


Abstract
Genetic therapy holds great promise for the treatment of inherited or acquired genetic diseases; however, its breakthrough is hampered by the lack of suitable gene delivery systems. Dumbbell-shaped DNA minimal vectors represent an attractive, safe alternative to the commonly used viral vectors which are fraught with risk, but dumbbell generation appears to be costly and time-consuming. We developed a new PCR-based method for dumbbell production which comprises only two steps. First, PCR amplification of the therapeutic expression cassette using chemically modified primers to form a ready-to-ligate DNA structure; and second, a highly efficient intramolecular ligation reaction. Compared with conventional strategies, the new method produces dumbbell vectors more rapidly, with higher yields and purity, and at lower costs. In addition, such produced small hairpin RNA expressing dumbbells triggered superior target gene knockdown compared with conventionally produced dumbbells or plasmids. Our novel method is suitable for large-scale dumbbell production and can facilitate clinical applications of this vector system.
INTRODUCTION
Various seminal therapeutic approaches depend on efficient delivery of recombinant DNA into target cells ex vivo or in vivo in order to trigger the expression of non-coding RNAs or proteins. These approaches include the genetic therapy of inherited and acquired genetic diseases, genetic vaccination, stem cell programming, somatic cell reprogramming, immunotherapy and protein expression (1). However, the biophysical and biochemical properties of naked DNA molecules, such as the polyanionic, hydrophilic character and the susceptibility to nuclease degradation, pose a major challenge with regard to extracellular transport including extravasation, diffusion through the extracellular matrix network, target cell binding, internalization and nuclear delivery. To overcome these physical including membrane barriers, researchers use non-viral or viral delivery vectors. Viral vectors are efficient but costly and their clinical application is limited either by severe immune responses due to viral pre-exposition of the patients or by the alarming possibility of uncontrolled genomic vector integration (2). Alternative non-viral delivery systems involving non-nucleic acid-based helper functions often are complex and toxic to the cells (3). The simplest non-viral vector system is direct delivery of naked plasmid DNA. However, although they are easy and cheap to manufacture, plasmid-based vectors harbor major disadvantages: first, the large size hampers cellular and nuclear delivery; second, the bacterial backbone can trigger side effects including immunotoxicity, antibiotics resistance and transgene silencing and third, plasmids are difficult to modify chemically.
To overcome these disadvantages, minimalistic dumbbell-shaped DNA vectors were invented, which have shown promising results in preclinical and clinical trials (4–8). These vectors are composed of a linear double-stranded DNA expression cassette which is covalently closed at both ends with single-stranded loop structures. The conventional method to synthesize these vectors is called enzymatic ligation assisted by nucleases (ELAN), which involves simultaneous intermolecular ligation and digestion of misligated off-pathway products (9). The ELAN method requires large amounts of enzymes and oligonucleotides and the yields are dissatisfactory. Recently, a more efficient method was reported whereby the expression cassette is amplified by PCR followed by nicking-enzyme cleavage to produce 5′-overhangs ready for intramolecular ligation to form the closing loops of the dumbbell vector (10,11). Though, producing higher yields, this method still depends on restriction-enzyme cleavage, and re-annealing of the generated oligonucleotides can impair loop formation.
MATERIALS AND METHODS
Primers and enzymes
Primers for cloning of plasmids
Oligodeoxyribonucleotides shR-luc-plus 5′-GATCCGAGCTGTTTCTGAGGAGCCTTCAAGAGAGGCTCCTCAGAAACAGCTCTTTTTC-3′, shR-luc-minus 5′-TCGAGAAAAAGAGCTGTTTCTGAGGAGCCTCTCTTGAAGGCTCCTCAGAAACAGCT-3', shR-gfp-plus 5′-GATCCGCTGACCCTGAAGTTCATCTTCAAGAGAGATGAACTTCAGGGTCAGCTTTTTC-3′ and shR-gfp-minus 5′-TCGAGAAAAAGCTGACCCTGAAGTTCATCTCTCTTGAAGATGAACTTCAGGGTCAGCG-3′ were synthesized by AITbiotech (Singapore).
Primers for db production
Unmodified oligodeoxyribonucleotides Fw-luc 5′-TAGAATTCATATTTGCATGTCGCTATGT-3′, and Rv-luc 5′-AACTCGAGAAAAAGAGCTGTTTCTGAG-3′ for the amplification of the linear shRNA expression cassette were synthesized by AITbiotech (Singapore). dSpacer1 (AP1) oligodeoxyribonucleotides Fw-AP1-hp 5′-pATCCAGTTTTCTGGA-AP1-TAGAATTCATATTTGCATGTCGCTATGT-3′, Rv-AP1-hp 5′-pAAGGTCTTTTGACCT-AP1-AACTCGAGAAAAAGAGCTGTTTCTGAG-3′, Fw-AP1-loop 5′-pATCCAGTTTTCAGCA-AP1-TAGAATTCATATTTGCATGTCGCTATGT-3′, Rv-AP1-loop 5′-pAAGGTCTTTTCAGCA-AP1-AACTCGAGAAAAAGAGCTGTTTCTGAG-3′ and dSpacer3 (AP3) oligodeoxyribonucleotides Fw-AP3-hp 5′-pATCTCCAGTTTTCTGGA-AP3-TAGAATTCATATTTGCATGTCGCTATGT-3′, Rv-AP3-hp 5′-pATCAGGTCTTTTGACCT-AP3-AACTCGAGAAAAAGAGCTGTTTCTGAG-3′, Fw-AP3-loop 5′-pATCTCCAGTTTTCAGCA-AP3-TAGAATTCATATTTGCATGTCGCTATGT-3′, Rv-AP3-loop 5′-pATCTCCAGTTTTCAGCA-AP3-AACTCGAGAAAAAGAGCTGTTTCTGAG-3′ as well as PEG-150 (S9) oligodeoxyribonucleotides Fw-S9-hp 5′-pATCGTCCAGTTTTCTGGA-S9-TAGAATTCATATTTGCATGTCGCTATGT-3′, Rv-S9-hp 5′-pATCGAGGTCTTTTGACCT-S9-AACTCGAGAAAAAGAGCTGTTTCTGAG-3′, Fw-S9-loop 5′-pATCGTCCAGTTTTCAGCA-S9-TAGAATTCATATTTGCATGTCGCTATGT-3′, and Rv-S9-loop 5′-pATCGAGGTCTTTTCAGCA-S9-AACTCGAGAAAAAGAGCTGTTTCTGAG-3′, all for gpPCR were synthesized by Integrated DNA Technologies (Coralville, USA). Fw-shGFP 5′-TTAGGAGTTTTCTCCTAAGCGAATTCATATTTGCATGTCGCTATGT-3′, Rv-shGFP 5′-TTAGGTCTTTTGACCTAAGCCTCGAGAAAAAGCTGACCCTGAA-3′, Fw-2nd 5′-pTTAGGAGTTTTCTCCTAAGC-3′ and Rv-2nd 5′-pTTAGGTCTTTTGACCTAAGC-3′ for the nicking enzyme method and loop-primers for the ELAN method L1 5′-pAATTGTCCAGTTTTCTGGAC-3′ and L2 5′-pTCGACAGGTCTTTTGACCTG-3′ were synthesized by AITbiotech.
Primers and probes for qPCR detection were synthesized by AITbiotech
Stem-loop primer for reverse transcription was 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAAGAGC-3′, universal Taqman probe was FAM-5′-TCGCACTGGATACG-3′-MGB, qPCR forward primer for shlucRNA was 5′-GAGCTGTTTCTGAGGAGCCTTC-3′, qPCR universal reverse primer for shlucRNA was 5′-GTGCAGGGTCCGAGGT-3′. Taqman probe for mH1 promoter was FAM-5′-TCTGGGAAATCACCATAAA-3′-BHQ-1, qPCR forward and reverse primers for mH1 promoter were 5′-TTCATATTTGCATGTCGCTATGTG-3′ and 5′-TCCCAAATCCAAAGACATTTCA-3′, respectively. qPCR forward and reverse primers for β-actin were 5′-CTGGCACCCAGCACAATG-3′ and 5′-GCCGATCCACACGGAGTACT-3′, respectively.
Taq DNA polymerase, pfu DNA polymerase, restriction enzymes, T4 DNA ligase and T7 DNA polymerase, if not specified otherwise, were purchased from Life technologies (Singapore).
Cloning of plasmids
The 227 bp human H1 promoter of the pSuperTM (Oligoengine) cloning vector was replaced by the 99 bp minimal H1 (mH1) promoter (12). Therefore, both DNA strands resembling the mH1 sequence were chemically synthesized, annealed, purified and inserted into the pSuper plasmid using EcoRI and BglII restriction sites to generate plasmid pSuper-mH1. The expression cassette for a firefly luciferase-targeting small hairpin RNA (shRNA) was cloned into the pSuper-mH1 vector. Therefore, oligodeoxyribonucleotides shR-luc-plus and shR-luc-minus were annealed and inserted into pSuper-mH1 using the BglII and XhoI restriction sites to generate plasmid pSuper-mH1-shR-luc. Similarly, oligodeoxyribonucleotides shR-gfp-plus and shR-gfp-minus were annealed and inserted into pSuper-mH1 using the BglII and XhoI restriction sites to generate plasmid pSuper-mH1-shR-gfp.
Dumbbell vector generation
gpPCR method. To generate the template for gap-primer PCR, pSuper-mH1-shR-luc was cleaved with restriction endonucleases KpnI and BamHI. Gap-primer PCR (gpPCR) was performed in a volume of 400 μl using 400 ng pSuper-mH1-shR-luc/KpnI/BamHI template, 0.2 mM of each dNTP, 0.3 μM of forward primer, 0.1 μM reverse primer, and mixture containing 8 U Taq DNA polymerase and 4 U Pfu DNA polymerase. PCR cycling was done as follows: initial denaturing at 95°C (3 min), then 30 cycles of denaturing at 95°C (30 s), primer annealing at 53°C (hp-primers) or 66°C (loop-primers) (30 s), and extension at 72°C (30 s) and final extension at 72°C (5 min). gpPCR products were converted into dumbbells by ligation. All the PCR products were purified by QIAquick PCR Purification Kit (Qiagen). Each 1 μg of gpPCR product was ligated as follows: AP1-hp products using 10 U T4 DNA ligase (Fermentas), AP3-hp with 20 U T4 DNA ligase and AP1-loop products with 417 U CircLigase (Epicentre). Ligation with T4 DNA ligase was performed for 15 h at 22°C, ligation with CircLigase for 4 h at 60°C. For exonuclease treatment, 10 U T7 DNA polymerase was added per μg PCR product and the reaction was incubated for 30 min at 37°C.
Nicking enzyme method. For nicking-enzyme-based production of egfp-targeting dumbbell db-Nick, we followed the protocol described by Taki et al. performing two rounds of PCR (10). Double-digested pSuper-mH1-shR-GFP plasmid was used as the PCR template, the sequences of forward and reverse primers for the first PCR reaction were Fw-shGFP and Rv-shGFP. The sense and antisense primer sequences for the second PCR reaction were Fw-2nd and Rv-2nd, respectively. Exonuclease treatment was done as described above.
ELAN method. For ELAN-based production of dumbbell db-ELAN we followed the protocol by Cost et al. (9). Two microgram PCR product was digested with each 2 U FD XhoI and FD EcoRI and each 25 pmol of the loop-sequences L1 and L2 was ligated using 10 U of T4 DNA ligase in the presence of 0.5 U of FD XhoI, FD EcoRI, FD SalI and FD MfeI. Exonuclease treatment was done as described above.
Knockdown assay
To monitor firefly luciferase target gene knockdown, HEK293T cells were co-transfected with 90 ng luciferase reporter vector pGL3 (Promega) and either 90 ng dumbbell or plasmid DNA (equimass) or 0.5 pmol of dumbbell or plasmid DNA (equimolar; pVAX1 plasmid was used as top-up DNA for dumbbell transfection) using Lipofectamine 2000 (Life Technologies) and luciferase knockdown was monitored 48 h post-transfection.
RNA extraction, reverse transcription and qPCR detection
105 HepG2 cells were seeded in 24-well plates and transfected with 0.25 pmol of dumbbell or plasmid DNA using Lipofectamine 2000. pVAX1 plasmid was used as control as well as a top-up DNA for dumbbell transfection. Twenty-four hours post-transfection, cells were harvested and total RNA was isolated using Trizol (Life Technologies) following the manufacturers protocol. Luciferase-targeting shRNA (shluc) was detected using the universal TaqMan-based RT-PCR protocol (13), and the fold change was determined by ΔΔCt quantification using β-actin RNA as an internal standard. qPCR was performed using the 7900HT Fast realtime PCR system (Applied Biosystems).
Nuclear import assay
5×105 HepG2 cells were seeded in 6-well plates and transfected with 1 pmol of dumbbell or plasmid DNA using Lipofectamine 2000. pVAX1 plasmid was used as control as well as a top-up DNA for dumbbell transfection. Twenty-four hours post-transfection, cells were harvested, incubated in hypotonic buffer for 15 min on ice and lysed by 20 times dounce homogenization in hypotonic buffer. After centrifugation at 3000 rpm for 10 min at 4°C, the supernatant (cytoplasmic fraction) was subsequently removed and the pellet (nuclear fraction) was further lysed by four freeze-thaw cycles using liquid nitrogen and a water bath. Total nuclear nucleic acids were extracted using Trizol and the absolute abundance of transfected vector DNA was determined by TaqMan qPCR quantification of the copy number of the minimal H1 promoter sequence using the 7900HT Fast realtime PCR system (Applied Biosystems). For qPCR quantification, respective rtPCR standard curves were used to measure db-AP1-hp, db-ELAN and the supercoiled plasmid DNA.
Capillary gel electrophoresis
High-resolution capillary electrophoresis was performed using a QIAxcel® DNA high-resolution gel cartridge (Qiagen) on a QIAxcel system (Qiagen) according to the manufacturer's instructions. QX DNA Size Marker pUC18/HaeIII (Qiagen) was used to determine dumbbell vector size using 5 ng/μl QX Alignment Marker 15 bp/1 kb (Qiagen) as internal standard. The OL800 method was used for analysis.
Statistical analysis
All data referring to target gene knockdown, transcriptional activity and nuclear vector abundance as reported in Figure 6 and Supplementary Figure S3 are mean values ± SEM of three independent experiments. The statistical analysis was performed using repeated one-way ANOVA plus a post-hoc Newman–Keuls test. The GraphPad Prism 5 software was used for the statistical analysis. The significance was denoted as ***p < 0.001; **p < 0.01; *p < 0.05.
Figure 6.
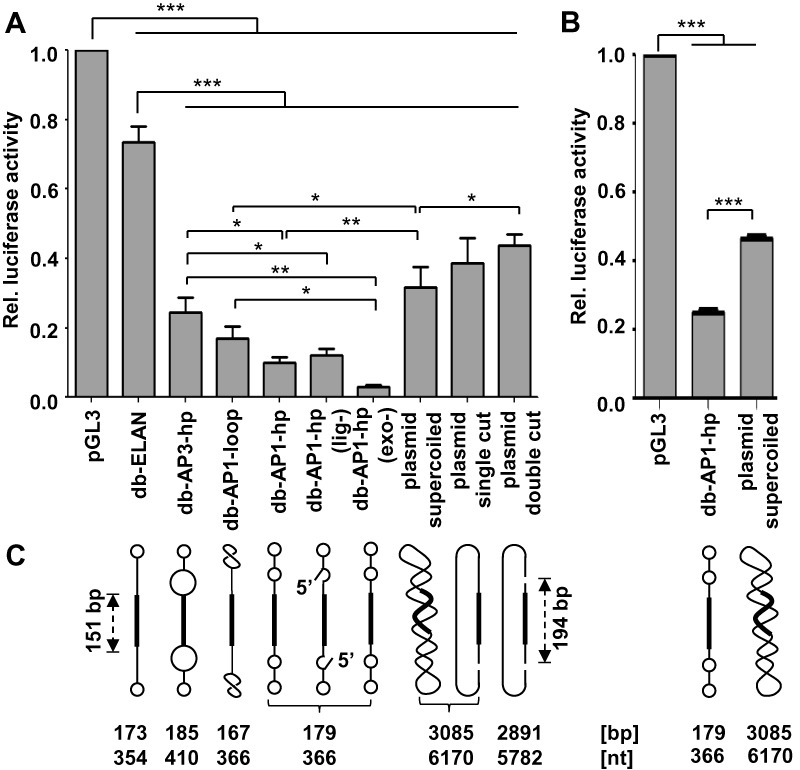
Luciferase target gene knockdown in HEK293T cells triggered by shRNA expressing dumbbells and plasmids. (A) Cells were co-transfected with 90 ng luciferase reporter vector pGL3 and 90 ng (equimass amounts) dumbbell or plasmid DNA. Firefly luciferase expression levels relative to the uninhibited negative control. Values are mean values ± SEM of two (db-ELAN) or three (rest) independent experiments. (B) Cells were co-transfected with 90 ng luciferase reporter vector pGL3 and 0.5 pmol (equimolar amounts) dumbbell or plasmid DNA. Firefly luciferase expression levels relative to the uninhibited negative control. Values are mean values ± SEM of three independent experiments. A and B, the statistical analysis was performed using repeated one-way ANOVA plus a post-hoc Newman–Keuls test. The significance was denoted as ***p < 0.001; **p < 0.01; *p < 0.05. (C) Structures/topology of the DNA vectors tested in A and B. All vectors harbor a 151 bp shRNA expression cassette. Hairpin loops and internal loops within the dumbbell (db) vectors are indicated as circles, i.e. small/large circles indicate small/large loops. All dumbbell vectors were tested after ligation and exonuclease treatment. Vector db-AP1-hp was additionally tested after skipping either the exonuclease treatment (exo-) or both ligation and exonuclease treatment (lig-). The pSuper plasmid vector was tested as supercoiled DNA, after linearization with KpnI (single cut), or after KpnI/BamHI digestion (double cut). The shRNA expression cassette was contained in the smaller 194 bp KpnI/BamHI fragment. AP1 or AP3 abasic site mimics were counted as 1 or 3 nt, respectively.
RESULTS
A novel fast two-step protocol for dumbbell vector production
To further simplify and cheapen dumbbell production and to increase the yields, we developed a two-step PCR-based method that involves chemically modified gap-primers (Figure 1). The new gap-primer-based PCR (gpPCR) method maximally reduces the number and amount of enzymes and oligonucleotides needed for the production of dumbbell-shaped DNA vectors. In step 1, the expression cassette of interest (coding or non-coding) is amplified by PCR with a pair of primers containing a 5′-phosphate, a central gap and a 3′-terminal target binding site with a 3′-hydroxyl group (3′-OH). The extension of both newly synthesized strands is terminated upon reaching the gap yielding PCR products with 5′-overhangs. In step 2, the phosphorylated 5′-overhangs are ligated intramolecularly to the 3′-OH groups to form the covalently closed dumbbell structure. The essential idea behind our method is that the abasic gaps cannot function as templates for base-pairing during primer extension forcing the polymerase to halt, thus directly yielding 5′-overhangs ready for efficient intramolecular ligation. The chemical nature of the abasic gaps may affect the efficiency of PCR amplification, ligation, or the biological function of the final dumbbell vector, and the length of the gap may impact polymerase halting. To achieve optimal efficiency and efficacy of dumbbell-production, we investigated oligonucleotides harboring abasic gaps of different chemistry and length (Figure 2): dSpacer1 (AP1) and dSpacer3 (AP3) are tetrahydrofuran-based mimics of one or three apurinic/apyrimidinic abasic sites; and PEG-150 (S9) is a triethylene glycol-based spacer with a molecular weight of 150 Da and a length of 1.3 nm which approximately corresponds to four base pairs (14,15). For each of the three gap variants we investigated two sets of primers. First, self-complementary hairpin (hp) primers causing the 5′-overhangs to fold back forming a stem-loop structure and positioning the 5′-phosphate close to the 3′-OH groups (Figure 1, left panel). To form linear dumbbell vectors, we bridged the AP1 or AP3 gaps in the opposing strand with one or three nucleotides (nt), and the S9 gap with 4 nt respectively, and ligated the ends using T4 DNA ligase. Second, we designed unstructured (loop) primers forming open 5′-overhangs which were ligated using the single-strand-specific Circligase (Figure 1, right panel) (16). Exonuclease resistance represents a characteristic feature of covalently closed dumbbell-shaped DNA (17). Thus, ligation reactions can be treated with T7 DNA polymerase, which exhibits a strong 3′ to 5′ exonuclease activity, to degrade all educts and by-products yielding purified dumbbell DNA (18).
Figure 1.
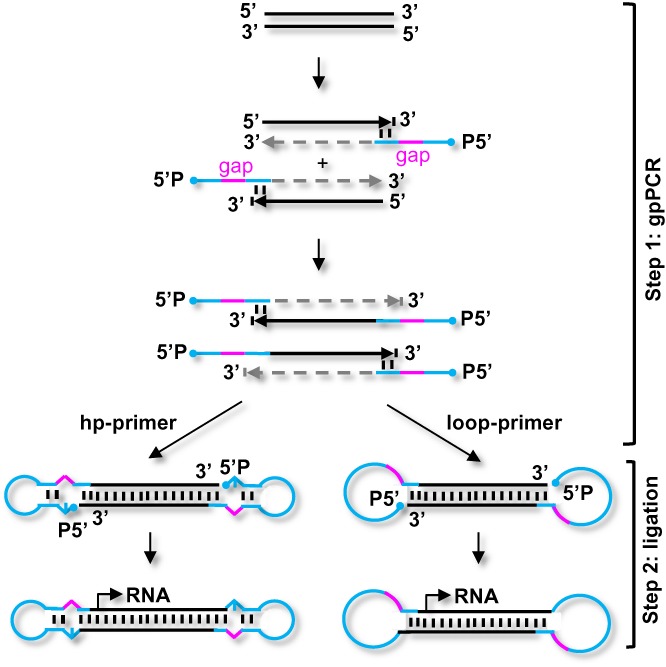
Schematic depiction of gap-primer-PCR (gpPCR)-based formation of DNA dumbbell vectors. In step 1, 5′-phosphorylated primers harboring abasic gaps are used to amplify the expression cassette of interest. The polymerase halts as soon as the gap is reached, yielding a PCR product with 5′-overhangs. Two kinds of gap-primers can be used: hairpin primers (hp) which prompt the 5′-overhangs to fold back and to position them close to the 3′-OH groups (left panel); or unstructured (loop) primers (right panel). In step 2, the gpPCR products are ligated to form the covalently closed dumbbell structure. Hp-primer products are ligated with the double-strand-specific T4 DNA ligase; loop-primers are ligated using the single-strand-specific CircLigase. Optionally, exonuclease treatment can be considered to purify the dumbbell DNA.
Figure 2.
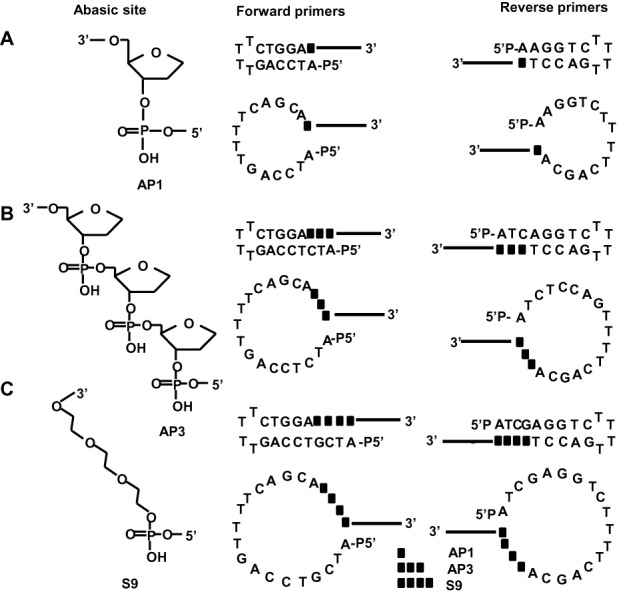
Design and structure of chemically modified gap-primers. Left panel: molecular structures of the abasic gaps; right panel: sequence and structure of hairpin and loop gap-primers. (A) AP1 (dSpacer1) primers harboring a single tetrahydrofuran-based abasic site mimic. (B) AP3 (dSpacer3) primers harboring three tetrahydrofuran-based abasic site mimics. (C) S9 (PEG-150) primers harboring a triethylene glycol-based spacer.
Hairpin primers harboring tetrahydrofuran-based mimics of one abasic site are most efficient
We investigated the efficiency of our new method in terms of dumbbell production using first the hp-primers together with T4 DNA Ligase (Figure 3). Therefore, hp-primer PCR products were treated with exonuclease either before or after ligation and the resulting DNA products were analyzed by 10% polyacrylamide gel electrophoresis (PAGE). All hp-primer PCR reactions yielded products of the expected size of 179–185 bp (lanes 2, 6 and 10) which were completely degraded during exonuclease treatment (lanes 3, 7 and 11) due to the availability of free 3′ ends. Ligase treatment triggered a detectable change in DNA topology reflected by an increase in mobility, which is characteristic for dumbbell DNA under the conditions of this analysis. Such a mobility shift was observed for PCR products generated using the AP1- (lane 4) or with lower yields the AP3-gap-primers (lane 8), but not with the S9-primers (lane 12). Accordingly, exonuclease treatment left highest dumbbell yields for the AP1-gpPCR (lane 5), followed by the AP3-gpPCR (lane 9); no dumbbell was produced based on S9-gpPCR (lane 13).
Figure 3.
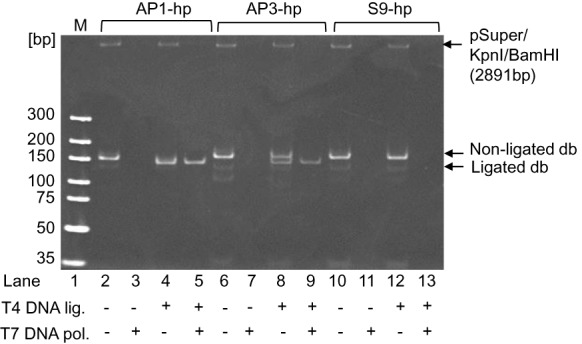
Dumbbell DNA vector formation using hairpin (hp)-gap-primer PCR. Products yielded from gpPCR using the AP1-hp, AP3-hp and S9-hp primers were analyzed before (lanes 2, 6 and 10) and after ligation (lanes 4, 8 and 12). The unligated (lanes 3, 7 and 11) and ligated products were subjected to T7 DNA polymerase (exonuclease) treatment (lanes 5, 9 and 13). Highest dumbbell yields were observed with AP1-hp primers (lane 5) followed by the AP3-hp primers (lane 9). No dumbbell DNA was detectable with the S9-hp primers (lane 13). Ethidium bromide stain of a 10% PAGE.
Gap-primer-based PCR produces dumbbells with highest conversion yields
Next, we evaluated the dumbbell conversion yields by determining the ratios of dumbbell vector DNA after ligation and exonuclease treatment divided by the PCR product yields prior to enzymatic treatment. Among the hp-primer series, highest conversion yields were obtained with the AP1-hp-primers (92%), followed by the AP3-hp-primers (64%), and no dumbbell DNA was produced with the S9-hp-primers (Figure 4A). Among the loop-primer series, only the AP1-loop-primers triggered dumbbell formation (75%) supporting the hypothesis that larger abasic gaps lead to difficult-to-ligate DNA substrates (Figure 4B). In comparison with our new method, the conventional dumbbell production techniques exhibited significantly lower conversion yields of 57% (nicking-enzyme method) or 18% (ELAN method), both lower than initially reported in the literature based on the same quantification method (Figure 4C and D).
Figure 4.
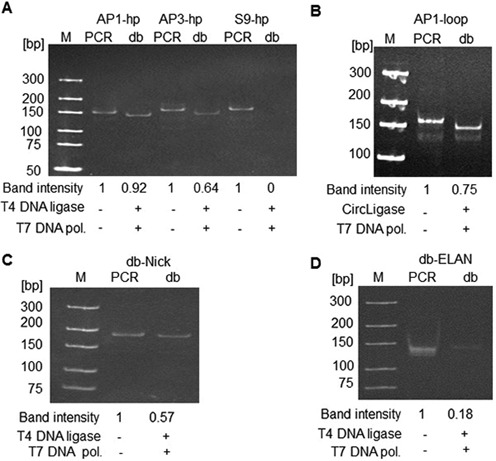
Conversion yields from linear gpPCR products to covalently closed dumbbell DNA. gpPCR products were either treated with ligase and exonuclease or not, and subjected to 10% PAGE. Band intensities of the ethidium bromide stained gels were quantified using ImageJ 1.37v software (NIH, USA). (A) hp-primer PCR products. (B) AP1-loop-primer PCR product. (C) Anti-GFP-shRNA expressing dumbbell db-Nick produced with the nicking-enzyme method. (D) Anti-luciferase-shRNA expressing dumbbell db-ELAN produced with the ELAN method.
Gap-primer-based PCR produces dumbbells with highest degree of purity
Dumbbell purity after exonuclease treatment was investigated using PAGE and high-resolution capillary gel electrophoresis (Figure 5). Both methods indicate that gap-primer PCR using AP1-hp-primers generates dumbbells with the highest level of purity which was calculated to be 93.5% by the QIAxcel system. AP3-hp gap-primer PCR as well as the ELAN or nicking-enzyme methods produced dumbbells with purity levels ranging between 82 and 85%.
Figure 5.
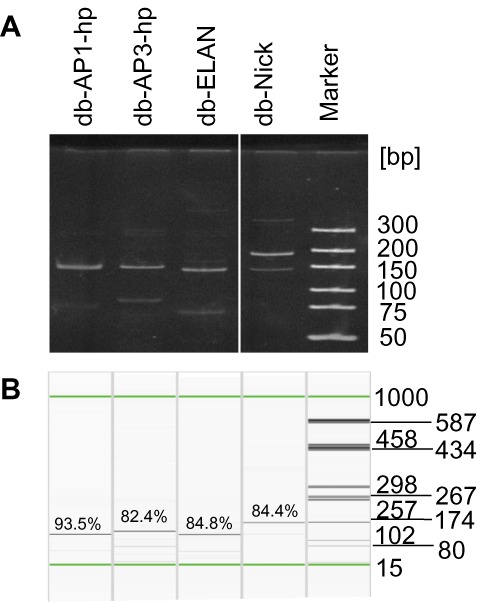
Purity of dumbbell vectors produced using different technologies. Db-AP1-hp and db-AP3-hp: gpPCR-produced dumbbells using primers AP1-hp or AP3-hp; db-ELAN: ELAN-produced dumbbell; db-Nick: egfp-targeting dumbbell produced using the nicking-enzyme method. (A) 10% PAGE analysis. (B) High-resolution capillary gel electrophoresis. The indicated purity in% refers to the total fraction of dumbbell vector DNA in the analyzed sample.
Gap-primer PCR-generated dumbbells trigger superior target gene knockdown
All dumbbell vectors investigated in this study harbor the expression cassette for a pre-validated firefly luciferase targeting shRNA that was designed according to earlier reported guide structure criteria (19,20). We tested the functionality of gpPCR-generated dumbbells, i.e. luciferase knockdown in human tissue culture cells, in comparison with a dumbbell produced using the ELAN method or a pSuper-based plasmid vector, all expressing the same shRNA driven by the minimal H1 promoter (Figure 6). To compare equimass amounts of vector DNA, HEK293T cells were co-transfected with 90 ng of dumbbell or plasmid DNA and 90 ng of the pGL3 luciferase reporter vector and luciferase knockdown was monitored 48 h post-transfection (Figure 6A). Under these conditions, all gpPCR-generated dumbbells and the plasmid vector triggered significantly stronger luciferase knockdown compared with the ELAN-produced dumbbell (db-ELAN; 17% knockdown; p < 0.001). Strongest knockdown (90%; p < 0.001) was measured for the AP1-hp-primer-produced dumbbell (db-AP1-hp), followed by the AP1-loop-primer-generated (db-AP1-loop) dumbbell (83%; p < 0.001), and db-AP3-hp (75%; p < 0.001). The non-ligated dumbbell db-AP1-hp-(lig-) was with 88% knockdown almost as effective as its ligated counterpart which might pretend that exonuclease resistance is not that relevant under the assay conditions. However, as db-AP1-hp-(lig-) was not exonuclease treated, it was contaminated by a small quantity the KpnI/BamHI-digested plasmid DNA which served as PCR template for dumbbell production. Hence, the best way to investigate the meaning of exonuclease resistance is to compare db-AP1-hp-(lig-) with the ligated but not exonuclease treated DNA db-AP1-hp-(exo-). The latter was observed to trigger a substantial stronger knockdown (97%) highlighting the importance of the ligation step. This assumption is further supported by the observation that single- (61% knockdown) or double-digestion (57% knockdown) of the supercoiled plasmid vector (68% knockdown) increasingly impaired its silencing activity. Nevertheless, it is unlikely that the small amount of undigested PCR template, which is not detectable on the gel, considerably contributes to the profound knockdown effect triggered by db-AP1-hp-(exo-). Thus, considering the high conversion yields of 92%, the exonuclease step may be skipped for db-AP1-hp production to further simplify the protocol. Considering limitations associated with some delivery strategies such as delivery volumes or toxicity triggered by liposomal compounds, together with the fact that the total mass of DNA that can be delivered can be limited, small db-vectors harbor the advantage that equimass amounts correspond to much higher (here ∼17-fold higher) equimolar amounts as compared with larger plasmids. To directly compare the activity of our best dumbbell db-AP1-hp with the corresponding supercoiled plasmid DNA, HEK293T cells were co-transfected with equimolar amounts, i.e. 0.5 pmol, of dumbbell or plasmid and 90 ng of the pGL3 luciferase reporter vector and luciferase knockdown was monitored 48 h post-transfection (Figure 6B). Even under these conditions, db-AP-1-hp triggered significantly (p < 0.001) stronger luciferase knockdown (74.7%) compared with the plasmid vector (53.7%) highlighting the advantage of the dumbbell size and/or structure.
Gap-primer PCR produces dumbbell vectors at lowest costs
Finally, we calculated and compared the overall input and expenses for the production of 1 μg dumbbell DNA produced either with the conventional methods or our new strategy (Table 1). Among all investigated protocols including the different gpPCR protocols, AP1-hp-primers generated the most active dumbbells at highest yields and lowest costs. In comparison, both the ELAN and the nicking-enzyme method require more and higher amounts of primers and enzymes. A major cost factor involved with the conventional methods is the need for restriction endonucleases. Though gpPCR depends on modified primers, AP1-hp-primer PCR is 10- or 5-fold cheaper compared with the ELAN method and 3.1- or 1.6-fold cheaper compared with the nicking-enzyme method, depending on whether the final exonuclease treatment is skipped or not.
Table 1. Calculation of costs, excluding those for column purification, for the production of 1 μg dumbbell DNA based on local prices in Singapore Dollar (SGD) converted into US Dollar (USD).
| ELAN db | Nicking-enzyme db | AP1-hp db | AP1-hp (exo-) db | AP3-hp db | AP1-loop db | |
|---|---|---|---|---|---|---|
| Oligodeoxyribonucleotides (pmol) | FwP: 330 | 1st round PCR | 5′P-FwP: 65 | 5′P-FwP: 65 | 5′P-FwP: 94 | 5′P-FwP: 94 |
| RvP: 110 | FwP: 5.6 | 5′P-RvP: 22 | 5′P-RvP: 22 | 5′P-RvP: 31 | 5′P-RvP: 31 | |
| Loop1: 70 | RvP: 5.6 | |||||
| Loop2: 70 | 2nd round PCR | |||||
| FwP: 70 | ||||||
| RvP: 70 | ||||||
| DNA polymerase (units) | Pfu: 11 | 1st round PCR | Pfu: 2.2 | Pfu: 2.2 | Pfu: 3.1 | Pfu: 3.1 |
| Taq: 22 | Taq: 0.7 | Taq: 4.4 | Taq: 4.4 | Taq: 6.3 | Taq: 6.3 | |
| 2nd round PCR | ||||||
| Taq: 8.8 | ||||||
| Restriction endonuclease (units) | XhoI: 9.7 | NB. Bpu 10I: 8.8 | ||||
| EcoRI: 9.7 | ||||||
| SalI: 4.2 | ||||||
| MfeI: 4.2 | ||||||
| DNA ligase (units) | T4 DNA ligase: 28 | T4 DNA ligase: 17.5 | T4 DNA ligase: 10.9 | T4 DNA ligase: 10.9 | T4 DNA ligase: 31.3 | CircLigase: 555 |
| T7 DNA polymerase (units) | 28 | 17.5 | 10.9 | 15.6 | 13.3 | |
| Total expensesa (USD) | 72.5 | 21.1 | 13.2 | 7.0 | 23.8 | 211.1 |
aBased on an exchange rate of 1 USD = 1.25 SGD.
DISCUSSION
We describe a novel two-step protocol for dumbbell generation that uses gap-primers to amplify ready-to-ligate PCR products. Among the two investigated gap-primer designs, hp-primers yielded PCR products that could efficiently be ligated with T4 DNA ligase. In comparison, loop-primer PCR products could not be ligated with T4 DNA ligase at all and ligation with the Circligase was rather inefficient. This might be explained by lower efficiency of ligating unstructured dangling overhangs. Our results further indicate that only those gap-primers harboring tetrahydrofuran-based gaps but not those with S9-gaps can trigger dumbbell formation. Currently the question cannot be answered as to whether S9-gaps are either skipped by the polymerase yielding double-stranded DNA ends which would be unsuitable for dumbbell formation, or alternatively trigger the formation of substrates which are difficult to ligate. Among the series of hp-primers furnished with tetrahydrofuran-based gaps, highest dumbbell yields were obtained for the smallest gap AP1. That is, a single abasic site efficiently pauses the polymerase without providing any evidence that the enzyme may jump over the gap. It is reasonable to assume that the larger AP3 gap halts the polymerase as effectively as the shorter AP1 gap. However, AP1-primer ligation likely is more efficient due to the more precise positioning of the 5′-phosphate for ligation with the 3′-OH group.
The yields of converting the expression cassette of interest into covalently closed exonuclease-resistant dumbbells as well as the purity of the obtained dumbbells were found to inversely correlate with the numbers of (i) manufacturing steps, (ii) involved enzymes and (iii) possible by-products that can be formed. Accordingly, highest yields of purest dumbbells were achieved with the new gpPCR-method, followed by the nicking-enzyme method and the ELAN-method. Notably, though the measured yields may vary depending on the used method of quantification or the batch of ligase, they represent a reliable indicator to compare the relative strengths of the methods.
The gpPCR-generated dumbbells triggered significantly (p < 0.001) stronger knockdown compared with the ELAN-produced dumbbell, the latter of which was equal in size and uses the same expression cassette to transcribe the same shRNA. Thus, the only difference between gpPCR- and ELAN-produced dumbbells relies in (i) abasic sites that trigger the formation of (ii) internal loops close to the ends of the gpPCR-generated dumbbells. Abasic sites are being cleaved by the apurinic/apyrimidinic enzyme 1 in human cells prior to base excision and/or nucleotide incision repair (Supplementary Figure S1) (21). Thus, these might not be considered advantageous for a genetic vector though the abasic site mimics used in this study, which are lacking the OH-group at the C-1 position of the 2’-deoxyribose, are considerably more stable. The internal loops as well as larger terminal loops, however, trigger increased flexibility of the gpPCR-dumbbell ends (Supplementary Figure S2A). While rigid rod-like ELAN-dumbbells might enter the nuclear pore complexes (NPCs) only in nearly perpendicular orientation to the nuclear membrane (Supplementary Figure S2B), the more flexible ends of the gpPCR-dumbbells could enable NPC threading even from sub-perpendicular angles, that way accelerating the trajectory through the NPCs (Supplementary Figure S2C). A recent study suggested that passive fluxes through NPCs are controlled by channels with three distinct radii of 1.74 nm (78%), 2.63 nm (22%) and 4.32 nm (0.07%) corresponding to diameters of 3.48 nm, 5.26 nm and 8.64 nm (22). The DNA double-helix effective diameter (deff) in solutions containing the physiological salt concentration was calculated to be 5 nm, which is significantly larger than the geometric diameter of 2 nm (23). That implies that base-paired double-helical DNA can freely pass through the medium-sized NPC channels; however, depending on their size, terminal and internal loops would rapidly enlarge deff beyond the cut-off value for passage through midsize meshes, thus significantly delaying the nuclear influx. Given a certain minimum size, however, terminal loops are more flexible than internal loops. This model is suitable to explain the reduced activity of dumbbells db-AP3-hp and db-AP3-loop as compared with db-AP1-hp (Supplementary Figure S2D).
To prove our hypothesis that gpPCR-dumbbells are more efficiently entering cellular nuclei, HepG2 cells were transfected with equimolar (1 pmol) amounts of db-AP1-hp, db-ELAN, or plasmid DNA and both nuclear vector abundance and transcriptional vector activity were monitored 24 h post-transfection using qPCR (Supplementary Figure S3). Since db-vectors and plasmids have different PCR amplification efficiencies, we used individual rtPCR standard curves for the absolute quantification of each of the respective vectors. For example, a db-AP1-hp standard curve was used to quantify db-AP1-hp and so forth. As shown in Supplementary Figure S3A, nuclear delivery of the gpPCR-produced dumbbell was 6.4-fold (p < 0.001) or 94.7-fold (p < 0.001) enhanced compared with the ELAN-produced dumbbell or the plasmid. The fact that db-AP1-hp entered the nucleus much more efficiently compared with the equally sized db-ELAN indicates that not only the vector size but also the vector structure matters in terms of nuclear delivery. In accordance with the measured nuclear vector abundancies, highest transcriptional activity was detected for db-AP1-hp followed by db-ELAN and the plasmid DNA (Supplementary Figure S3B). These findings are consistent with our suggested model that the flexible ends of gpPCR-produced dumbbells might accelerate the trajectory through the nuclear pores. However, one has to keep in mind that the observed nuclear vector abundance is the result of a multi-step process and depends not only on diffusion through the nuclear pores but also on cellular delivery, endosomal escape, cytoplasmic and nuclear DNA stability, and the efficiency of vector DNA isolation from the nuclear compartment. To fully understand the phenomenon of facilitated nuclear targeting by gpPCR-produced dumbbells, more detailed studies of the delivery kinetics will be required.
In summary, our novel two-step gpPCR method produces higher yields of superior and cleaner dumbbells at lower costs within a shorter period of time. The protocol is scalable and may facilitate large-scale production of RNA or protein expressing dumbbell vectors for preclinical and clinical investigation toward efficient and safe genetic therapy. The current focus and future challenge lies in covalent linkage of RNA, peptide or protein helper functions to the loops of the dumbbells for targeted delivery in vivo.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Footnotes
Present address:
Kar Tong Tan, Cancer Science Institute, National University of Singapore, Singapore 119260, Singapore.
Liting Hang, Neurodegeneration Research Laboratory, National Neuroscience Institute, Singapore 308433, Singapore.
FUNDING
NUS-Cambridge Start-up Grant from National University of Singapore (NUS), [NIG/1058/2011] from National Medical Research Council (NMRC); Academic Research Fund (AcRF) Tier 1 Faculty Research Committee (FRC) Grants [T1–2011Sep-04] and [T1–2014Apr-02] from the Ministry of Education (MOE) of Singapore.
Conflict of interest statement. The authors declare competing financial interests. A patent application covering major parts of the work is pending.
REFERENCES
- 1.Kay M.A. State-of-the-art gene-based therapies: the road ahead. Nat. Rev. Genet. 2011;12:316–328. doi: 10.1038/nrg2971. [DOI] [PubMed] [Google Scholar]
- 2.Biasco L., Baricordi C., Aiuti A. Retroviral integrations in gene therapy trials. Mol. Ther. 2012;20:709–716. doi: 10.1038/mt.2011.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yin H., Kanasty R.L., Eltoukhy A.A., Vegas A.J., Dorkin J.R., Anderson D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014;15:541–555. doi: 10.1038/nrg3763. [DOI] [PubMed] [Google Scholar]
- 4.Mok P.L., Cheong S.K., Leong C.F., Chua K.H., Ainoon O. Extended and stable gene expression via nucleofection of MIDGE construct into adult human marrow mesenchymal stromal cells. Cytotechnology. 2012;64:203–216. doi: 10.1007/s10616-011-9413-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaur T., Slavcev R.A., Wettig S.D. Addressing the challenge: current and future directions in ovarian cancer therapy. Curr. Gene Ther. 2009;9:434–458. doi: 10.2174/156652309790031148. [DOI] [PubMed] [Google Scholar]
- 6.López-Fuertes L., Pérez-Jiménez E., Vila-Coro A.J., Sack F., Moreno S., Konig S.A., Junghans C., Wittig B., Timón M., Esteban M. DNA vaccination with linear minimalistic (MIDGE) vectors confers protection against Leishmania major infection in mice. Vaccine. 2002;21:247–257. doi: 10.1016/s0264-410x(02)00450-4. [DOI] [PubMed] [Google Scholar]
- 7.Schakowski F., Gorschlüter M., Junghans C., Schroff M., Buttgereit P., Ziske C., Schöttker B., König-Merediz S.A., Sauerbruch T., Wittig B., et al. A novel minimal-size vector (MIDGE) improves transgene expression in colon carcinoma cells and avoids transfection of undesired DNA. Mol. Ther. 2001;3:793–800. doi: 10.1006/mthe.2001.0322. [DOI] [PubMed] [Google Scholar]
- 8.Zanta M.A., Belguise-Valladier P., Behr J.-P. Gene delivery: a single nuclear localization signal peptide is sufficient to carry DNA to the cell nucleus. Proc. Natl. Acad. Sci. U.S.A. 1999;96:91–96. doi: 10.1073/pnas.96.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cost G.J. Enzymatic ligation assisted by nucleases: simultaneous ligation and digestion promote the ordered assembly of DNA. Nat. Protoc. 2007;2:2198–2202. doi: 10.1038/nprot.2007.325. [DOI] [PubMed] [Google Scholar]
- 10.Taki M., Kato Y., Miyagishi M., Takagi Y., Taira K. Small-interfering-RNA expression in cells based on an efficiently constructed dumbbell-shaped DNA. Angew. Chem., Int. Ed. 2004;43:3160–3163. doi: 10.1002/anie.200353445. [DOI] [PubMed] [Google Scholar]
- 11.Taki M., Kato Y., Miyagishi M., Takagi Y., Sano M., Taira K. A direct and efficient synthesis method for dumbell-shaped linear DNA using PCR in vitro. Nucleic Acids Symp. Ser. 2003;3:191–192. doi: 10.1093/nass/3.1.191. [DOI] [PubMed] [Google Scholar]
- 12.Myslinski E., Amé J.-C., Krol A., Carbon P. An unusually compact external promoter for RNA polymerase III transcription of the human H1RNA gene. Nucleic Acids Res. 2001;29:2502–2509. doi: 10.1093/nar/29.12.2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jung U., Jiang X., Kaufmann S.H.E., Patzel V. A universal stem-loop primer-based TaqMan RT-PCR protocol for cost efficient detection of small non-coding RNA. RNA. 2013;19:1864–1873. doi: 10.1261/rna.040501.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rumney S., Kool E.T. Structural optimization of non-nucleotide loop replacements for duplex and triplex DNAs. J. Am. Chem. Soc. 1995;117:5635–5646. doi: 10.1021/ja00126a004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takeshita M., Chang C.N., Johnson F., Will S., Grollman A.P. Oligodeoxynucleotides containing synthetic abasic sites. Model substrates for DNA polymerases and apurinic/apyrimidinic endonucleases. J. Biol. Chem. 1987;262:10171–10179. [PubMed] [Google Scholar]
- 16.Lin C., Xie M., Chen J.J.L., Liu Y., Yan H. Rolling-circle amplification of a DNA nanojunction. Angew. Chem., Int. Ed. 2006;45:7537–7539. doi: 10.1002/anie.200602113. [DOI] [PubMed] [Google Scholar]
- 17.Chu B.C.F., Orgel L.E. The stability of different forms of double-stranded decoy DNA in serum and nuclear extracts. Nucleic Acids Res. 1992;20:5857–5858. doi: 10.1093/nar/20.21.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hori K., Mark D.F., Richardson C.C. Deoxyribonucleic acid polymerase of bacteriophage T7. Characterization of the exonuclease activities of the gene 5 protein and the reconstituted polymerase. J. Biol. Chem. 1979;254:11604–11598. [PubMed] [Google Scholar]
- 19.Patzel V., Rutz S., Dietrich I., Köberle C., Scheffold A., Kaufmann S.H.E. Design of siRNAs producing unstructured guide-RNAs results in improved RNA interference efficiency. Nat. Biotechnol. 2005;23:1440–1444. doi: 10.1038/nbt1151. [DOI] [PubMed] [Google Scholar]
- 20.Köberle C., Kaufmann S.H.E., Patzel V. Selecting effective siRNAs based on guide RNA structure. Nat. Protoc. 2006;1:1832–1839. doi: 10.1038/nprot.2006.206. [DOI] [PubMed] [Google Scholar]
- 21.Li M., Wilson D.M. Human apurinic/apyrimidinic endonuclease 1. Antioxid. Redox Signal. 2013;20:678–707. doi: 10.1089/ars.2013.5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mohr D., Frey S., Fischer T., Güttler T., Görlich D. Characterisation of the passive permeability barrier of nuclear pore complexes. EMBO J. 2009;28:2541–2553. doi: 10.1038/emboj.2009.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rybenkov V.V., Cozzarelli N.R., Vologodskii A.V. Probability of DNA knotting and the effective diameter of the DNA double helix. Proc. Natl. Acad. Sci. U.S.A. 1993;90:5307–5311. doi: 10.1073/pnas.90.11.5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.