0.02 | 0.06±0.02 | 0.02±0.01 | 0.02±0.01 | 0.01±0.01 |
Note: Results are presented as mean ± standard deviation, n=4.
Abbreviations: h, hours; sb-DMC, free dimethoxycurcumin (DMC) dissolved in dimethyl sulfoxide.
Table 6.
Pharmacokinetic parameters of dimethoxycurcumin (DMC) in main tissues from nude mice intravenously administering DMC-PMs or sb-DMC at a dose of 50 mg/kg
| Tissue | t1/2 (h) | AUC0–∞ (μg·h·g−1) | MRT0–∞ (h) | Cl (g·h−1·g−1) |
|---|---|---|---|---|
| Heart | ||||
| DMC-PMs | 0.87±0.04* | 5.70±0.07* | 1.25±0.86* | 8.77±0.01* |
| sb-DMC | 0.20±0.01 | 0.89±0.01 | 0.29±0.02 | 56.18±0.31 |
| Liver | ||||
| DMC-PMs | 0.38±0.01* | 2.78±0.08* | 0.54±0.02* | 17.99±0.11* |
| sb-DMC | 0.91±0.04 | 1.50±0.04 | 1.32±0.05 | 33.37±0.11 |
| Spleen | ||||
| DMC-PMs | 1.31±0.06 | 1.69±0.03* | 1.89±0.09 | 29.59±0.11* |
| sb-DMC | 1.33±0.05 | 1.57±0.03 | 1.91±0.08 | 31.81±0.20 |
| Lung | ||||
| DMC-PMs | 1.19±0.04 | 3.18±0.01* | 1.71±0.06 | 15.72±0.13* |
| sb-DMC | 1.19±0.11 | 1.90±0.02 | 1.71±0.16 | 26.35±0.09 |
| Kidney | ||||
| DMC-PMs | 0.39±0.03* | 1.05±0.03* | 0.56±0.05* | 47.62±0.22* |
| sb-DMC | 0.50±0.02 | 0.76±0.03 | 0.73±0.03 | 65.60±0.22 |
| Brain | ||||
| DMC-PMs | 0.87±0.12 | 0.36±0.32 | 1.25±0.15 | 138.89±1.44 |
| sb-DMC | 1.77±0.71 | 0.64±0.13 | 2.55±1.02 | 79.94±0.35 |
| Tumor | ||||
| DMC-PMs | 1.94±1.25* | 1.09±0.20* | 2.80±2.80* | 45.87±0.31* |
| sb-DMC | 0.22±0.01 | 0.38±0.03 | 0.32±0.02 | 130.74±1.43 |
Notes: Statistical analysis was performed using a one-way ANOVA followed by a Student’s t-test. Results are presented as mean ± standard deviation, n=4.
P<0.05 versus sb-DMC.
Abbreviations: h, hours; DMC-PMs, dimethoxycurcumin loaded polymeric micelles; sb-DMC, free dimethoxycurcumin dissolved in dimethyl sulfoxide; t1/2, plasma half-life; AUC0–∞, area under the curve; MRT0–∞, mean residence time; Cl, clearance; ANOVA, analysis of variance.
Conclusion
In recent times, self-assembly nanoparticles, especially amphiphilic block copolymer micelles, have been receiving increasing attention because of their advantageous properties. In this study, we successfully encapsulated DMC using an mPEG-PCL-Phe(Boc) copolymer carrier to form micelles by a one-step thin-film hydration method. The amphiphilic copolymer itself had the ability of passively targeting tumors, and it was easy to form micelles at very low concentrations. DMC-PMs have typical shell–core structure with small particle sizes (17.9±0.4 nm) and a narrow particle size distribution (PDI, 0.045±0.011), which could realize long circulation and passive targeting goals. High EE and DL characteristics demonstrated that the copolymer had obviously increased the solubility of DMC in our current study, which could meet clinical use requirements. DMC-PMs could be maintained for up to 10 days at room temperature, and showed a sustained drug release characteristic both in vitro and in vivo. Moreover, the micelles also altered most tissue distribution characteristics of DMC in vivo, especially in tumor, liver and heart tissues, which revealed significant tumor targeting, liver and heart disease cure potential. In conclusion, DMC-loaded mPEG-PCL-Phe(Boc) micelles could be regarded as an effective drug delivery system to solve the problem of DMC administration.
Acknowledgments
This work was supported by the National Science and Technology Major Projects of China for Major New Drugs Innovation and Development (grant number 2014ZX09301306-006), the Natural Science Foundation of Shandong Province (grant number ZR2013HZ004), and the Youth Scholar Backbone Training Plan Project of Yantai University (2012–2016).
Footnotes
Disclosure
The authors report no conflicts of interest in relation to this work.
References
- 1.Newman DJ, Cragg GM. Natural products as sources of new drugs over the last 25 years. J Nat Prod. 2007;70(3):461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 2.Kuttan R, Bhanumathy P, Nirmala K, George MC. Potential anticancer activity of turmeric (Curcuma longa) Cancer Lett. 1985;29(2):197–202. doi: 10.1016/0304-3835(85)90159-4. [DOI] [PubMed] [Google Scholar]
- 3.Sharma RA, Gescher AJ, Steward WP. Curcumin: the story so far. Eur J Cancer. 2005;41(13):1955–1968. doi: 10.1016/j.ejca.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 4.Naksuriya O, Okonoqi S, Schiffelers RM, Hennink WE. Curcumin nanoformulations: a review of pharmaceutical properties and preclinical studies and clinical data related to cancer treatment. Biomaterials. 2014;35(10):3365–3383. doi: 10.1016/j.biomaterials.2013.12.090. [DOI] [PubMed] [Google Scholar]
- 5.Kamat AM, Tharakan ST, Sung B, Aggarwal BB. Curcumin potentiates the antitumor effects of Bacillus Calmette-Guerin against bladder cancer through the downregulation of NF-κB and upregulation of TRAIL receptors. Cancer Res. 2009;69(23):8958–8966. doi: 10.1158/0008-5472.CAN-09-2045. [DOI] [PubMed] [Google Scholar]
- 6.Yoon MJ, Kim EH, Lim JH, Kwon TK, Choi KS. Superoxide anion and proteasomal dysfunction contribute to curcumin-induced paraptosis of malignant breast cancer cells. Free Radic Biol Med. 2010;48(5):713–726. doi: 10.1016/j.freeradbiomed.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 7.Syng-Ai C, Kumari AL, Khar A. Effect of curcumin on normal and tumor cells: role of glutathione and bcl-2. Mol Cancer Ther. 2004;3(9):1101–1108. [PubMed] [Google Scholar]
- 8.Dhillon N, Aggarwal BB, Newman RA, et al. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin Cancer Res. 2008;14(14):4491–4499. doi: 10.1158/1078-0432.CCR-08-0024. [DOI] [PubMed] [Google Scholar]
- 9.Wang YJ, Pan MH, Cheng AL, et al. Stability of curcumin in buffer solutions and characterization of its degradation products. J Pharm Biomed Anal. 1997;15(12):1867–1876. doi: 10.1016/s0731-7085(96)02024-9. [DOI] [PubMed] [Google Scholar]
- 10.Pan MH, Huang TM, Lin JK. Biotransformation of curcumin through reduction and glucuronidation in mice. Drug Metab Dispos. 1999;27(4):486–494. [PubMed] [Google Scholar]
- 11.Asai A, Miyazawa T. Occurrence of orally administered curcuminoid as glucuronide and glucuronide/sulfate conjugates in rat plasma. Life Sci. 2000;67(23):2785–2793. doi: 10.1016/s0024-3205(00)00868-7. [DOI] [PubMed] [Google Scholar]
- 12.Sundaryono A, Nourmamode A, Gardrat C, et al. Studies on the photochemistry of 1,7-diphenyl-1,6-heptadiene-3,5-dione, a non-phenolic curcuminoid model. Photochem Photobiol Sci. 2003;2(9):914–920. doi: 10.1039/b301229h. [DOI] [PubMed] [Google Scholar]
- 13.Amolins MW, Peterson LB, Blagg BS. Synthesis and evaluation of electron-rich curcumin analogues. Bioorg Med Chem. 2009;17(1):360–367. doi: 10.1016/j.bmc.2008.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khanna S, Park HA, Sen CK, et al. Neuroprotective and antiinflammatory properties of a novel demethylated curcuminoid. Antioxid Redox Signal. 2009;11(3):449–468. doi: 10.1089/ars.2008.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krackov MH, Bellis HE, inventors Gene Print Inc, assignee. Process for the synthesis of curcumin-related compounds. 5679864. United States patent US. 1997 Oct 21;
- 16.Ohtsu H, Xiao Z, Ishida J, et al. Antitumor agents. 217. Curcumin analogues as novel androgen receptor antagonists with potential as anti-prostate cancer agents. J Med Chem. 2002;45(23):5037–5042. doi: 10.1021/jm020200g. [DOI] [PubMed] [Google Scholar]
- 17.Tamvakopoulos C, Dimas K, Sofianos ZD, et al. Metabolism and anticancer activity of the curcumin analogue, dimethoxycurcumin. Clin Cancer Res. 2007;15(13):1269–1277. doi: 10.1158/1078-0432.CCR-06-1839. [DOI] [PubMed] [Google Scholar]
- 18.Markatou E, Gionis V, Chryssikos GD, Hatziantoniou S, Georgopoulos A, Demetzos C. Molecular interactions between dimethoxycurcumin and Pamam dendrimer carriers. Int J Pharm. 2007;339(1–2):231–236. doi: 10.1016/j.ijpharm.2007.02.037. [DOI] [PubMed] [Google Scholar]
- 19.Hadjidemetriou M, Pippa N, Pispas S, Demetzos C. Incorporation of dimethoxycurcumin into charged liposomes and the formation kinetics of fractal aggregates of uncharged vectors. J Liposome Res. 2013;23(2):94–100. doi: 10.3109/08982104.2012.747534. [DOI] [PubMed] [Google Scholar]
- 20.Wicki A, Witzigmann D, Balasubramanian V, Huwyler J. Nanomedicine in cancer therapy: challenges, opportunities, and clinical applications. J Control Release. 2015;200:138–157. doi: 10.1016/j.jconrel.2014.12.030. [DOI] [PubMed] [Google Scholar]
- 21.Ren J, Fang Z, Yao L, et al. A micelle-like structure of poloxamer–methotrexate conjugates as nanocarrier for methotrexate delivery. Int J Pharm. 2015;487(1–2):177–186. doi: 10.1016/j.ijpharm.2015.04.014. [DOI] [PubMed] [Google Scholar]
- 22.Jindal N, Mehta SK. Nevirapine loaded poloxamer 407/pluronic P123 mixed micelles: optimization of formulation and in vitro evaluation. Colloids Surf B Biointerfaces. 2015;129:100–106. doi: 10.1016/j.colsurfb.2015.03.030. [DOI] [PubMed] [Google Scholar]
- 23.Torchilin VP. Micellar nanocarriers: pharmaceutical perspectives. Pharm Res. 2007;24(1):1–16. doi: 10.1007/s11095-006-9132-0. [DOI] [PubMed] [Google Scholar]
- 24.Qiu JF, Gao X, Wang BL, et al. Preparation and characterization of monomethoxy poly(ethylene glycol)-poly(ε-caprolactone) micelles for the solubilization and in vivo delivery of luteolin. Int J Nanomedicine. 2013;8:3061–3069. doi: 10.2147/IJN.S45062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Croy SR, Kwon GS. Polymeric micelles for drug delivery. Curr Pharm Des. 2006;12(36):4669–4684. doi: 10.2174/138161206779026245. [DOI] [PubMed] [Google Scholar]
- 26.Guo P, Song S, Li Z, et al. In vitro and in vivo evaluation of APRPG-modified angiogenic vessel targeting micelles for anticancer therapy. Int J Pharm. 2015;486(1–2):356–366. doi: 10.1016/j.ijpharm.2015.03.067. [DOI] [PubMed] [Google Scholar]
- 27.Cai L, Qiu N, Xiang M, et al. Improving aqueous solubility and antitumor effects by nanosized gambogic acid-mPEG2000 micelles. Int J Nanomedicine. 2014;9:243–255. doi: 10.2147/IJN.S54050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maiti S, Mukherjee S. Controlled drug delivery attributes of co-polymer micelles and xanthan-O-carboxymethyl hydrogel particles. Int J Biol Macromol. 2014;70:37–43. doi: 10.1016/j.ijbiomac.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 29.Kim MS, Seo KS, Khang G, Lee HB. Ring-opening polymerization of ε-caprolactone by poly (ethylene glycol) by an activated monomer mechanism. Macromol Rapid Commun. 2005;26(8):643–648. [Google Scholar]
- 30.Ouahab A, Shen Y, Tu JS. Novel oral delivery system of indomethacin by solidified mPEG-PDLLA micelles: in vivo study. Drug Deliv. 2012;19(4):232–237. doi: 10.3109/10717544.2012.690109. [DOI] [PubMed] [Google Scholar]
- 31.Zhang CY, Xiong D, Sun Y, Zhao B, Lin WJ, Zhang LJ. Self-assembled micelles based on pH-sensitive PAE-g-MPEG-cholesterol block copolymer for anticancer drug delivery. Int J Nanomedicine. 2014;9:4923–4933. doi: 10.2147/IJN.S69493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Daman Z, Ostad S, Amini M, Gilani K. Preparation, optimization and in vitro characterization of stearoyl-gemcitabine polymeric micelles: a comparison with its self-assembled nanoparticles. Int J Pharm. 2014;468(1–2):142–151. doi: 10.1016/j.ijpharm.2014.04.021. [DOI] [PubMed] [Google Scholar]
- 33.Cho H, Kwon GS. Polymeric micelles for neoadjuvant cancer therapy and tumor-primed optical imaging. ACS Nano. 2011;5(11):8721–8729. doi: 10.1021/nn202676u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cho H, Indig GL, Weicchert J, Shin HC, Kwon GS. In vivo cancer imaging by poly(ethylene glycol)-b-poly(ε-caprolactone) micelles containing a near-infrared probe. Nanomedicine. 2012;8(2):228–236. doi: 10.1016/j.nano.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gong C, Deng S, Wu Q, et al. Improving antiangiogenesis and anti-tumor activity of curcumin by biodegradable polymeric micelles. Biomaterials. 2013;34(4):1413–1432. doi: 10.1016/j.biomaterials.2012.10.068. [DOI] [PubMed] [Google Scholar]
- 36.Dash TK, Konkimalla VB. Poly-ε-caprolactone based formulations for drug delivery and tissue engineering: a review. J Control Release. 2012;158(1):15–33. doi: 10.1016/j.jconrel.2011.09.064. [DOI] [PubMed] [Google Scholar]
- 37.Gou M, Men K, Shi H, et al. Curcumin-loaded biodegradable polymeric micelles for colon cancer therapy in vitro and in vivo. Nanoscale. 2011;3(4):1558–1567. doi: 10.1039/c0nr00758g. [DOI] [PubMed] [Google Scholar]
- 38.Wang BL, Shen YM, Zhang QW, et al. Codelivery of curcumin and doxorubicin by MPEG-PCL results in improved efficacy of systemically administered chemotherapy in mice with lung cancer. Int J Nanomedicine. 2013;8:3521–3531. doi: 10.2147/IJN.S45250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gill KK, Kaddoumi A, Nazzal S. Mixed micelles of PEG(2000)-DSPE and vitamin-E TPGS for concurrent delivery of paclitaxel and parthenolide: enhanced chemosenstization and antitumor efficacy against non-small cell lung cancer (NSCLC) cell lines. Eur J Pharm Sci. 2012;46(1–2):64–71. doi: 10.1016/j.ejps.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 40.Zheng N, Dai W, Du W, et al. A novel lanreotide-encoded micelle system targets paclitaxel to the tumors with overexpression of somatostatin receptors. Mol Pharm. 2012;9(5):1175–1188. doi: 10.1021/mp200464x. [DOI] [PubMed] [Google Scholar]
- 41.United States Food and Drug Administration . Guidance for Industry: Bioanalytical Method Validation. Bethesda, MD: United States Food and Drug Administration; 2001. [Accessed November 24, 2014]. Available from: http://www.fda.gov/downloads/Drug/GuidanceComplianceRegulatoryInformation/Guidances/ucm070107.pdf. [Google Scholar]
- 42.Fan Y, Chen G, Tanaka J, Tateishi T. L-Phe end-capped poly(L-lactide) as macroinitiator for the synthesis of poly(L-lactide)-b-poly(L-lysine) block copolymer. Biomacromolecules. 2005;6(6):3051–3056. doi: 10.1021/bm050368u. [DOI] [PubMed] [Google Scholar]
- 43.Mezghrani O, Tang Y, Ke X, et al. Hepatocellular carcinoma dually-targeted nanoparticles for reduction triggered intracellular delivery of doxorubicin. Int J Pharm. 2015;478(2):553–568. doi: 10.1016/j.ijpharm.2014.10.041. [DOI] [PubMed] [Google Scholar]
- 44.Cao J, Su T, Zhang L, et al. Polymeric micelles with citraconic amide as pH-sensitive bond in backbone for anticancer drug delivery. Int J Pharm. 2014;471(1–2):28–36. doi: 10.1016/j.ijpharm.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 45.Shi Y, Huang W, Liang R, et al. Improvement of in vivo efficacy of recombinant human erythropoietin by encapsulation in PEG-PLA micelle. Int J Nanomedicine. 2013;8:1–11. doi: 10.2147/IJN.S38648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen L, Sha X, Jiang X, Chen Y, Ren Q, Fang X. Pluronic P105/F127 mixed micelles for the delivery of docetaxel against Taxol-resistant non-small cell lung cancer: optimization and in vitro, in vivo evaluation. Int J Nanomedicine. 2013;8:73–84. doi: 10.2147/IJN.S38221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Choi KY, Min KH, Yoon HY, et al. PEGylation of hyaluronic acid nanoparticles improves tumor targetability in vivo. Biomaterials. 2011;32(7):1880–1889. doi: 10.1016/j.biomaterials.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 48.Huang ZJ, Li T, Guo XJ, et al. Technical study of vinpocetine micelles prepared by thin-film hydration method. Zhong Yao Cai. 2012;35(11):1850–1854. Chinese. [PubMed] [Google Scholar]
- 49.Li J, Chen Q, Zha Z, et al. Ternary polyplex micelles with PEG shells and intermediate barrier to complexed DNA cores for efficient systemic gene delivery. J Control Release. 2015;209:77–87. doi: 10.1016/j.jconrel.2015.04.024. [DOI] [PubMed] [Google Scholar]
- 50.Abouzeid AH, Patel NR, Torchilin VP. Polyethylene glycolphosphatidylethanolamine (PEG-PE)/vitamin E micelles for co-delivery of paclitaxel and curcumin to overcome multi-drug resistance in ovarian cancer. Int J Pharm. 2014;464(1–2):178–184. doi: 10.1016/j.ijpharm.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang Y, Wang F, Xu H, Liu H, Meng Q, Liu W. Development of andrographolide loaded PLGA microspheres: optimization, characterization and in vitro–in vivo correlation. Int J Pharm. 2014;475(1–2):475–484. doi: 10.1016/j.ijpharm.2014.09.016. [DOI] [PubMed] [Google Scholar]
- 52.Hoehle SI, Pfeiffer E, Sólyom AM, Metzler M. Metabolism of curcuminoids in tissue slices and subcellular fractions from rat liver. J Agric Food Chem. 2006;54(3):756–764. doi: 10.1021/jf058146a. [DOI] [PubMed] [Google Scholar]